
Abstract
Crohn's disease (CD) is a complex autoimmune disorder presumed to be driven by complex interactions of genetic, immune, microbial and even environmental factors. Intrinsic molecular mechanisms in CD, however, remain poorly understood. The identification of novel biomarkers in CD cases based on larger samples through machine learning approaches may inform the diagnosis and treatment of diseases. A comprehensive analysis was conducted on all CD datasets of Gene Expression Omnibus (GEO); our team then used the robust rank aggregation (RRA) method to identify differentially expressed genes (DEGs) between controls and CD patients. PPI (protein‒protein interaction) network and functional enrichment analyses were performed to investigate the potential functions of the DEGs, with molecular complex detection (MCODE) identifying some important functional modules from the PPI network. Three machine learning algorithms, support vector machine-recursive feature elimination (SVM-RFE), random forest (RF), and least absolute shrinkage and selection operator (LASSO), were applied to determine characteristic genes, which were verified by ROC curve analysis and immunohistochemistry (IHC) using clinical samples. Univariable and multivariable logistic regression were used to establish a machine learning score for diagnosis. Single-sample GSEA (ssGSEA) was performed to examine the correlation between immune infiltration and biomarkers. In total, 5 datasets met the inclusion criteria: GSE75214, GSE95095, GSE126124, GSE179285, and GSE186582. Based on RRA integrated analysis, 203 significant DEGs were identified (120 upregulated genes and 83 downregulated genes), and MCODE revealed some important functional modules in the PPI network. Machine learning identified LCN2, REG1A, AQP9, CCL2, GIP, PROK2, DEFA5, CXCL9, and NAMPT; AQP9, PROK2, LCN2, and NAMPT were further verified by ROC curves and IHC in the external cohort. The final machine learning score was defined as [Expression level of AQP9 × (2.644)] + [Expression level of LCN2 × (0.958)] + [Expression level of NAMPT × (1.115)]. ssGSEA showed markedly elevated levels of dendritic cells and innate immune cells, such as macrophages and NK cells, in CD, consistent with the gene enrichment results that the DEGs are mainly involved in the IL-17 signaling pathway and humoral immune response. The selected biomarkers analyzed by the RRA method and machine learning are highly reliable. These findings improve our understanding of the molecular mechanisms of CD pathogenesis.
Similar content being viewed by others

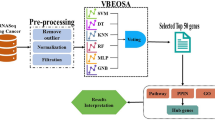

Introduction
Crohn's disease (CD) is a complex genetic disorder likely caused by genetic, microbial, environmental, and immune factors1,2,3, with chronic diarrhea, abdominal pain, and weight loss being among the most common symptoms. Currently, there is no curative medical approach for CD4. Therefore, the importance of understanding the cellular and molecular mechanisms involved in CD pathogenesis, exploring novel intervention targets, and identifying potential biomarkers as diagnostic indicators cannot be overlooked.
Several immune-associated cell types present in the intestinal mucosa are reported to contribute to CD pathophysiology, including dendritic cells5,6,7 and lymphocytes8,9. However, the inherent complexity of CD, as manifested by a widely variable clinical course, makes it difficult to dissect disease mechanisms and to identify biomarkers that play a key role in disease progression. With the development of genomic sequencing technology, an increasing number of microarray datasets have been reported, providing an ideal source to investigate various molecular roles in CD. In recent years, several studies utilizing microarray technology have been published to identify CD biomarkers10,11,12. However, as these studies included relatively small sample sizes or lacked verification in external datasets, representing the molecular characteristics of this complex disease is difficult. The identification of novel biomarkers in CD cases based on larger samples through machine learning approaches may inform the diagnosis and treatment of diseases.
In this study, we constructed a CD cohort with the largest sample size to date through the robust rank aggregation (RRA) method13. A network of protein‒protein interactions (PPIs) was then built, and several functional modules were detected after identifying differentially expressed genes (DEGs). In addition, three machine learning algorithms, namely, support vector machine-recursive feature elimination (SVM-RFE), random forest (RF), and least absolute shrinkage and selection operator (LASSO), were applied to determine characteristic genes among multiple CD cohorts. We further illustrate the immune molecule-related functions in CD. This work reveals the key role of different immune molecules in the occurrence and development of CD.
Materials and methods
Search strategy for CD microarray datasets
A total of 73 datasets were collected from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) by systematic retrieval using the following keywords: ("Crohn Disease"[MeSH Terms] OR Crohn Disease [All Fields]) AND "Homo sapiens"[porgn] AND ("Expression profiling by array"[Filter] AND (“2012/01/01”[PDAT]: “2022/01/01”[PDAT])). Inclusion criteria were (1) sample size > 50, (2) both cases and controls included, (3) "ileum/colon" as sample source, and (4) available gene annotation information (listed in Fig. 1).

Flowchart of the overall study design.
Identification of DEGs in each CD dataset
The gene expression profiles of all datasets in the final analysis were downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). For multiple probes targeting the same gene, the scaled expression values were averaged. The matrix file was extracted using PERL software (PERL version 5.6.1), and quantile normalization was performed using the normalizeBetweenArrays function14 in R software (R version 4.2.0). Due to the large fluctuations of the values in GSE179285, all values in these data were logarithmically transformed (e.g., "a" to "log(a + 1)") to make them conform to the requirements of a normal distribution and facilitate downstream data analysis. LogFC (log fold change) > 0.7 and adjusted P < 0.05 were set as the criteria for identifying DEGs.
RRA analysis and identification of DEGs in the integrated cohort
Using RRA, all genes were sorted for each dataset and ranked based on their logFC with the limma package. The DEGs were then ranked using the ranked list and aggregated using the "RobustRankAggreg" package13 of R software. In this method, an adjusted P value determines the likelihood that DEGs will be identified in datasets with highly ranked genes. LogFC (log fold change) > 0.7 and adjusted P < 0.05 were set as the criteria for identifying DEGs.
Functional and pathway enrichment analyses
We performed Gene Ontology biological process (GO-BP) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis15,16,17 on the DEGs identified by RRA using the limma and clusterProfiler packages. For enrichment analysis of DEGs, adjusted P < 0.05 was used18.
PPI (protein‒protein interactions) network analysis
For the DEGs obtained by RRA analysis, a PPI network was constructed using the STRING website (https://cn.string-db.org/) with a parameter of confidence > 0.4. Visualization of the PPI network was performed by Cytoscape (Cytoscape version 3.7.2), and molecular complex detection (MCODE) (a plug-in in Cytoscape) was used to identify functional modules19.
Analysis of CD- and immune-associated genes
A list of 2483 immune-associated genes was obtained from the Immunology Database and Analysis Portal (https://www.immport.org/shared/genelists). Candidate genes were determined by the intersection of DEGs and immune-related genes, followed by the elimination of gene symbols that do not exist in the five RRA analysis-associated datasets.
Feature selection of characteristic biomarkers via three machine learning methods
We used LASSO20, SVM-RFE21, and RF22 to perform feature selection for diagnostic biomarkers for CD. The LASSO algorithm was applied with a turning/penalty parameter using tenfold cross-validation via the glmnet package23. With SVM-RFE, relevant characteristics are selected, and redundant characteristics are removed more effectively than with linear discriminant analysis or the mean squared error method. By using tenfold cross-validation, SVM-RFE was applied for feature selection, and the top 10 genes were determined as characteristic genes24. The RF algorithm is a randomization algorithm to reduce overfitting of a single decision tree and promote model accuracy based on numerous relevant decision trees from one training set; the top 10 genes were determined as the characteristic genes25.
For every dataset included in RRA, the genes obtained from the intersection of genes selected by the three methods were identified as characteristic genes. The area under the receiver operating characteristic (ROC) curve (AUC) was used to estimate diagnostic efficacy.
Batch correction, data merging and PCA dimensionality reduction
With the combat function by the sva package, we reduced the batch differences and merged 3 cohorts: GSE75214 (GPL6244, Affymetrix), GSE126124 (GPL6244, Affymetrix), and GSE186582 (GPL570, Affymetrix) from the same company platform. The dataset was named the combined dataset. A principal component analysis (PCA) was performed to evaluate the magnitude of batch differences before and after correction.
Construction of the diagnostic machine learning score
Univariate logistic regression analysis was performed to identify diagnostic genes in combined dataset patients (p < 0.05). The identified genes were further included in a multivariate logistic regression analysis to construct a potential machine learning score in CD. Finally, a formula for the risk score was established, and we calculated the risk score of each case as follows:
Coefi indicates the correlation coefficient of each gene, and X indicates the level of gene expression.
Landscape of immune cell infiltration
To combine the datasets, the limma package and the combat function of the sva package26 were applied to preprocess and remove the batch effects of these three datasets. Based on the expression profiles of 29 immunity-relevant signatures, the single-sample gene set enrichment analysis (ssGSEA) method was utilized to determine the degree of immune cell infiltration.
Gene set enrichment analysis
GSEA was performed on characteristic genes to elucidate their biological significance27. To achieve a normalized enrichment score for each analysis, 1000 gene set permutations were conducted. A false discovery rate (FDR) < 0.05 was regarded as significant enrichment to identify significant KEGG pathways.
Verification of CD-associated characteristic genes by immunohistochemical (IHC) staining
To validate the results of genetic analysis at the transcriptional level, 6 patients with chronic colitis or CD were consecutively recruited between March and May 2021 in the Department of Gastroenterology at the Second Hospital of Hebei Medical University. Written informed consent was obtained from all individuals. In addition, the study was approved by the Ethics Committee of Second Hospital of Hebei Medical University. All research was performed in accordance with relevant guidelines/regulations. A total of 3 chronic colitis samples and 3 CD samples from human intestinal mucosal tissues were collected to perform histopathologic diagnosis by two pathologists.
The collected intestinal mucosa samples were fixed with 4% PFA and embedded in paraffin. IHC staining was performed as previously described28. Antibodies against the following were used: AQP9 (A8540; 1:200 dilution) and PROK2 (A6705; 1:200 dilution). HRP-labeled goat anti-rabbit antibody (AS014; 1:200 dilution, all from ABclonal, Wuhan, China) was used as the secondary antibody.
Statistical analysis
All statistical tests were implemented using R software 4.1.3. The Wilcoxon test was applied to analyze the significant difference between two groups, and Spearman's correlation test was used to determine the correlation between the variables. A statistically significant P value was regarded as 0.05.
Results
Characteristics of the microarrays included in RRA analysis
The flow diagram of the CD dataset search strategy and inclusion and exclusion criteria is described in Fig. 1. According to the criteria described in the methods, a total of five datasets were included in further analysis: GSE7521429, GSE95095, GSE12612430, GSE17928531, and GSE18658232. Among these five datasets, 671 CD cases (including 308 inflamed samples of CD) were enrolled in the CD group, and 109 were enrolled in the control group. An overview of the microarray datasets included in the study is shown in Table 1.
RRA integrated analysis
Before RRA analysis, all included cohorts were standardized to reduce batch differences among multicenter data (displayed in Supplementary Fig. 1). DEGs in each dataset were identified; volcano maps are illustrated in Fig. 2A–E.

Identification of DEGs from a single dataset and RRA analysis. Volcano plots of DEG distributions in GSE75214 (A), GSE95095 (B), GSE126124 (C), GSE179285 (D), and GSE186582 (E). Upregulated genes are marked by red points, and downregulated genes are marked by green points; genes with no significant differences are marked by black points. (F) Heatmap of the top 15 DEGs (upregulated or downregulated) identified in RRA.
After the RRA method, a total of 203 DEGs (83 downregulated and 120 upregulated) were identified. A heatmap including the top 15 DEGs (upregulated or downregulated) is illustrated in Fig. 2F. According to the analysis, the top 10 significant genes expressed in CD included five upregulated genes [REG1B (P = 2.76E−11), REG1A (P = 7.43E−10), MMP1 (P = 3.60E−09), S100A8 (P = 3.39E−08), and LCN2 (P = 7.78E−08)] and five downregulated genes [HMGCS2 (P = 3.60E−08), AQP8 (P = 1.55E−07), SLC16A9 (P = 1.55E−07), TRPM6 (P = 8.21E−07), and CA1 (P = 9.62E−07)]. Interestingly, the top 10 significant genes were somewhat similar to another RRA analysis from our previous study in ulcerative colitis (UC) (five upregulated genes: DUOX2, SLC6A14, MMP3, REG1A, and REG1B; and five downregulated genes: AQP8, HMGCS2, PCK1, SLC26A2, and ABCG2), indicating high homogeneity in clinical inflammatory bowel disease (IBD)33. Supplementary Table 1 lists the overall RRA results.
DEG-based functional enrichment analysis
The DEGs, including 120 upregulated and 83 downregulated genes, were subjected to GO-BP analysis and KEGG analysis, and the top five results are listed in Fig. 3A,B. The results showed humoral immune response, antimicrobial humoral immune response mediated by antimicrobial peptide, leukocyte migration, antimicrobial humoral response, and leukocyte chemotaxis to be the top five enriched BPs. The IL-17 signaling pathway, the TNF signaling pathway, viral protein interaction with cytokine and cytokine receptor, rheumatoid arthritis, and Staphylococcus aureus infection were found to be the top five enriched KEGG pathways (Fig. 3A). The detailed results are listed in Supplementary Table 2. Summarizing the results of our gene enrichment analysis and those in similar studies, we found that the results of KEGG analysis were similar to those of previously reported articles, i.e., IL-17 and TNF-α were significantly enriched in CD11,34; however, in BP analysis, our study found that a variety of humoral immunities, especially microbial-associated humoral immunity, were significantly enriched in CD, which has not been previously reported in the literature.

Functional enrichment analysis of DEGs. (A) GO-BP analysis and (B) KEGG pathway analysis.
PPI network analysis and identification of characteristic genes
Using the STRING website, a visual network of PPIs based on RRA analysis was constructed, including 203 nodes and 940 edges. The network was then imported into Cytoscape software for further analysis, with upregulated genes shown with orange markers and downregulated genes with blue markers (Fig. 4A). The top two modules with the highest scores were determined by MCODE. Module 1 comprised MMP9, CCL2, SERPINE1, MMP10, PLAU, MMP7, CCL25, MMP12, CD274, CHI3L1, LCN2, SPP1, MMP1, SELE, MMP3, CXCL5, CXCL6, CXCL9, CCL20, CXCL2, CXCL1, CXCL10, IL6, CXCL8, and IL1RN, with the seed gene IL1β (Fig. 4B). Module 2 comprised S100A9, NCF2, FPR1, MNDA, AQP9, S100A12, VNN2, and S100A8, with the seed gene CSF3R (Fig. 4C). The detailed scores of each module are shown in Supplementary Table 3.

Visualization and module identification of the PPI network. (A) A total of 203 DEGs were mapped using Cytoscape software. Two PPI network modules were identified using the MCODE plug-in. (B) Module 1 comprised MMP9, CCL2, SERPINE1, MMP10, PLAU, MMP7, CCL25, MMP12, CD274, CHI3L1, LCN2, SPP1, MMP1, SELE, MMP3, CXCL5, CXCL6, CXCL9, CCL20, CXCL2, CXCL1, CXCL10, IL6, CXCL8, and IL1RN, with the seed gene IL1β. (C) Module 2 comprised S100A9, NCF2, FPR1, MNDA, AQP9, S100A12, VNN2, and S100A8, with the seed gene CSF3R. In functional enrichment analyses of the genes in module 1 (D) and module 2 (E), the red points represent upregulated genes, and the blue points represent downregulated genes.
Based on GO-BP enrichment analysis of module 1, the genes are mainly involved in cellular response to lipopolysaccharide, myeloid leukocyte migration, and cellular response to molecules of bacterial origin. KEGG analysis showed that these genes play a major role in the IL-17 signaling pathway, the TNF signaling pathway, and rheumatoid arthritis (Fig. 4D).
GO enrichment analysis of module 2 revealed the DEGs to be mainly related to neutrophil chemotaxis, neutrophil migration, and granulocyte chemotaxis. KEGG analysis revealed that these genes are mainly involved in the IL-17 signaling pathway (Fig. 4E).
Determination of characteristic genes
Considering that the immune response is an important factor leading to the occurrence, progression and prognosis of CD, we next extracted immune-related genes among the DEGs, eliminated those not present in the five datasets, and obtained a total of 46 candidate immune genes (Supplementary Table 4).
In the following investigation, three different machine learning methods (LASSO, SVM-RFE and RF) were employed for feature selection and to determine characteristic genes in each dataset. As described above, when a gene was selected by the three methods at the same time, the gene was identified as a characteristic gene in each dataset.
The results were as follows. For GSE75214, CXCL1, STAT1, CXCL6, AQP9, LCN2, REG1A, GIP, and VIPR1 were selected by LASSO; LCN2, MMP12, VIPR1, REG1A, CCL20, FPR2, AQP9, IL1β, PYY, and CXCL2 were selected by SVM-RFE; and NOS2, IDO1, DMBT1, STAT1, AQP9, CXCL1, REG1A, LCN2, SOCS3, and CXCL6 were selected by RF. For GSE95095, PYY, SOCS3, CXCL1, SPP1, PROK2, DEFA5, CXCL10, LCN2, REG1A, PLAU, GIP, CCL2, and VIPR1 were selected by LASSO; CCL2, LCN2, VIPR1, PROK2, DEFA5, PYY, DES, CXCL6, GIP, and CXCL2 were selected by SVM-RFE; and CCL25, S100A12, DEFA5, SOCS3, SERPINA3, PROK2, GIP, CCL2, CXCL6, and PLAU were selected by RF. For GSE126124, STAT1, PROK2, LCN2, REG1A, CXCL11, and CXCL9 were selected by LASSO; IDO1, MMP12, PROK2, GREM1, CXCL5, AQP9, CXCL9, S100A8, LCN2, and CHP2 were selected by SVM-RFE; and AQP9, CXCL6, S100A8, IDO1, CSF3R, CXCL11, CXCL9, LCN2, CXCL1, and CXCL5 were selected by RF. For GSE179285, FPR1, SPP1, NOS2, CXCL10, GREM1, CHP2, LCN2, GIP, DES, CCL20, DMBT1, and VIPR1 were selected by LASSO; PROK2, CXCL10, LCN2, VIPR1, SPP1, FPR1, CSF3R, SERPINA3, S100A9, and DES were selected by SVM-RFE; and DEFA5, DEFA6, REG1A, LCN2, PROK2, CXCL1, SERPINA3, DUOX2, NOS2, and S100A8 were selected by RF. For GSE186582, PYY, CSF3R, SPP1, STAT1, NOS2, GREM1, LCN2, NAMPT, REG1A, GIP, DES, CCL2, and MMP9 were selected by LASSO; NAMPT, GIP, STAT1, LCN2, PROK2, SPP1, CXCL1, GZMB, PLAU, and PYY were selected by SVM-RFE; and NAMPT, GIP, STAT1, LCN2, PROK2, SPP1, CXCL1, GZMB, PLAU, and PYY were selected by RF. These results are detailed in Supplementary Table 5.
Characteristic genes were finally obtained by taking the intersection of the results of the three feature selection methods, as depicted in Fig. 5. The selected characteristic genes were LCN2, REG1A, and AQP9 for GSE75214; CCL2, GIP, PROK2, and DEFA5 for GSE95095; LCN2 and CXCL9 for GSE126124; LCN2 for GSE179285; and GIP and NAMPT for GSE186582.

Determination of characteristic genes in GSE75214 (A), GSE95095 (B), GSE126124 (C), GSE179285 (D), and GSE186582 (E).
Verification of characteristic genes in RRA datasets and clinical samples
To further verify the value of the characteristic genes as diagnostic markers, we explored ROC curves in different cohorts included in the RRA analysis (Fig. 6). The results showed that the AUCs of AQP9 and PROK2 were greater than 0.75 and those of LCN2 and NAMPT were greater than 0.70 in all five cohorts included in the RRA. Therefore, these genes have good application value as biomarkers in CD diagnosis and treatment.

Diagnostic efficacy of characteristic genes in the prediction of CD progression and external verification of the expression of characteristic genes. ROC curves estimating the diagnostic performance of characteristic genes, including LCN2, REG1A, AQP9, CCL2, GIP, PROK2, DEFA5, CXCL9, and NAMPT, in the identification of CD patients in the GSE75214 (A), GSE95095 (B), GSE126124 (C), GSE179285 (D), and GSE186582 (E) datasets.
To verify the accuracy of the results, chronic colitis and CD tissue specimens were examined by IHC for the expression levels of AQP9 and PROK2. Representative images of IHC staining are illustrated in Fig. 7A–D. We found that IHC expression of AQP9 and PROK2 in CD tissues was higher than that in chronic colitis tissues.

Validation of the expression of two prognostic genes in tissue samples. The IHC staining figures show representative images of the expression levels of AQP9 (A,B) and PROK2 (C,D) in chronic colitis and CD colon samples. The black arrow indicates the positive area of the IHC test.
Establishment of a machine learning score with 3 screened genes
To better explore the role of the four genes in the diagnosis of CD, we further constructed a machine learning score using machine learning scores for the four genes AQP9, LCN2, NAMPT, and PROK2. First, three datasets from the array platform of Affymetrix: GSE75214 (GPL6244. Affymetrix), GSE126124 (GPL6244, Affymetrix), and GSE186582 (GPL570, Affymetrix) were batch corrected and merged by the Combat function of sva packages, and the merged datasets were named Combined Datasets. We explored the batch differences of the 3 datasets before and after processing by PCA (Fig. 8A,B). The results showed that the batch differences were significantly reduced after Combat function treatment. We then performed univariable logistic regression analysis with the combined datasets as the training set and selected genes with P < 0.05 for multivariable logistic regression analysis (Table 2). The results showed that all four genes had P < 0.05 in the univariable logistic regression, and a total of three of these genes were included in the further multivariable regression analysis. The final machine learning score was defined as [Expression level of AQP9 × (2.644)] + [Expression level of LCN2 × (0.958)] + [Expression level of NAMPT × (1.115)].

Establishment of a machine learning score with 3 screened genes. The GSE75214, GSE126124 and GSE186582 datasets were merged, and batch effects were further removed. PCA plots of different datasets are illustrated before (A) and after (B) batch effects were removed. To investigate the effects of the machine learning score, ROC curves were illustrated in the combined datasets (C) and GSE179285 (D) and GSE95095 (E).
We then performed an ROC curve analysis and compared it with three single gene examinations, AQP9, LCN2, and NAMPT. The results showed an AUC of 0.969 for diagnosis using machine learning scores in the combined datasets (training group) (Fig. 8C), compared to 0.833 in GSE95095 (validation set 1) (Fig. 8D) and 0.838 in GSE179285 (validation set 2) (Fig. 8E). The diagnostic value of the scores was compared with other single-gene AQP9, LCN2, and NAMPT3, and the results showed good robustness of the machine learning scores across cohorts in multiple centers.
Signaling pathways involving characteristic genes in the combined cohort
The signaling pathways associated with the selected characteristic genes were evaluated using GSEA in the combined cohort (Fig. 9A–D). Our analysis indicated that AQP9, PROK2, LCN2, and NAMPT were positively linked to the IL-17/IL-17-associated signaling pathway (rheumatoid arthritis, systemic lupus erythematosus, and type 1 diabetes mellitus), immune-related disorders (allograft rejection and graft-versus-host disease) or infection (leishmaniasis and malaria)8.

GSEA identifies signaling pathways involving characteristic genes. The main signaling pathways significantly enriched in highly expressed characteristic genes (A) AQP9, (B) LCN2, (C) PROK2, and (D) NAMPT.
Analysis of correlation between related biomarkers and infiltrating immune cells
By performing ssGSEA, we compared the infiltration levels of most immune cell populations between CD and normal samples in the combined cohort. The results showed several immune cell types, including dendritic cells, macrophages, regulatory T cells and NK cells, to be more abundant in CD patients than in controls in most of the datasets included in the RRA analysis (Fig. 10A). Further analysis showed that AQP9, PROK2, LCN2, and NAMPT correlated mainly positively with the differentially expressed cell types, indicating their value as biomarkers in CD (Fig. 10B).

Immune cell infiltration analysis. (A) Level of immune infiltration between CD and normal samples. (B) Correlation heatmap showing the correlation between 23 kinds of infiltrating immune cells and 4 characteristic genes. Red denotes a positive correlation, blue denotes a negative correlation, and the size of the colored square denotes the correlation intensity.
Discussion
CD is a chronic and relapsing inflammatory condition of the gastrointestinal tract that occurs following immune system dysregulation35,36. In this study, we integrated multiple datasets by RRA analysis to thoroughly identify DEGs and several immune-related genes via three machine learning methods. Currently, the pathogenesis of CD is still unclear, and relapse easily occurs after treatment37. Therefore, identifying novel intervention targets and investigating potential biomarkers as diagnostic indicators are essential to clarifying the cellular and molecular mechanisms contributing to CD pathogenesis38.
Our RRA analysis was conducted using five CD cohorts, identifying 203 DEGs. To the best of our knowledge, this study involves one of the largest sample sizes in current research on CD, including 671 cases of CD and 109 controls. The advantage of RRA analysis is that it can integrate different sequencing platforms and multicenter studies and effectively reduce batch differences to better identify DEGs13. In this study, most of the datasets published to date were retrieved and reviewed, and the retrieval strategies of the included and excluded datasets were clearly defined, which improves the credibility of the conclusions.
Analysis of PPI networks was performed for all DEGs, and MCODE was used to identify key functional gene modules39. Functional enrichment analysis revealed that the IL-17 signaling pathway plays an important role in the progression of CD for both DEGs and the top two modules with the highest score. IL-17 is the key cytokine produced by Th17 cells and has versatile functions8. Although Th1 and Th2 cells were once considered to be the only T-cell lineages resulting from progenitor CD4+ helper T cells, the discovery of Th17 cells as a distinct lineage of CD4+ helper T cells has changed our understanding of chronic inflammatory diseases such as CD and indicated a new way in which immune responses trigger intestinal tissue damage8,40,41. Studies have reported the role of IL-17 in a group of seemingly unrelated diseases that are triggered by or result from dysregulation of the innate and adaptive immune systems, including CD, UC, psoriasis42,43, rheumatoid arthritis44,45,46,47,48, and systemic lupus erythematosus49,50,51, among others. Interestingly, although overexpression of IL-17 in CD has been reported, its exact role in IBD is still controversial8,40,52,53. Nevertheless, similar to our results, high IL-17 mRNA expression levels have been detected in intestinal mucosa samples of patients with active CD as well as those with UC54,55. Mice deficient in IL-17 or treated with anti-IL-17 exhibit severe epithelial damage in the colon, indicating that IL-17 acts as a protective factor56,57. KEGG analysis also highlighted the role of the TNF pathway in CD. In the clinic, anti-TNF therapy was approved for use in CD in 1998, leading to increased rates of response and remission58. Our GO-BP analysis showed the DEGs and two modules to be mainly involved in inflammation. Notably, gene enrichment analysis showed the DEGs to mainly participate in the humoral immune response/antimicrobial humoral immune response. A widely accepted theory about the etiopathogenesis of CD is that the disorder is caused by an aggressive immune response to microorganisms of the intestinal microbiota in genetically predisposed individuals59. Immunoglobulin A (IgA) and immunoglobulin M (IgM) antibodies generated by the immune system are essential for maintaining mutualism between our bodies and the microbes that colonize our mucosal surfaces60. In addition, although intestinal IgG responses are limited in the healthy adult gut, several studies have demonstrated the importance of B cells and IgG in the pathogen containment and elimination effects that dominate those of IgA and IgM61,62,63,64,65,66.
We selected immune-related genes from among 203 DEGs. Traditionally, the multifactor logistic regression model is utilized for model building, the premise of which is that all included factors are independent of each other. However, this is certainly difficult to achieve for high-dimensional data such as array datasets, which may lead to overfitting of the model due to serious gene multicollinearity, resulting in reduced generalization ability of the model. Therefore, three machine learning methods, namely, LASSO, SVM-RFE, and RF, were applied to select characteristic genes, and the intersecting genes were further examined by ROC curve and IHC analyses. Four characteristic genes, AQP9, PROK2, LCN2, and NAMPT, exhibited superior diagnostic value in multiple cohorts.
Aquaporin 9 (AQP9) belongs to the aquaporin family of water-selective membrane channels that play a role in specialized leukocyte functions such as immunological response and bactericidal activity. Several studies have reported that AQP9 is a promising biomarker in CD patients12,67,68. Interestingly, AQP9 has also been reported as a prognostic indicator of many cancers in recent years and is related to immune infiltration69,70,71,72. Similar to CD, AQP9 expression is significantly increased in colon cancer69. In view of the correlation between long-term chronic IBD and AQP9, determining whether AQP9 plays a role in the transformation of IBD into cancer is a very valuable research direction. Although the mechanism of AQP9 in CD is not clear at present, it has been reported that AQP9 is required for inflammatory responses and DC maturation and that its expression level is markedly elevated by LPS exposure73. The inflammatory cytokine PROK2 (prokineticin 2) is produced primarily by macrophages and neutrophils invading sites of tissue damage, and increased levels of PROK2 have been reported in gut inflammation74,75. It remains to be determined exactly how elevated PROK2 causes visceral nociception. Several reports have shown that PROK2 released by inflammatory cells may cause inflammatory pain by attracting monocytes and macrophages as well as by stimulating the secretion of inflammatory and analgesic cytokines76,77,78,79,80,81.
LCN2 (lipocalin 2) belongs to the lipocalin family. LCN2 has been reported as a biomarker of IBD not only in the intestinal mucosa but also in feces82,83,84. In our research, LCN2 was selected using multiple CD samples in datasets including GSE75214, GSE126124 and GSE179285. Mechanistically, LCN2 is produced by a variety of cell types, including myeloid and intestinal epithelial cells, which seem to be particularly important in IBD. In IBD remission, persistent mucosal overexpression of LCN2 makes it a promising candidate for molecular inflammation that warrants investigation85. NAMPT (nicotinamide phosphoribosyltransferase) belongs to the nicotinic acid phosphoribosyltransferase (NAPRTase) family, and by catalyzing the rate-limiting step of NAD salvage, it is critical for maintenance of the cellular nicotinamide adenine dinucleotide (NAD) supply86. Considering that NAD is a major coenzyme in bioenergetic processes, NAMPT is biologically indispensable, and it has been implicated in a variety of inflammatory disorders, such as tumorigenesis, diabetes, rheumatoid arthritis and sepsis87,88,89,90. Moreover, NAMPT overexpression has been identified as a marker of severity in pediatric IBD91,92. A small molecule inhibitor, FK866, inhibits NAMPT enzymatic activity with little toxicity, making it a potentially useful drug for various inflammatory conditions89,93,94.
There are also some deficiencies in this study. (1) Although RRA analysis reduces batch differences in different study combinations, the inclusion criteria for cases, sample size and treatment received by the patients in these studies differ, which introduces bias in the final results. (2) Due to differences in gene probes between different technical platforms, some key genes may not be detected in a cohort, which results in a significant reduction in the number of candidate genes for analysis in the combined cohort, thus omitting some important biomarkers. (3) Larger-scale experimental validation is needed to prove the clinical value of these markers.
Conclusion
In conclusion, our analysis reveals putative key biomarkers in CD, i.e., AQP9, PROK2, LCN2, and NAMPT. ssGSEA showed obviously elevated levels of DCs and innate immune cells, such as macrophages and NK cells, in CD, consistent with the gene enrichment results that the DEGs are mainly involved in the IL-17 signaling pathway and humoral immune response. Importantly, the identified biomarkers were validated by multiple external datasets and by IHC in independent clinical samples. Finally, the identified biomarkers correlate with elevated immune cell types, representing key features of the immune response, which might—in addition to serving as biomarkers for diagnostic purposes—prove to be efficient indicators of disease risk or improvement.
Data availability
The datasets analyzed during the current research are all available in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/). The data used to support the findings of this study are available from the corresponding author upon request.
References
Morgan, X. C. et al. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 16, 67. https://doi.org/10.1186/s13059-015-0637-x (2015).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. https://doi.org/10.1038/nature11582 (2012).
Sartor, R. B. & Wu, G. D. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 152, 327–39.e4. https://doi.org/10.1053/j.gastro.2016.10.012 (2017).
Kim, M. J. & Choe, Y. H. Change in the treatment strategy for pediatric Crohn’s disease. Korean J. Pediatr. 53, 830–833. https://doi.org/10.3345/kjp.2010.53.9.830 (2010).
Despalatović, B. R., Babić, M., Bratanić, A., Tonkić, A. & Vilović, K. Difference in presence and number of CD83(+) dendritic cells in patients with ulcerative colitis and Crohn’s disease. Sci. Rep. 10, 10055. https://doi.org/10.1038/s41598-020-67149-5 (2020).
Mitsialis, V. et al. Single-cell analyses of colon and blood reveal distinct immune cell signatures of ulcerative colitis and Crohn’s disease. Gastroenterology 159, 591-608.e10. https://doi.org/10.1053/j.gastro.2020.04.074 (2020).
Aliberti, J. Immunity and tolerance induced by intestinal mucosal dendritic cells. Mediators Inflamm. 2016, 3104727. https://doi.org/10.1155/2016/3104727 (2016).
Bunte, K. & Beikler, T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20143394 (2019).
Jaeger, N. et al. Single-cell analyses of Crohn’s disease tissues reveal intestinal intraepithelial T cells heterogeneity and altered subset distributions. Nat. Commun. 12, 1921. https://doi.org/10.1038/s41467-021-22164-6 (2021).
Chen, Y., Li, H., Feng, Q. & Shen, J. Development and validation of an interleukin-6 nomogram to predict primary non-response to infliximab in Crohn’s disease patients. Front. Pharmacol. 12, 654985. https://doi.org/10.3389/fphar.2021.654985 (2021).
Chen, H. et al. Identification of immune cell landscape and construction of a novel diagnostic nomogram for Crohn’s disease. Front. Genet. 11, 423. https://doi.org/10.3389/fgene.2020.00423 (2020).
Yu, B. et al. Diagnostic and predictive value of immune-related genes in Crohn’s disease. Front. Immunol. 12, 643036. https://doi.org/10.3389/fimmu.2021.643036 (2021).
Kolde, R., Laur, S., Adler, P. & Vilo, J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28, 573–580. https://doi.org/10.1093/bioinformatics/btr709 (2012).
Liu, R. et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 43, e97 (2015).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951 (2019).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16(5), 284–287 (2012).
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L. & Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 27(3), 431–432 (2011).
Alhamzawi, R. & Ali, H. The Bayesian adaptive lasso regression. Math. Biosci. 303, 75–82. https://doi.org/10.1016/j.mbs.2018.06.004 (2018).
Lin, X. et al. A support vector machine-recursive feature elimination feature selection method based on artificial contrast variables and mutual information. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 910, 149–155. https://doi.org/10.1016/j.jchromb.2012.05.020 (2012).
Alakwaa, F. M., Chaudhary, K. & Garmire, L. X. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J. Proteome Res. 17, 337–347. https://doi.org/10.1021/acs.jproteome.7b00595 (2018).
Engebretsen, S. & Bohlin, J. Statistical predictions with glmnet. Clin. Epigenet. 11(1), 123 (2019).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: Selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19, 432. https://doi.org/10.1186/s12859-018-2451-4 (2018).
Segal, M. R., Dahlquist, K. D. & Conklin, B. R. Regression approaches for microarray data analysis. J. Comput. Biol. 10, 961–980. https://doi.org/10.1089/106652703322756177 (2003).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 102(43), 15545–15550 (2005).
Zhang, H. et al. The molecular feature of macrophages in tumor immune microenvironment of glioma patients. Comput. Struct. Biotechnol. J. 19, 4603–4618. https://doi.org/10.1016/j.csbj.2021.08.019 (2021).
Vancamelbeke, M. et al. Genetic and transcriptomic bases of intestinal epithelial barrier dysfunction in inflammatory bowel disease. Inflamm. Bowel Dis. 23(10), 1718–1729 (2017).
Palmer, N. P. et al. Concordance between gene expression in peripheral whole blood and colonic tissue in children with inflammatory bowel disease. PLoS ONE 14(10), e0222952 (2019).
Keir, M. E. et al. Regulation and role of αe integrin and gut homing integrins in migration and retention of intestinal lymphocytes during inflammatory bowel disease. J. Immunol. 207(9), 2245–2254 (2021).
Ngollo, M. et al. Identification of gene expression profiles associated with an increased risk of post-operative recurrence in Crohn’s disease. J. Crohns Colitis. 16, 1269–1280 (2022).
Chen, Z. A. et al. Integrated analysis of multiple microarray studies to identify novel gene signatures in ulcerative colitis. Front. Genet. 12, 697514 (2021).
Sun, Y., Cai, D., Hu, W. & Fang, T. Identifying hub genes and miRNAs in Crohn’s disease by bioinformatics analysis. Front. Genet. 13, 950136. https://doi.org/10.3389/fgene.2022.950136 (2022).
Baumgart, D. C. & Carding, S. R. Inflammatory bowel disease: Cause and immunobiology. Lancet 369(9573), 1627–1640 (2007).
Zundler, S. et al. Immune cell trafficking and retention in inflammatory bowel disease: Mechanistic insights and therapeutic advances. Gut 68(9), 1688–1700 (2019).
Veauthier, B. & Hornecker, J. R. Crohn’s disease: Diagnosis and management. Am. Fam. Physician 98, 661–669 (2018).
Usman, O. L., Muniyandi, R. C., Omar, K. & Mohamad, M. Advance machine learning methods for dyslexia biomarker detection: A review of implementation details and challenges. IEEE Access PP, 1 (2021).
Bader, G. D. & Hogue, C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4, 2 (2003).
Schmitt, H., Neurath, M. F. & Atreya, R. Role of the IL23/IL17 pathway in Crohn’s disease. Front. Immunol. 12, 622934 (2021).
Ashton, J. J. et al. Ileal transcriptomic analysis in paediatric Crohn’s disease reveals IL17- and NOD-signalling expression signatures in treatment-naïve patients and identifies epithelial cells driving differentially expressed genes. J. Crohns Colitis 15(5), 774–786 (2021).
Chhabra, S. et al. Circulating T-helper 17 cells and associated cytokines in psoriasis. Clin. Exp. Dermatol. 41(7), 806–810 (2016).
Mease, P. J. Inhibition of interleukin-17, interleukin-23 and the TH17 cell pathway in the treatment of psoriatic arthritis and psoriasis. Curr. Opin. Rheumatol. 27(2), 127–133 (2015).
Hirota, K. et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J. Exp. Med. 204(12), 2803–2812 (2007).
Lubberts, E. et al. Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm. Res. 51(2), 102–104 (2002).
Nakae, S. et al. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 171(11), 6173–6177 (2003).
Hirota, K. et al. T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J. Exp. Med. 204(1), 41–47 (2007).
Jaller Char, J. J. et al. The role of IL-17 in the human immune system and its blockage as a treatment of rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. J. Drugs Dermatol. 17(5), 539–542 (2018).
Koga, T., Ichinose, K. & Tsokos, G. C. T cells and IL-17 in lupus nephritis. Clin. Immunol. 185, 95–99 (2017).
Apostolidis, S. A., Crispín, J. C. & Tsokos, G. C. IL-17-producing T cells in lupus nephritis. Lupus 20(2), 120–124 (2011).
Kluger, M. A. et al. RORγt expression in T(regs) promotes systemic lupus erythematosus via IL-17 secretion, alteration of T(reg) phenotype and suppression of Th2 responses. Clin. Exp. Immunol. 188(1), 63–78 (2017).
Kobayashi, T. et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 57(12), 1682–1689. https://doi.org/10.1136/gut.2007.135053 (2008).
Siakavellas, S. I. & Bamias, G. Role of the IL-23/IL-17 axis in Crohn’s disease. Discov. Med. 14(77), 253–262 (2012).
Song, L. et al. High intestinal and systemic levels of interleukin-23/T-helper 17 pathway in Chinese patients with inflammatory bowel disease. Mediators Inflamm. 2013, 425915. https://doi.org/10.1155/2013/425915 (2013).
Sugihara, T. et al. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin. Exp. Immunol. 160(3), 386–393. https://doi.org/10.1111/j.1365-2249.2010.04093.x (2010).
Ogawa, A. et al. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 110(1), 55–62. https://doi.org/10.1016/j.clim.2003.09.013 (2004).
Ito, R. et al. Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem. Biophys. Res. Commun. 377(1), 12–16. https://doi.org/10.1016/j.bbrc.2008.09.019 (2008).
Adegbola, S. O. et al. Anti-TNF therapy in Crohn’s disease. Int. J. Mol. Sci. 19(8), 2244 (2018).
Carrière, J., Darfeuille-Michaud, A. & Nguyen, H. T. Infectious etiopathogenesis of Crohn’s disease. World J. Gastroenterol. 20(34), 12102–12117 (2014).
Pabst, O. New concepts in the generation and functions of IgA. Nat. Rev. Immunol. 12(12), 821–832 (2012).
Masuda, A. et al. Fcgamma receptor regulation of Citrobacter rodentium infection. Infect. Immun. 76(4), 1728–1737 (2008).
Maaser, C. et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 72(6), 3315–3324 (2004).
Castro-Dopico, T. & Clatworthy, M. R. Mucosal IgG in inflammatory bowel disease—A question of (sub)class?. Gut Microbes 12(1), 1–9 (2020).
Caballero-Flores, G. et al. Maternal immunization confers protection to the offspring against an attaching and effacing pathogen through delivery of IgG in breast milk. Cell Host Microbe 25(2), 313-323.e4 (2019).
Yoshida, M. et al. Neonatal Fc receptor for IgG regulates mucosal immune responses to luminal bacteria. J. Clin. Investig. 116(8), 2142–2151 (2006).
Kamada, N. et al. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe 17(5), 617–627 (2015).
Taman, H. et al. Transcriptomic landscape of treatment-naïve ulcerative colitis. J. Crohns Colitis 12(3), 327–336. https://doi.org/10.1093/ecco-jcc/jjx139 (2018).
Mesko, B. et al. Peripheral blood gene expression patterns discriminate among chronic inflammatory diseases and healthy controls and identify novel targets. BMC Med. Genomics 3, 15. https://doi.org/10.1186/1755-8794-3-15 (2010).
Liu, X. et al. Integrated analysis identifies AQP9 correlates with immune infiltration and acts as a prognosticator in multiple cancers. Sci. Rep. 10(1), 20795. https://doi.org/10.1038/s41598-020-77657-z (2020).
Huang, D. et al. AQP9-induced cell cycle arrest is associated with RAS activation and improves chemotherapy treatment efficacy in colorectal cancer. Cell Death Dis. 8(6), e2894. https://doi.org/10.1038/cddis.2017.282 (2017).
Jing, J. et al. AQP9 is a prognostic factor for kidney cancer and a promising indicator for M2 TAM polarization and CD8+ T-cell recruitment. Front. Oncol. 11, 770565. https://doi.org/10.3389/fonc.2021.770565 (2021).
Gao, C. et al. Low expression of AQP9 and its value in hepatocellular carcinoma. Transl. Cancer Res. 10(4), 1826–1841. https://doi.org/10.21037/tcr-20-3158 (2021).
De Santis, S. et al. Aquaporin 9 contributes to the maturation process and inflammatory cytokine secretion of murine dendritic cells. Front. Immunol. 9, 2355. https://doi.org/10.3389/fimmu.2018.02355 (2018).
Li, X. et al. Meta-analysis of expression profiling data indicates need for combinatorial biomarkers in pediatric ulcerative colitis. J. Immunol. Res. 2020, 8279619. https://doi.org/10.1155/2020/8279619 (2020).
Watson, R. P. et al. Increased prokineticin 2 expression in gut inflammation: Role in visceral pain and intestinal ion transport. Neurogastroenterol. Motil 24(1), 65-75.e12. https://doi.org/10.1111/j.1365-2982.2011.01804.x (2012).
Franchi, S. et al. The prokineticin receptor agonist Bv8 decreases IL-10 and IL-4 production in mice splenocytes by activating prokineticin receptor-1. BMC Immunol. 9, 60. https://doi.org/10.1186/1471-2172-9-60 (2008).
LeCouter, J. et al. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc. Natl. Acad. Sci. USA 101(48), 16813–16818. https://doi.org/10.1073/pnas.0407697101 (2004).
Monnier, J. & Samson, M. Cytokine properties of prokineticins. FEBS J. 275(16), 4014–4021. https://doi.org/10.1111/j.1742-4658.2008.06559.x (2008).
Martucci, C. et al. Bv8, the amphibian homologue of the mammalian prokineticins, induces a proinflammatory phenotype of mouse macrophages. Br. J. Pharmacol. 147(2), 225–234. https://doi.org/10.1038/sj.bjp.0706467 (2006).
Giannini, E. et al. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc. Natl. Acad. Sci. USA 106(34), 14646–14651. https://doi.org/10.1073/pnas.0903720106 (2009).
Dorsch, M. et al. PK1/EG-VEGF induces monocyte differentiation and activation. J. Leukoc. Biol. 78(2), 426–434. https://doi.org/10.1189/jlb.0205061 (2005).
Bakke, I. et al. Mucosal and faecal neutrophil gelatinase-associated lipocalin as potential biomarkers for collagenous colitis. J. Gastroenterol. 56(10), 914–927. https://doi.org/10.1007/s00535-021-01814-y (2021).
Muthas, D. et al. Neutrophils in ulcerative colitis: A review of selected biomarkers and their potential therapeutic implications. Scand. J. Gastroenterol. 52(2), 125–135. https://doi.org/10.1080/00365521.2016.1235224 (2017).
Thorsvik, S. et al. Fecal neutrophil gelatinase-associated lipocalin as a biomarker for inflammatory bowel disease. J. Gastroenterol. Hepatol. 32(1), 128–135. https://doi.org/10.1111/jgh.13598 (2017).
Zollner, A. et al. Faecal biomarkers in inflammatory bowel diseases: Calprotectin versus lipocalin-2-a comparative study. J. Crohns Colitis 15(1), 43–54. https://doi.org/10.1093/ecco-jcc/jjaa124 (2021).
Gerner, R. R. et al. NAD metabolism fuels human and mouse intestinal inflammation. Gut 67(10), 1813–1823. https://doi.org/10.1136/gutjnl-2017-314241 (2018).
Galli, U. et al. Medicinal chemistry of nicotinamide phosphoribosyltransferase (NAMPT) inhibitors. J. Med. Chem. 56(16), 6279–6296. https://doi.org/10.1021/jm4001049 (2013).
Jia, S. H. et al. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Investig. 113(9), 1318–1327. https://doi.org/10.1172/JCI19930 (2004).
Meier, F. M. et al. Visfatin/pre-B-cell colony-enhancing factor (PBEF), a proinflammatory and cell motility-changing factor in rheumatoid arthritis. J. Biol. Chem. 287(34), 28378–28385. https://doi.org/10.1074/jbc.M111.312884 (2012).
El-Mesallamy, H. O. et al. Vaspin and visfatin/Nampt are interesting interrelated adipokines playing a role in the pathogenesis of type 2 diabetes mellitus. Metabolism 60(1), 63–70. https://doi.org/10.1016/j.metabol.2010.04.008 (2011).
Moschen, A. R. et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 178(3), 1748–1758. https://doi.org/10.4049/jimmunol.178.3.1748 (2007).
Starr, A. E. et al. Proteomic analysis of ascending colon biopsies from a paediatric inflammatory bowel disease inception cohort identifies protein biomarkers that differentiate Crohn’s disease from UC. Gut 66(9), 1573–1583. https://doi.org/10.1136/gutjnl-2015-310705 (2017).
Moschen, A. R. et al. A key role for Pre-B cell colony-enhancing factor in experimental hepatitis. Hepatology 54(2), 675–686. https://doi.org/10.1002/hep.24416 (2011).
Bruzzone, S. et al. Catastrophic NAD+ depletion in activated T lymphocytes through Nampt inhibition reduces demyelination and disability in EAE. PLoS ONE 4(11), e7897. https://doi.org/10.1371/journal.pone.0007897 (2009).
Acknowledgements
This study was funded by the Medical Science Research Projects of Hebei Province (Grant No. 20220990), Hebei Natural Science Foundation (Grant No. H2020206337), Medical Applicable Technology Tracking Project of Hebei Province (Grant No. GZ2022012), and Key Scientific and Technological Research Programs of Hebei Province (Grant No. 20200930, Grant No. 20210531).
Author information
Authors and Affiliations
Contributions
Z.C. and H.M. collected the papers and analyzed data, analyzed the conclusions, and drafted the manuscript. Y.W., H.T. and J.M. reviewed the data and contributed to writing. C.Y. and D.Y. presented the idea of this manuscript, supported the funding, analyzed the conclusions, and drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, ZA., Ma, Hh., Wang, Y. et al. Integrated multiple microarray studies by robust rank aggregation to identify immune-associated biomarkers in Crohn's disease based on three machine learning methods. Sci Rep 13, 2694 (2023). https://doi.org/10.1038/s41598-022-26345-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26345-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
