
Abstract
Infectious diseases are known to cause a wide variety of post-infection complications. However, it’s been challenging to identify which diseases are most associated with a given pathogen infection. Using the recently developed LeMeDISCO approach that predicts comorbid diseases associated with a given set of putative mode of action (MOA) proteins and pathogen-human protein interactomes, we developed PHEVIR, an algorithm which predicts the corresponding human disease comorbidities of 312 viruses and 57 bacteria. These predictions provide an understanding of the molecular bases of complications and means of identifying appropriate drug targets to treat them. As an illustration of its power, PHEVIR is applied to identify putative driver pathogens and corresponding human MOA proteins for Type 2 diabetes, atherosclerosis, Alzheimer’s disease, and inflammatory bowel disease. Additionally, we explore the origins of the oncogenicity/oncolyticity of certain pathogens and the relationship between heart disease and influenza. The full PHEVIR database is available at https://sites.gatech.edu/cssb/phevir/.
Similar content being viewed by others

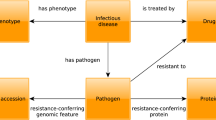

Introduction
Infectious diseases have been a source of widespread, fatal outcomes throughout history1. As typified by COVID-19, pandemics in recent decades have become more frequent and deadly1,2. Yet, host–pathogen interactions are poorly characterized, and how they result in post-infection complications are not well-understood3,4,5,6,7. Host–pathogen interactome data provides an opportunity to assess putative diseases that can be at least partly attributed to a given set of host proteins that interact with a given pathogen. Characterizing such pathogen-disease associations can yield new areas of research and opportunities to develop targeted preventatives and therapeutics to not only treat the pathogenic infection itself but also prevent its potential downstream disease consequences8.
The COVID-19 pandemic has graphically illustrated numerous associations between SARS-CoV-2 and post-infection complications, such as loss of smell or unusual neurological symptoms3,9. Thus, it is obvious that infectious diseases can impact human health well beyond the initial virus infection. Although there is evidence that individuals with some common complex diseases are more susceptible to certain infectious diseases10, the contrary hasn’t been widely explored. That is, there has been limited research undergone to assess the association between viral or bacterial infections and the subsequent development of common complex diseases such as Type 2 diabetes, atherosclerosis, Alzheimer’s disease, and inflammatory bowel disease (IBD). For example, some cases of Alzheimer’s disease could be seeded by pathogen infection11. Furthermore, little is known about the post-infection complications associated with prevalent infectious diseases such as Influenza A and B, E. coli, Herpes simplex 1 and 2, salmonella, Epstein-Barr Virus (EBV) and clostridium difficile. For example, EBV infection increases the risk of developing autoimmune diseases such as IBD, Type 1 diabetes, and celiac disease12. More generally, perhaps pathogen infections play a greater role in causing complex human diseases than was previously appreciated.
Research has provided significant evidence that viruses and bacteria have oncogenic (cancer causing) and oncolytic (cancer treating) potential13,14. Indeed, eleven pathogens are now classified as carcinogenic according to the International Agency for Research on Cancer (IARC)14,15. Currently, approximately 12% of cancers have a known oncovirus association14. Both DNA and RNA viruses can contribute to cancer. For example, Epstein-Barr virus, a dsDNA virus, and human T-cell lymphotropic virus-1 (HTLV-1), an ssRNA-RT virus, are both implicated in some cancers. Some strains of human papillomavirus (HPV) cause cervical cancer16. There has also been speculation that SARS-CoV-2 might be an oncovirus17. Other pathogens could help in treating rather than causing cancers13. For examples, H5N1 influenza can induce cellular apoptosis18; measles viruses are oncolytic19, and herpes simplex virus 1(HSV-1) kills cancer cells20. However, despite such clear associations, it is unknown whether other pathogens also have a significant oncogenic/oncolytic potential. The problem is that oncogenic viruses might not give rise to cancers until a decade or longer following initial infection. As such, establishing the clinical connection between viral infection and the subsequent development of cancer is challenging.
To enhance the understanding of the mode of action (MOA) proteins driving the down-stream consequences associated with pathogen infection, we have developed the PHEVIR algorithm: disease comorbidities Predicted using Human–pathogEn interactomes for VIRulence. Here, we employ LeMeDISCO21, a recently developed tool that predicts on a proteomic scale human disease comorbidities, comorbidity enriched human MOA proteins and pathways given a pathogenic gene-human interactome set. At present, the pathogen-human interactome is provided by the HPIDB database22 but in practice any set of human–pathogen protein–protein interactions may be used. This work exploits the proteomic scale prediction of human disease MOA proteins for diseases identified by the artificial intelligence (AI) based method MEDICASCY23. The results of this analysis for 312 viruses and 57 bacteria are compiled in the PHEVIRdb whose goal is to guide and encourage research on human diseases that may be at least partly driven by pathogen infection. It is possible that it may take years post-infection for such complications to occur, or on the contrary, the predicted comorbidities explain how a preexisting disease might make one more susceptible to the particular comorbid infectious disease.
Results
Overview
The PHEVIR algorithm works as follows: Previously we employed the LeMeDISCO21 algorithm to predict disease co-morbidities based on a common set of mode of action (MOA) proteins. We assert that if a viral or bacterial protein interacts with these MOA proteins, it helps cause the corresponding comorbid diseases. The precision and recall rate of LeMeDISCO co-morbidity prediction on a large set of clinical observation data (~ 200,000 pairs of diseases) are 77.2 and 37.1%, respectively. On a variety of consensus datasets, in comparison to other molecular methods24,25, LeMeDISCO has an order of magnitude larger recall rate with similar precision21. For pathogen-cancer associated (either oncogenic or oncolytic) virus prediction, on a set of 13 viruses including 9 known oncogenic viruses, the recall rate is 66.7% with a precision of 100%26. We then examine the overall network of pathogen-diseases and focused our analysis on penetrant disease groups. Subsequently, pathogen-cancer and heart disease-flu relationships were examined in detail. For many of our predictions, we found literature evidence to support the predictions.
Bacterial and viral induced human disease networks
A total of 39,393 significant pathogen-disease connections were identified (q-value < 0.05), of which 32,694 were virus—disease connections and 6699 were bacteria—disease connections. Of 3608 human diseases that might partially arise due to pathogen infections, 3285 unique diseases have at least one strong viral comorbidity. Similarly, 2405 unique diseases have at least one significant bacterial comorbidity. The top five viruses most connected to human diseases were Molluscum contagiosum virus subtype 1 that is comorbid to 1381 human diseases, Influenza A virus (strain A/Puerto Rico/8/1934 H1N1) that is comorbid to 1183 human diseases, Rubella virus (strain RA27/3 vaccine) that is comorbid to 1183 human diseases, Influenza A virus (strain A/Wilson-Smith/1933 H1N1) that is comorbid to 1137 human diseases, and Human immunodeficiency virus type 1 group M subtype B (isolate HXB2) that is comorbid to 1137 human diseases. The top five most connected bacteria in the network were Helicobacter pylori (strain ATCC 700392/26695) that is comorbid to 1080 human diseases, Yersinia pestis that is comorbid to 855 human diseases, Staphylococcus aureus that is comorbid to 578 human diseases, Streptococcus pyogenes serotype M1 that is comorbid to 428 human diseases, and Mycoplasma pneumoniae strain ATCC 29342/M129) that is comorbid to 406 human diseases. These results indicate that some pathogens are associated with up to one third of the diseases considered.
Penetrant disease groups across pathogens
Tables 1 and 2 provide the numbers of comorbidities and their fractions in total pathogen-comorbidities for each ICD-10 main classification. The top three disease groups/classes with the greatest numbers of comorbid diseases to viruses were Neoplasms; Diseases of the eye and adnexa; and Diseases of the nervous system. The top three disease groups with the greatest numbers of comorbid diseases to bacteria were Certain infectious and parasitic diseases, Neoplasms and Diseases of the eye and adnexa. Apparently, the group of Certain infectious and parasitic diseases has the largest overall relative risk (RR, see Eq. 1 for definition) (1.22 with a p value of 6.46e−04 for viruses and 2.94 with a p value of 1.41e−113 for bacteria) that is consistent with its definition. The other frequent human diseases that are comorbid to both viruses and bacteria were Neoplasms. Both have a RR greater than 1 (RR = 1.54 with a p value of 1.90e−32 for viruses, and RR = 1.25 with a p value of 1.10e−06 for bacteria) compared to background causes of the diseases (see Tables 1 and 2). This association will be further addressed below. Another predicted high frequent comorbid disease group are Diseases of the eye and adnexa. However, it is not significantly risker than the background (RR = 1.001 with a p value of 0.98 for viruses and 1.06 with a p value of 0.33 for bacteria). Viral infections may cause irreversible neurological damage (RR = 1.12, p value = 0.06 for Diseases of the nervous system), possibly due to some of them being able to penetrate the blood brain barrier following an increased inflammatory response. This may lead to oxidative stress and dysregulation in producing sufficient energy27,28. It has also been speculated that viruses might contribute to or cause autoimmune diseases. Such viruses include Influenza A virus, Coxsackie B virus, rotavirus and herpes viruses29. Thus, assessing viral-induced autoimmunity is critical to preventing post-infection downstream complications.
Common complex diseases and pathogens
Common complex diseases are diseases that are penetrant in the population and typically follow non-Mendelian patterns. They typically arise from a series of genetic and environmental factors that perhaps include infectious diseases. Four common complex diseases, Type 2 diabetes (T2D), Atherosclerosis, Inflammatory bowel disease (IBD) and Alzheimer’s diseases and their associations to infectious diseases were assessed. Table 3 demonstrates the top 10 pathogens for each disease. The complete lists including the MOA human proteins of all pathogens for these four diseases can be found in Supplementary Materials, Table S1.1–S1.4.
Insulin resistance, a major characteristic of Type 2 diabetes, may be the consequence of frequent bouts of pathogen exposure and mild inflammatory response30. 6 significant viruses were predicted to be associated with Type 2 diabetes (q-value < 0.05) (see Table S1.1). Human immunodeficiency virus type 1 (HIV) is predicted to be the most significant virus associated with T2D. HIV-infected adults have a 3.8% higher incidence of diabetes mellitus than the general adult population31. Of the viruses associated with Type 2 diabetes, Epstein-Barr virus (strain AG876) (EBV) is prominent. The literature supports a link between Type 1 diabetes and EBV32. To identify the possible MOA proteins of EBV’s association with Type 2 diabetes, we examine the shared proteins of the EBV interactome with the MOA proteins of Type 2 diabetes predicted by MEDICASCY23. A total of 11 proteins are shared between them. MGST1 is associated with tissue damage that are part of diabetes33. All 6 viruses interact with CTSB. CTSB was found to contribute to Autophagy-related 7 (Atg7)-induced inflammatory response resulting in aggravation of lipotoxicity34 and increased T2D risk35.
Atherosclerosis is characterized by the formation of cholesterol plaque(s) in the walls of the arteries. 5 significant viruses and 1 significant bacteria are predicted to be associated with Atherosclerosis (see Table S1.2). The most significant is helicobacter pylori. Interestingly, it is significantly associated with subclinical coronary atherosclerosis in healthy subjects36. Human papillomavirus (HPV) is associated with increased prevalence of cardiovascular disease post-infection. This may be due to HPV increasing pro-inflammatory activity and altered lipid metabolism37. Three hepatitis C virus (HCV) strains were predicted to be associated with Atherosclerosis; HCV infection is known to be a risk factor for Atherosclerosis38. 4 of the 6 pathogens interact with human protein ITGB1. A bioinformatics study suggests that ITGB1 is a key gene associated with carotid atherosclerosis39.
Inflammatory Bowel Disease is an umbrella condition represented by Crohn’s disease and ulcerative colitis primarily characterized by intestinal inflammation. There are 10 significant viruses and 3 significant bacteria predicted to be associated with IBD (see Table S1.3). Literature suggests that dysregulation of intestinal mucosa may contribute to the pathogenesis of IBD40. Furthermore, gut microbiota play a major role in the pathogenesis of IBD as it may promote inflammation41. Some infectious diseases can alter the homeostasis of the gut microbiota, thus, contributing to the intestinal inflammation42. Influenza A virus (H1N1) is predicted to be significantly associated with IBD, a prediction supported by literature evidence43. Additionally, HIV is predicted to be significantly associated with IBD; indeed, HIV infection causes onset of Crohn’s disease44. Among the 90 unique MOA proteins of pathogens’ association with IBD (see Table S1.3, union of all MOA proteins), ITGB1 and GSN have the largest numbers (6 and 5 of 13) of interacting pathogens. ITGB1 plays an important role in the pathogenesis of IBD45; GSN is a potential biomarker for ulcerative colitis46.
Alzheimer’s disease (AD), a neurodegenerative disease characterized by memory loss and cognitive impairment, may result from the amyloid cascade or the tau hyperphosphorylation47. There is a theory that infections can seed some cases of AD11. Indeed, there are 10 significant viruses predicted to be associated with Alzheimer’s disease (see Table S1.4). Two strains of H1N1 are predicted to be significantly associated with AD. It has been shown by three studies that at least one flu vaccination is associated with a 17% decrease in AD incidence48. Among the 43 proteins interacting with H1N1, PPIA plays role in tau oligomerization and amyloid processing in AD49; RBBP7 is a mediator against neuronal loss in AD50. Simian virus 40 is predicted to be associated to AD. One study found that its antigen expression induces AD like pathology in mice51. Simian virus interacts with FBXW11 that is found to be related to AD alleviation52.
It is possible that infectious diseases may encourage damaging molecular processes in the specific human body/tissue yielding key characteristics of some common complex diseases such as persistent inflammation. It may also be that individuals have to encounter a series of these pathogens before subsequently developing a common complex disease.
Prevalent pathogens and diseases
Next, we will explore the disease comorbidities associated with the following prevalent infectious pathogens: EBV (strain B95-8) (taxid 10377), Influenza A virus (strain A/New York/1682/2009(H1N1)) (taxid 643960), Escherichia coli (taxid 562) and Salmonella typhimurium (taxid 90371). Table 4 shows the top 10 comorbidities for each pathogen. Full lists including the MOA proteins are found in Supplementary Table S2.1–S2.4.
There were 408 significant comorbidities associated with EBV (strain B95-8) (see Table S2.1). Of these, 167 and 130 involve Endocrine nutritional and metabolic diseases and Neoplasms, respectively. Oligospermia, characterized by a low sperm count is the top associated comorbidity with EBV. Viral infections may contribute to male infertility via initiating inflammatory reactions that yield an increase in reactive oxygen species causing testicular damage53. Lipid metabolism disorder and carbohydrate metabolic disorder were predicted to be significant diseases associated with EBV. As another example, we examined the EBV interactome proteins of association with Gestational diabetes. We found 266 proteins that interact with EBV. The top 5 proteins prioritized by LeMeDISCO are STX7, STX10, CDIPT, GLIPR2, SNRPA. STX7 is upregulated in T2D54, and STX10 is differentially methylated in the offspring of women with maternal diabetes55. CDIPT was hypomethylated and up-regulated in the fetus of mice with Maternal Gestational Diabetes56. SNRPA was found to be associated with metabolic syndromes57.
There were 211 significant comorbidities associated with Influenza A virus (strain A/New York/1682/2009(H1N1) (see Table S2.2). The top 2 groups of diseases involve 33 Endocrine, nutritional and metabolic diseases and 26 Diseases of the blood and blood-forming organs and certain disorders involving the immune mechanism. The top 2 significant human comorbid diseases for Influenza A virus (flu) were Tay-Sachs disease (mutations in HEXA) and Sandhoff disease (mutations in HEXB) that are very similar rare genetic diseases. There is no direct interaction of flu with those two proteins. However, the human proteins in the flu-human interactome of HK1 protein interacts with both HEXA and HEXB58, and they share a pathway involving carbohydrate metabolism59. Another significant comorbid disease is coronary stenosis. Studies have suggested an association between influenza and cardiovascular diseases due to the activation of inflammatory pathways7. We shall examine this in more detail in the following “Heart disease and flu” section. There were several other rare diseases predicted to be associated with Influenza A. Some involve the immune mechanism and result in a fatal outcome from contracting the flu. For example, thrombocytopenia can be induced by flu60 and flu vaccination61. However, we do not know which genes are responsible for thrombocytopenia. Rather, we list 66 candidate genes in Table S2.2 for future investigation.
There were 380 significant comorbidities associated with Escherichia coli (taxid 562) (see Table S2.3). 289 involve Certain infectious and parasitic diseases. The top predicted significant comorbid disease for Escherichia coli (E. coli) was Uveitis. Amazingly, E.coli is beneficial for treating Uveitis62. Among the 5 interactomes of E.coli for Uveitis, deficiency of the top ranked SERPINA1 is a uveitis risk factor63. Hypertension, renal tubular acidosis and cardiovascular syphilis were predicted to be other significant comorbidities to E. coli. In fact, E. coli contamination in drinking water increases the risk of hypertension, renal impairment and cardiovascular disease64.
There were 50 significant comorbidities associated with Salmonella typhimurium (see Table S2.4). 14 and 10 of them, respectively, are Endocrine, nutritional and metabolic diseases & Diseases of the genitourinary system. The third most significant comorbid disease is psoriasis and Salmonella typhimurium is proposed as photochemotherapy agent for psoriasis65. Another example of significant comorbidity is hyperglycemia, which is characterized by high blood sugar. Salmonella infection causes hyperglycemia in pigs66.
On the oncogenic/oncolytic potential of pathogens
The above results (see Tables 1 and 2) show that Neoplasms have the largest relative risk as being caused by viruses and the second largest relative risk as being caused by bacterial infection compared to general causes. Thus, it is worthwhile to examine the oncogenic/oncolytic potential of pathogens in detail.
We define an onco_index, a p value characterizing the overlap of pathogen’s interactome with the 723 cancer drivers given in the COSMIC database67 to detect pathogens associated with cancers (either oncogenic or oncolytic) from others. An onco_index < 0.05 is considered to have oncogenic/oncolytic potential. Table 5 shows the top 10 cancer associated viruses and bacteria. A complete list of predicted oncogenic/oncolytic pathogens (109 viruses and 15 bacteria) whose onco-index p value < 0.05 is given in Supplementary Tables S3 and S4 along with possible MOA proteins, denoted as onco_MOA protein. These are defined as those pathogen proteins that interact with the 723 cancer drivers that have documented literature evidence of oncogenic/oncolytic potential. Of the 124 pathogens 93(75%) have literature evidence, 55(38%) of having oncogenic(oncolytic) potential.
For those predictions with literature evidence, we have the putative molecular basis of associations that can be validated by experimentalists. About ~ 1/4 of the predictions are novel and worthy of further investigation. To demonstrate how the MOA proteins explain the cancers a given pathogen is associated with, we present the example of the first discovered human tumor virus associated with Epstein-Barr virus68. There are 885 interactome proteins for EBV(strain AG876), 64 proteins are cancer drivers according to COSMIC67. Among these are EGFR and ERBB2, which are well-known tumor drivers69.
Distinguishing oncolytic from oncogenic pathogens
While the above onco_index distinguishes pathogens associated with cancers from others, some of the cancer associated pathogens can be oncolytic. To distinguish oncolytic from oncogenic potential of pathogens, we examined the interactomes of the 93 pathogens having literature evidence of being oncogenic (total 55) or oncolytic (total 38) and derived oncolytic and oncogenic propensities for each pathogen. First, the oncolytic/oncogenic propensity of an interactome protein of pathogen is derived by calculating a p value of its association with oncolytic/oncogenic pathogens. Then the oncolytic/oncogenic propensity of pathogen is calculated as the sum of its interactomes’ propensity (see “Methods” for addition details). A jackknife test was performed on these 93 pathogens by excluding self in deriving oncolytic and oncogenic propensity. We classify a pathogen as being oncolytic if its oncolytic propensity is greater than its oncogenic propensity. The resulting Matthews correlation coefficient (MCC) of this analysis on the 93 pathogens is 0.77, the recall rate (sensitivity) is 94.7%, accuracy 88.2%, and precision is 80.0%. The oncolytic/oncogenic propensity of possible cancer associated pathogens is found in Tables S3 and S4. We then apply the oncolytic/oncogenic propensity to all the pathogens (not limited to those that are predicted cancer associated) to discover possible pathogens that might be used for cancer treatment. The oncolytic_MOA proteins are those human proteins in the given pathogen-human interactome ranked by their oncolytic propensities (see Eq. 3a). 136 pathogens with oncolytic > oncogenic propensity not included in Tables S3 and S4 are listed in Table S5.1 ranked by oncolytic minus oncogenic propensity. Among the top, many are various strains of flu; we note that subtype H5N1 has already shown to have a curative effect on cancer18.
In a recent work26 we predicted that SARS-CoV-2 is likely associated with cancers by applying the 332 interactomes from ref70. Using the same interactomes and the above method, we now additionally predict that SARS-CoV-2 is likely to be oncolytic with a propensity score of 0.017 that will rank 13th in Table S5.1’s 136 predictions. Its significant oncolytic MOA proteins (propensity p value < 0.05, see Eq. 3a) along with literature supports are given in Table S5.2. For 10 of the 12 proteins, we have literature evidence of their associations with cancers. For example, MIN, NUP214, PABPC1, LARP4B & DDX10 are established cancer drivers in the COSMIC database67. The top protein, MIN, is associated with the risk of colorectal cancer71. Knockdown of the second protein MOV10 leads to upregulation of INK4, a tumor suppressor72. Inhibition of the third protein NUP214 leads to cell death73. PABPC4 plays role in the pathogenesis of colorectal cancer74.
While the oncolytic effect might be due to collective effect of the oncolytic proteins, many (48/147) of the top unique pathways (different from those of the oncogenic significant proteins with p value < 0.05) of the significant proteins involve PIK3R1 (see Table S6 for unique pathways and proteins involved): e.g., CD28 dependent PI3K/Akt signaling, Signaling by cytosolic FGFR1 fusion mutants, Signaling by PDGFR in disease, etc. PIK3R1 is a known tumor suppressor75. Another frequent protein in unique pathways is PIK3CA and its mutations cause a variety of common human tumor types76. The above SARS-CoV-2 interactome MOV10 is involved in 12 pathways. Although it does not directly involve apoptotic pathways, its interacting partners ACIN1 and SLC25A558 involve Apoptosis59. The colorectal cancer related protein PABPC4 of the SARS-CoV-2 interactome interacts with YWHAQ, YWHAZ & TNFRSF10D that all involve Apoptosis59.
Heart disease and flu
Studies show that heart disease is one of the most common chronic conditions of adults hospitalized with flu77,78; it also increases the incidence of strokes. To understand the molecular bases of this observation, we examined the significant comorbidities (q-value < 0.05) belonging to the class “Diseases of the circulatory system” associated with various strains of flu. In total, we predicted 79 pairs of flu virus—Diseases of the circulatory system involving 20 strains of flu and 25 cardiovascular diseases. The 79 pairs and related putative MOA proteins are listed in Table S7.
The 25 comorbid diseases ranked by the number of associated flu strains along with literature evidence are given in Table S8. The top 5 diseases are: intracranial vasospasm, Dressler's syndrome, brain stem infarction, brain ischemia79, lymphatic system disease. We found 12 of the diseases have supporting literature evidence for their associations with flu. The novel predictions of disease associations are useful in guiding clinicians for disease diagnosis.
Next, we analyze the most frequent MOA proteins and their pathways59. For each human protein that interacts with flu, we count its frequency as a MOA protein in the 79 flu-heart disease pairs, each of which may contain multiple human proteins that interact with the given strain of flu. The top 100 ranked most frequent proteins are listed in Table S9 along with literature evidence of their association with heart disease. For the top 20 proteins, we find evidence for 9 proteins. For example, the top 1st protein, PIK3R1, is a cardiac regulator80. The 2nd protein, GSN, is critical for heart disease81.
The 39 significant pathways (q-value < 0.05) involving the above top 100 proteins are given in Table S10. For 22 we found literature evidence of their associations with heart disease. Many of them involve protein synthesis. The top 5 pathways are Translation82, Mitochondrial translation initiation83, Mitochondrial translation elongation83, Mitochondrial translation termination83, Mitochondrial translation83. There are number of ribosomal proteins (RPS19/RPL8/RPL30A/RPL3/RPL23/RPL19/RPL15/RPL11) in these pathways that the flu viruses interact with. Studies have shown that mutations in many ribosomal proteins result in a Minute phenotype in Drosophila and Cardiomyopathy is correlated with the Minute phenotype84. With all the literature evidence, our novel predictions of MOA proteins and pathways are useful in guiding experimentalists for further investigations.
PHEVIRdb web application
The PHEVIRdb web application allows researchers to access disease comorbidities and the corresponding MOA proteins associated with interactions with the respective pathogen. With multiple input options (keyword and exact search), one can input a keyword for a pathogen name or disease name or select a pathogen name and disease name from the pull down menu. The web service is freely available for academic users at https://sites.gatech.edu/cssb/phevir/. Figure 1 shows screenshots of the web interface and an output example. The keyword search provides a fuzzy search that matches pathogen name or disease name containing the keyword. From the pull down menu, the user can select the pathogen name and disease name for an exact match search. The output can be saved and is searchable by keywords in the search box.

Screenshots of PHEVIR webserver. (A) Web interface of PHEVIR. (B) Sample output of PHEVIR.
Discussion
PHEVIR, with 77.2% recall and 37.1% precision based on large scale benchmarking, has predicted post-infection complications of 369 pathogens. Consistent with its quite general definition, our prediction that Certain infectious and parasitic diseases have the largest overall relative risk for pathogens. We also predict that Neoplasms are the only other group of diseases, on average, having a significant relative risk compared to general causes. By examining some common complex diseases associated with pathogens, oncogenic/oncolytic pathogens and heart disease association with flu, we found that many of PHEVIR’s predictions have literature evidence (which is unknown to the algorithm which views these as bona fide predictions). For all predictions, PHEVIR provides the molecular basis of each human disease-pathogen association. In addition, the onco_index and oncolytic propensity can tell whether a pathogen is potentially oncogenic or oncolytic. Importantly, the oncolytic/oncogenic propensity can distinguish oncolytic from oncogenic viruses at 88.2% accuracy and 80.0% precision. Oncolytic pathogens are a useful means of treating cancers and their MOA proteins could be targeted by small molecules or antibodies. Furthermore, PHEVIR predicted 25 heart diseases (Disease of circulatory system) associated with flu for which ~ 50% have literature evidence. These predictions as well as their corresponding MOA proteins are useful for guiding further experimental investigations on disease etiology and for clinic diagnosis. The goal is to eventually find better prevention and treatments of these diseases.
On another note, PHEVIR strongly suggests that many non-Mendelian diseases have a viral component. Indeed, 91% (3285) of the 3608 diseases we consider which cover almost all disease types have a least one viral pathogen associated with it. For the 57 bacteria, 66.7% (2405) of diseases have bacterial associated human disease comorbidities. What is important to realize is that we merely considered 312 strains of viruses and 57 different bacteria. This is clearly a tiny minority of bacterial and viral pathogens. At present, we cannot definitively differentiate whether the pathogen’s infection induces the onset of the complex disease or merely exacerbates its progression. But what this study strongly suggests is that infectious diseases and complex noninfectious diseases are not disjoint and non-interacting. Rather, their interaction is likely to be the rule rather than the exception. Some infections such as an oncolytic virus might be antagonists to the given disease while others, (e.g. oncogenic viruses) might be agonists. This does suggest that one possible means of eliminating/preventing severe diseases such as IBD, AD, and some cancers might be by eliminating the infectious diseases that might be a major contributory factor. In the case of viruses, this suggests that the broader development of antiviral vaccines or antiviral drugs is clearly needed.
Materials and methods
A flowchart of PHEVIR method is given in Fig. 2. We detail each of the steps below.

General PHEVIR methodology. (A) Comorbidity pipeline; (B) Onco-index calculation and oncogenic/oncolytic prediction pipeline.
Curating human–pathogen interactomes
Host–pathogen interactome data were extracted from the HPIDB 3.0 database (https://hpidb.igbb.msstate.edu)22. Interactions with homo sapiens (taxid: 9606) and homo sapiens proteins with known UniProtKB IDs were obtained. Next, the pathogens were mapped to their corresponding taxonomy IDs (taxids) using the NCBI taxid file from: https://www.uniprot.org/taxonomy/# which contained 2,658,466 organism entries. Bacteria and viruses were extracted, and those with “No lineage” were removed. The bacteria and viruses were mapped to their corresponding KEGG infectious disease classification from https://www.genome.jp/brite/br08401. If there were less than two homo sapiens proteins that interacted with a given pathogen, they were removed as a minimum of 2 proteins are required for the subsequent analysis. There were 312 viruses and 57 bacteria that remaining after filtration.
Comorbidity predictions
Comorbidities were predicted by LeMeDISCO; we refer the reader to Ref.21 for details. Following determination of the significant comorbidities associated with the 312 viruses and 57 bacteria curated from HPIDB, a virus-disease network and a bacteria-disease network were constructed. Following determination of the significant comorbidities associated with the 312 viruses and 57 bacteria curated from HPIDB, a virus-disease network and a bacteria-disease network were constructed.
Pan-virus and pan-bacteria assessment
We first assessed the frequency of each ICD-10 disease group across all the significant (q-value < 0.05) comorbidities. Then, to find out which ICD-10 disease groups are affected most by pathogens, we define a relative risk (RR) of disease group after infection with respect to background (all possible source of causes):
and calculate a corresponding p value using Fisher’s exact test85.
Oncogenic/oncolytic index
For each pathogen an oncogenic/oncolytic index (onco_index) characterized by its p value was computed. The p value is calculated by Fisher’s exact test85 of the overlapped cancer drivers of the COSMIC 723 census proteins67 with the interactome proteins. The p value is calculated on the following contingency table:
Here \({N}^{overlapped}\) is the number of overlapped proteins between \({N}^{interatome}\) of interactomes of the given pathogen and the \({N}^{driver}\) = 723 cancer drivers, \({N}^{total}=\mathrm{18,663}\) is the total number of human proteins considered in the work. Onco_MOA proteins are defined as those of the pathogen’s interactomes overlapped with the 723 drivers.
Oncogenic/oncolytic distinguishing and propensity
To distinguish an oncolytic from oncogenic pathogen, we examine the possible difference between the interactomes (the human partner proteins a pathogen interacts with) of the oncogenic or oncolytic pathogens as evident from literature. We first collect all the interactomes of oncogenic or oncolytic pathogens and then count the frequencies that each human protein is part of the given pathogen-human interactome. Then, using the frequencies of these two lists, we calculate a p value85 of each protein T’s relative risk for oncolytic or oncogenic effects using the following contingency tables:
where (3a) for oncolytic effect, (3b) for oncogenic effect; \({N}_{lytic}^{T}\),\({N}_{genic}^{T}\) are numbers of oncolytic, oncogenic pathogens targeting the protein T; \({N}_{lytic}^{all}\), \({N}_{genic}^{all}\) are total number of oncolytic, oncogenic pathogens. We then define the oncolytic or oncogenic propensity of a pathogen as
The oncolytic propensity is obtained using the p value from (3a) and \({N}_{onco}^{all}\) is the total number of unique interactomes of oncolytic pathogens; and the oncogenic propensity from (3b) and \({N}_{onco}^{all}\) is the total number of unique interactomes of oncogenic pathogens. When a pathogen’s oncolytic propensity > oncogenic propensity, we predict it to be an oncolytic pathogen; otherwise it is classified as an oncogenic pathogen. Oncolytic_MOA proteins are defined as the overlap of interactomes of given pathogen with the union of those from literature supported oncolytic pathogens.
Data availability
The datasets generated and/or analysed during the current study are not publicly available due to the tools generating them are independently licensed, but are available from the corresponding author on reasonable request.
References
Piret, J. & Boivin, G. Pandemics throughout history. Front. Microbiol. https://doi.org/10.3389/fmicb.2020.631736 (2021).
Shaw-Taylor, L. An introduction to the history of infectious diseases, epidemics and the early phases of the long-run decline in mortality. Econ Hist Rev 73, E1–E19. https://doi.org/10.1111/ehr.13019 (2020).
Cates, J. et al. Risk for in-hospital complications associated with covid-19 and influenza—Veterans health administration, United States, October 1, 2018–May 31, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020(69), 1528–1534 (2020).
Chu, C. & Selwyn, P. A. Complications of HIV infection: A systems-based approach. Am. Fam. Physician 83, 395–406 (2011).
Rothberg, M. B., Haessler, S. D. & Brown, R. B. Complications of viral influenza. Am. J. Med. 121, 258–264. https://doi.org/10.1016/j.amjmed.2007.10.040 (2008).
Long, S. S. Neurologic complications of common respiratory tract virus infections. J. Pediatr. 239, 1–4. https://doi.org/10.1016/j.jpeds.2021.10.003 (2021).
Hebsur, S., Vakil, E., Oetgen, W. J., Kumar, P. N. & Lazarous, D. F. Influenza and coronary artery disease: Exploring a clinical association with myocardial infarction and analyzing the utility of vaccination in prevention of myocardial infarction. Rev. Cardiovasc. Med. 15, 168–175 (2014).
Kwok, A. J., Mentzer, A. & Knight, J. C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 22, 137–153. https://doi.org/10.1038/s41576-020-00297-6 (2021).
Gysi, D. M. et al. Network medicine framework for identifying drug repurposing opportunities for COVID-19. ArXiv https://arxiv.org/abs/2004.07229 (2020).
Kwiatkowski, D. Susceptibility to infection. BMJ 321, 1061–1065. https://doi.org/10.1136/bmj.321.7268.1061 (2000).
Abbott, A. Are infections seeding some cases of Alzheimer’s disease?. Nature 587, 22–25 (2020).
Harley, J. B. et al. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 50, 699–707. https://doi.org/10.1038/s41588-018-0102-3 (2018).
Alemany, R. Viruses in cancer treatment. Clin. Transl. Oncol. 15, 182–188. https://doi.org/10.1007/s12094-012-0951-7 (2013).
Mui, U. N., Haley, C. T. & Tyring, S. K. Viral oncology: Molecular biology and pathogenesis. J. Clin. Med. 6, 111. https://doi.org/10.3390/jcm6120111 (2017).
Bouvard, V. et al. A review of human carcinogens–Part B: Biological agents. Lancet Oncol. 10, 321–322. https://doi.org/10.1016/s1470-2045(09)70096-8 (2009).
Burd, E. M. Human papillomavirus and cervical cancer. Clin. Microbiol. Rev. 16, 1–17. https://doi.org/10.1128/cmr.16.1.1-17.2003 (2003).
Stingi, A. & Cirillo, L. SARS-CoV-2 infection and cancer. BioEssays 43, 2000289. https://doi.org/10.1002/bies.202000289 (2021).
Huo, C. et al. H5N1 influenza a virus replicates productively in pancreatic cells and induces apoptosis and pro-inflammatory cytokine response. Front. Cell Infect. Microbiol. 8, 386. https://doi.org/10.3389/fcimb.2018.00386 (2018).
Aref, S., Bailey, K. & Fielding, A. Measles to the rescue: A review of oncolytic measles virus. Viruses https://doi.org/10.3390/v8100294 (2016).
Shen, Y. & Nemunaitis, J. Herpes simplex virus 1 (HSV-1) for cancer treatment. Cancer Gene Ther. 13, 975–992. https://doi.org/10.1038/sj.cgt.7700946 (2006).
Astore, C., Zhou, H., Ilkowski, B., Forness, J. & Skolnick, J. LeMeDISCO is a computational method for large-scale prediction & molecular interpretation of disease comorbidity. Commun. Biol. 5, 870. https://doi.org/10.1038/s42003-022-03816-9 (2022).
Ammari, M. G., Gresham, C. R., McCarthy, F. M. & Nanduri, B. HPIDB 2.0: A curated database for host–pathogen interactions. Database https://doi.org/10.1093/database/baw103 (2016).
Zhou, H. et al. MEDICASCY: A machine learning approach for predicting small molecule drug side effects, indications, efficacy and mode of action. Mol. Pharm. 17, 1558–1574. https://doi.org/10.1021/acs.molpharmaceut.9b01248 (2020).
Menche, J. et al. Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347, 1257601 (2015).
Ko, Y., Cho, M., Lee, J.-S. & Kim, J. Identification of disease comorbidity through hidden molecular mechanisms. Sci. Rep. 6, 39433 (2016).
Astore, C., Zhou, H., Jacob, J. & Skolnick, J. Prediction of severe adverse events, modes of action and drug treatments for COVID-19’s complications. Sci. Rep. 11, 20864. https://doi.org/10.1038/s41598-021-00368-6 (2021).
Wouk, J., Rechenchoski, D. Z., Rodrigues, B. C. D., Ribelato, E. V. & Faccin-Galhardi, L. C. Viral infections and their relationship to neurological disorders. Arch. Virol. 166, 733–753. https://doi.org/10.1007/s00705-021-04959-6 (2021).
Mattson, M. P. Infectious agents and age-related neurodegenerative disorders. Ageing Res. Rev. 3, 105–120. https://doi.org/10.1016/j.arr.2003.08.005 (2004).
Smatti, M. K. et al. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 11, 762. https://doi.org/10.3390/v11080762 (2019).
Fernández-Real, J.-M. et al. Burden of infection and insulin resistance in healthy middle-aged men. Diabetes Care 29, 1058. https://doi.org/10.2337/dc05-2068 (2006).
Hernandez-Romieu, A. C., Garg, S., Rosenberg, E. S., Thompson-Paul, A. M. & Skarbinski, J. Is diabetes prevalence higher among HIV-infected individuals compared with the general population? Evidence from MMP and NHANES 2009–2010. BMJ Open Diabetes Res. Care 5, e000304. https://doi.org/10.1136/bmjdrc-2016-000304 (2017).
Fujiya, A. et al. Fulminant type 1 diabetes mellitus associated with a reactivation of Epstein-Barr virus that developed in the course of chemotherapy of multiple myeloma. J. Diabetes Investig. 1, 286–289. https://doi.org/10.1111/j.2040-1124.2010.00061.x (2010).
Schaffert, C. S. Role of MGST1 in reactive intermediate-induced injury. World J. Gastroenterol. 17, 2552–2557. https://doi.org/10.3748/wjg.v17.i20.2552 (2011).
Li, S. et al. Cathepsin B contributes to autophagy-related 7 (Atg7)-induced nod-like receptor 3 (NLRP3)-dependent proinflammatory response and aggravates lipotoxicity in rat insulinoma cell line. J. Biol. Chem. 288, 30094–30104. https://doi.org/10.1074/jbc.M113.494286 (2013).
DeFronzo, R. A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 53, 1270–1287. https://doi.org/10.1007/s00125-010-1684-1 (2010).
Lee, M. et al. Current Helicobacter pylori infection is significantly associated with subclinical coronary atherosclerosis in healthy subjects: A cross-sectional study. PLoS ONE 13, e0193646–e0193646. https://doi.org/10.1371/journal.pone.0193646 (2018).
Tonhajzerova, I. et al. Novel biomarkers of early atherosclerotic changes for personalised prevention of cardiovascular disease in cervical cancer and human papillomavirus infection. Int. J. Mol. Sci. 20, 3720. https://doi.org/10.3390/ijms20153720 (2019).
Adinolfi, L. E. et al. Chronic hepatitis C virus infection and atherosclerosis: clinical impact and mechanisms. World J. Gastroenterol. 20, 3410–3417. https://doi.org/10.3748/wjg.v20.i13.3410 (2014).
Mao, Z., Wu, F. & Shan, Y. Identification of key genes and miRNAs associated with carotid atherosclerosis based on mRNA-seq data. Medicine 97, e9832. https://doi.org/10.1097/md.0000000000009832 (2018).
Swidsinski, A. et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 122, 44–54. https://doi.org/10.1053/gast.2002.30294 (2002).
Khan, I. et al. Alteration of gut microbiota in inflammatory bowel disease (IBD): Cause or consequence? IBD treatment targeting the gut microbiome. Pathogens 8, 126. https://doi.org/10.3390/pathogens8030126 (2019).
Ramos, G. P. & Papadakis, K. A. Mechanisms of disease: Inflammatory bowel diseases. Mayo Clin. Proc. 94, 155–165. https://doi.org/10.1016/j.mayocp.2018.09.013 (2019).
Maddika, S. et al. 791 morbidity and mortality of influenza virus in inflammatory bowel disease. Off. J. Am. Coll. Gastroenterol. ACG 114, S458 (2019).
Lautenbach, E. & Lichtenstein, G. R. Human immunodeficiency virus infection and Crohn’s disease: the role of the CD4 cell in inflammatory bowel disease. J. Clin. Gastroenterol. 25, 456–459. https://doi.org/10.1097/00004836-199709000-00013 (1997).
Dotan, I. et al. The role of integrins in the pathogenesis of inflammatory bowel disease: Approved and investigational anti-integrin therapies. Med. Res. Rev. 40, 245–262. https://doi.org/10.1002/med.21601 (2020).
Maeda, K. et al. Gelsolin as a potential biomarker for endoscopic activity and mucosal healing in ulcerative colitis. Biomedicines 10, 872. https://doi.org/10.3390/biomedicines10040872 (2022).
Fan, L. et al. New insights into the pathogenesis of Alzheimer’s disease. Front. Neurol. https://doi.org/10.3389/fneur.2019.01312 (2020).
Amran, A. et al. Influenza vaccination is associated with a reduced incidence of Alzheimer’s disease. Alzheimer’s Dement. 16, e041693. https://doi.org/10.1002/alz.041693 (2020).
Blair, L. J., Baker, J. D., Sabbagh, J. J. & Dickey, C. A. The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer’s disease. J. Neurochem. 133, 1–13. https://doi.org/10.1111/jnc.13033 (2015).
Dave, N. et al. Identification of retinoblastoma binding protein 7 (Rbbp7) as a mediator against tau acetylation and subsequent neuronal loss in Alzheimer’s disease and related tauopathies. Acta Neuropathol. 142, 279–294. https://doi.org/10.1007/s00401-021-02323-1 (2021).
Park, K. H., Hallows, J. L., Chakrabarty, P., Davies, P. & Vincent, I. Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J. Neurosci. 27, 2969–2978. https://doi.org/10.1523/jneurosci.0186-07.2007 (2007).
Sun, J. et al. FBXW11 deletion alleviates Alzheimer’s disease by reducing neuroinflammation and amyloid-β plaque formation via repression of ASK1 signaling. Biochem. Biophys. Res. Commun. 548, 104–111. https://doi.org/10.1016/j.bbrc.2020.12.081 (2021).
Henkel, R., Offor, U. & Fisher, D. The role of infections and leukocytes in male infertility. Andrologia 53, e13743. https://doi.org/10.1111/and.13743 (2021).
Andersson, S. A. et al. Reduced insulin secretion correlates with decreased expression of exocytotic genes in pancreatic islets from patients with type 2 diabetes. Mol. Cell. Endocrinol. 364, 36–45. https://doi.org/10.1016/j.mce.2012.08.009 (2012).
West, N. A., Kechris, K. & Dabelea, D. Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism 1, 1–9. https://doi.org/10.2478/immun-2013-0001 (2013).
Luo, S.-S. et al. Integrated multi-omics analysis reveals the effect of maternal gestational diabetes on fetal mouse hippocampi. Front. Cell Dev. Biol. https://doi.org/10.3389/fcell.2022.748862 (2022).
Louis, J. M., Agarwal, A., Mondal, S. & Talukdar, I. A global analysis on the differential regulation of RNA binding proteins (RBPs) by TNF–α as potential modulators of metabolic syndromes. BBA Adv. 2, 100037. https://doi.org/10.1016/j.bbadva.2021.100037 (2022).
Alanis-Lobato, G., Andrade-Navarro, M. A. & Schaefer, M. H. HIPPIE v2.0: Enhancing meaningfulness and reliability of protein–protein interaction networks. Nucleic Acids Res. 45, D408–D414. https://doi.org/10.1093/nar/gkw985 (2016).
Jassal, B. et al. The reactome pathway knowledgebase. Nucleic Acids Res. 48, D498-d503. https://doi.org/10.1093/nar/gkz1031 (2020).
Jansen, A. J. G. et al. Influenza-induced thrombocytopenia is dependent on the subtype and sialoglycan receptor and increases with virus pathogenicity. Blood Adv. 4, 2967–2978. https://doi.org/10.1182/bloodadvances.2020001640 (2020).
Yamamoto, Y. et al. Influenza vaccination-associated acute thrombocytopenia and diffuse alveolar hemorrhage. Intern. Med. 59, 1633–1637. https://doi.org/10.2169/internalmedicine.3991-19 (2020).
Dusek, O. et al. Severity of experimental autoimmune uveitis is reduced by pretreatment with live probiotic Escherichia coli Nissle 1917. Cells https://doi.org/10.3390/cells10010023 (2020).
Ghavami, S. et al. Alpha-1-antitrypsin phenotypes and HLA-B27 typing in uveitis patients in southeast Iran. Clin. Biochem. 38, 425–432. https://doi.org/10.1016/j.clinbiochem.2005.02.006 (2005).
Clark, W. F. et al. Long term risk for hypertension, renal impairment, and cardiovascular disease after gastroenteritis from drinking water contaminated with Escherichia coli O157:H7: A prospective cohort study. BMJ 341, c6020. https://doi.org/10.1136/bmj.c6020 (2010).
Venturini, S., Tamaro, M., Monti-Bragadin, C. & Carlassare, F. Mutagenicity in Salmonella typhimurium of some angelicin derivatives proposed as new monofunctional agents for the photochemotherapy of psoriasis. Mutat. Res. 88, 17–22. https://doi.org/10.1016/0165-1218(81)90085-9 (1981).
Zong, Y., Chen, W., Zhao, Y., Suo, X. & Yang, X. Salmonella infection causes hyperglycemia for decreased GLP-1 content by Enteroendocrine L cells pyroptosis in pigs. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23031272 (2022).
Tate, J. G. et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947. https://doi.org/10.1093/nar/gky1015 (2019).
Pagano, J. S. Epstein-Barr virus: The first human tumor virus and its role in cancer. Proc. Assoc. Am. Physicians 111, 573–580. https://doi.org/10.1046/j.1525-1381.1999.t01-1-99220.x (1999).
Mitsudomi, T. & Yatabe, Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. Febs J. 277, 301–308. https://doi.org/10.1111/j.1742-4658.2009.07448.x (2010).
Gordon, D. E. et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature https://doi.org/10.1038/s41586-41020-42286-41589 (2020).
Yasuda, Y. et al. Human NINEIN polymorphism at codon 1111 is associated with the risk of colorectal cancer. Biomed. Rep. 13, 45. https://doi.org/10.3892/br.2020.1352 (2020).
El Messaoudi-Aubert, S. et al. Role for the MOV10 RNA helicase in polycomb-mediated repression of the INK4a tumor suppressor. Nat. Struct. Mol. Biol. 17, 862–868. https://doi.org/10.1038/nsmb.1824 (2010).
Bhattacharjya, S. et al. Inhibition of nucleoporin member Nup214 expression by miR-133b perturbs mitotic timing and leads to cell death. Mol. Cancer 14, 42. https://doi.org/10.1186/s12943-015-0299-z (2015).
Liu, D. et al. Cytoplasmic poly(A) binding protein 4 is highly expressed in human colorectal cancer and correlates with better prognosis. J. Genet. Genom. 39, 369–374. https://doi.org/10.1016/j.jgg.2012.05.007 (2012).
Liu, Y. et al. Pan-cancer analysis on the role of PIK3R1 and PIK3R2 in human tumors. Sci. Rep. 12, 5924. https://doi.org/10.1038/s41598-022-09889-0 (2022).
Samuels, Y. & Waldman, T. Oncogenic mutations of PIK3CA in human cancers. Curr. Top. Microbiol. Immunol. 347, 21–41. https://doi.org/10.1007/82_2010_68 (2010).
Kwong, J. C. et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N. Engl. J. Med. 378, 345–353. https://doi.org/10.1056/NEJMoa1702090 (2018).
Chow, E. J. et al. Acute cardiovascular events associated with influenza in hospitalized adults. Ann. Intern. Med. 173, 605–613. https://doi.org/10.7326/M20-1509 (2020).
Honorat, R. et al. Influenza A(H1N1)-associated ischemic stroke in a 9-month-old child. Pediatr. Emerg. Care 28, 368–369. https://doi.org/10.1097/PEC.0b013e31824dcaa4 (2012).
Oudit, G. Y. & Penninger, J. M. Cardiac regulation by phosphoinositide 3-kinases and PTEN. Cardiovasc. Res. 82, 250–260. https://doi.org/10.1093/cvr/cvp014 (2009).
Patel, V. B. et al. PI3Kα-regulated gelsolin activity is a critical determinant of cardiac cytoskeletal remodeling and heart disease. Nat. Commun. 9, 5390. https://doi.org/10.1038/s41467-018-07812-8 (2018).
Zeitz, M. J. & Smyth, J. W. Translating translation to mechanisms of cardiac hypertrophy. J. Cardiovasc. Dev. Dis. https://doi.org/10.3390/jcdd7010009 (2020).
Wang, F., Zhang, D., Zhang, D., Li, P. & Gao, Y. Mitochondrial protein translation: Emerging roles and clinical significance in disease. Front. Cell Dev. Biol. https://doi.org/10.3389/fcell.2021.675465 (2021).
Casad, M. E. et al. Cardiomyopathy is associated with ribosomal protein gene haplo-insufficiency in Drosophila melanogaster. Genetics 189, 861–870. https://doi.org/10.1534/genetics.111.131482 (2011).
Fisher, R. A. On the interpretation of χ2 from contingency tables, and the calculation of P. J. R. Stat. Soc. 85, 87–94 (1922).
Acknowledgements
We thank Bartosz Ilkowski for internal computing support and Jessica Forness for proof-reading the manuscript.
Funding
This project was funded by R35GM118039 of the Division of General Medical Sciences of the NIH.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.S. Methodology: H.Z., C.A., J.S. Investigation: H.Z. Visualization: C.A. Funding acquisition: J.S. Project administration: J.S. Supervision: J.S. Writing—original draft: H.Z., C.A., J.S. Writing—subsequent drafts, review & editing: H.Z., J.S., C.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, H., Astore, C. & Skolnick, J. PHEVIR: an artificial intelligence algorithm that predicts the molecular role of pathogens in complex human diseases. Sci Rep 12, 20889 (2022). https://doi.org/10.1038/s41598-022-25412-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-25412-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
