
Abstract
Gram-negative bacteria possess intrinsic resistance to glycopeptide antibiotics so these important antibacterial medications are only suitable for the treatment of Gram-positive bacterial infections. At the same time, polymyxins are peptide antibiotics, structurally related to glycopeptides, with remarkable activity against Gram-negative bacteria. With the aim of breaking the intrinsic resistance of Gram-negative bacteria against glycopeptides, a polycationic vancomycin aglycone derivative carrying an n-decanoyl side chain and five aminoethyl groups, which resembles the structure of polymyxins, was prepared. Although the compound by itself was not active against the Gram-negative bacteria tested, it synergized with teicoplanin against Escherichia coli, Pseudomonas aeruginosa and Acinetobacter baumannii, and it was able to potentiate vancomycin against these Gram-negative strains. Moreover, it proved to be active against vancomycin- and teicoplanin-resistant Gram-positive bacteria.
Similar content being viewed by others


Introduction
Glycopeptide antibiotics are used for treating several types of infections caused by Gram-positive bacteria, such as methicillin resistant Staphylococcus aureus (MRSA), vancomycin resistant enterococci (VRE) etc. either in monotherapy or in combination with other antibiotics1,2. Vancomycin is also used per os in C. difficile infections3,4. Semisynthetic glycopeptides such as telavancin or oritavancin are indicated for the treatment of acute bacterial skin and skin structure infections (ABSSSI)5,6. The fundamental mechanism of action of these compounds is the inhibition of bacterial cell-wall synthesis by binding to the d-Ala-d-Ala termini of peptidoglycan precursors with hydrogen bonds7,8.
According to several healthcare sources (e.g. CDC, Centers for Disease Control and Prevention)9 and clinical experience, Gram-negatives are at the top of the list of the most dangerous bacteria today due to a very broad spectrum of resistance mechanisms, which sometimes makes it impossible to treat patients effectively10,11. These pathogens are intrinsically resistant to glycopeptide antibiotics because of the presence of an outer membrane, which consists of phospholipids, negatively charged lipopolysaccharides (LPS), porins and Braun's lipoprotein. The relatively large glycopeptides are unable to cross the outer membrane either by diffusion or by transport through the porins to reach the peptidoglycan.
Our goal was to prepare a semisynthetic glycopeptide which can overcome this intrinsic resistance. Some research groups have already synthesized such derivatives12,13,14 (e.g. several amide derivatives of teicoplanin and its aglycone, where different polyamines were coupled to the C-terminal carboxyl group)15. The aglycone derivatives were notably more effective, possibly because of their smaller molecular size. Other research groups prepared C-terminal amide derivatives of vancomycin using alkyl amines with quaternary ammonium groups16,17, a single amino acid arginine18,19 or LPS-binding peptides20 to vancomycin and these manipulations have successfully endowed the compounds with activity against Gram-negative bacteria.
Our idea was considerably different than those above. The synthetic goal was a compound that resembles an antibiotic which is already proven to be effective against Gram-negative bacteria.
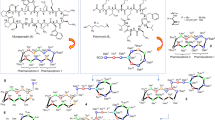
Among currently used antibiotics, polymyxins21,22 (Fig. 1A) are the most relevant structurally related compounds to glycopeptides as—despite significant differences—both are peptides in the 1000–2000 Dalton MW range. These polycationic antibiotics are remarkably effective against Gram-negative bacteria, however they are also highly toxic, which is the reason why they are used only as a last resort22. The primary mechanism of action of polymyxins is related to the bacterial outer membrane. We anticipated that this cellular structure should be an ideal target for a polycationic glycopeptide derivative as well. As a matter of fact, targeting intracellular structures with glycopeptides would be less reasonable, as despite any synthetic modifications, the derivatives would be most likely still too large to cross both the outer and inner bacterial membranes. Furthermore, these antibiotics are inherent cell-wall synthesis inhibitors having no significant action on intracellular targets.

(A) The structure of polymyxins. (B) Target compound with five primary amino groups and a fatty acyl side chain.
Polymyxins have two key structural components that are essential to their activity23. The five aliphatic amino groups in protonated form allow the molecules to bind to the LPS of the Gram-negative outer membrane, displacing the stabilizing Ca2+- and Mg2+-ions, while the simultaneous presence of a fatty acyl side chain provides an amphiphilic character, which leads to the disruption of the membrane. Glycopeptides possess a substantially dissimilar structure with much less flexibility and different spatial organization of amino acids, which seem to be key in the antibacterial activity of polymyxins. However, even lipophilic cholic acid derivatives with multiple primary amino/guanidine groups were reported to permeabilize the membrane24. This effect may be comparable to polymyxin B nonapeptide (polymyxin B missing the fatty acyl side chain), depending on the structure of said compounds. Polymyxin B nonapeptide, despite not being active, has been shown to help otherwise ineffective antibiotics to cross the membrane and act accordingly, thanks to its cationic structure12. Considering these, it is probably not surprising, that glycopeptides mentioned above with a positively charged polyamine chain or quaternary ammonium group showed activity against Gram-negative strains12,16,17.
We hypothesized that the incorporation of amino groups on multiple sites and an acyl chain into a glycopeptide antibiotic might broaden its activity spectrum toward Gram-negative strains. In addition, based on the literature25,26 and our own results27,28, we assumed that such compound, which can be considered a polymyxin analog on the one hand, and a cationic glycopeptide derivative on the other hand, can also be effective against Gram-positive bacteria. We have shown that the introduction of cationic guanidine groups endows the glycopeptide antibiotic teicoplanin with strong activity against resistant Gram-positive strains27,28. In addition, there is increasing evidence that polymyxins and related compounds are suitable for the treatment of Gram-positive bacterial infections, presumably because negatively charged teichoic acids produced by Gram-positive bacteria can be targeted by polymyxins23,25,26.
Results and discussion
For the synthetic modification, the glycopeptide of choice was vancomycin, as it is a relatively inexpensive and easily accessible starting material. The target molecule we planned to prepare (Fig. 1B) has the structural elements that could hypothetically provide antibacterial activity against Gram-negative bacteria: (a) multiple amino groups to interact with the LPS as seen in the case of polymyxins or polyamines, (b) a lipophilic side chain, separated from amino groups in space, to make the compound sufficiently amphiphilic to permeabilize the membrane. In theory, this could result in an antibiotic with a “2-in-1” action, interacting with the outer membrane in the first step, making it permeable. Then by self-promoted uptake (like polymyxins) it could further disturb the membrane, and most importantly, reach the cell-wall and exert its inherent cell-wall synthesis inhibitor effect.
The first two relevant modifications were the removal of the disaccharide moiety to decrease the molecular size and the attachment of the fatty acyl side chain. These were carried out in a four-step procedure (Figs. 2 and 3).

Synthesis of vancomycin aglycone hexapeptide (VAHP) 2.

Acylation of VAHP.
First, vancomycin (1) was submitted to the two-step Edman degradation to remove the N-Me-d-Leu moiety, as direct acylation of the N-terminus proved to be ineffective. After the reaction of 1 with phenylisothiocyanate (PITC), the crude phenylthiocarbamoyl derivative was treated with TFA to remove the N-terminal amino acid, then the disaccharide was hydrolyzed in the same flask at elevated temperature to yield vancomycin aglycone hexapeptide (VAHP) 229,30. After workup the crude product was suitable for further use (Fig. 2).
Next, d-leucine 3 was N-acylated with decanoyl chloride 5, prepared from decanoic acid 4, to yield N-decanoyl-d-leucine 6. From this material, its N-hydroxysuccinimide (NHS) active ester was prepared using a standard procedure with 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride (EDCI.HCl) and N-hydroxysuccinimide. The active ester was suitable for acylation without any purification. The reaction of crude VAHP 2 and the NHS ester of 6 yielded the acylated derivative 7, which was used in the next step after simple workup (Fig. 3).
The following task was to link the amine-containing units to the peptide. As the polymyxins have five amino groups, we decided that the vancomycin derivative should carry five primary amino groups, as well. First, N-Boc-ethylenediamine 9 was prepared using an excess of ethylenediamine 8 and Boc2O31. After workup, PyBOP mediated amide formation was carried out on 7 using 9 as the amine component (Fig. 4). At this point, the amide product 10 was purified by column chromatography.

Amide formation at the C-terminal of vancomycin aglycone derivative 7.
Next, N-Boc-bromoethylamine 12 was prepared from bromoethylamine hydrobromide 11 using Boc2O32. The alkylation33 of 10 in the presence of a large excess of 12 and Cs2CO3 gave the tetraalkylated product 13 (Fig. 5). After column chromatography, further purification was accomplished by preparative TLC to remove residual impurities from the previous reactions. In the final step, the protecting groups were removed by TFA, and the product 14 was isolated as the trifluoroacetate salt after reversed phase column chromatography.

The alkylation of phenolic OH-groups and global deprotection.
The structure of compound 14 was confirmed by different NMR techniques, including COSY, HSQC, HMBC and NOESY experiments. (The NMR data are collected in Tables 1 and 2, atom numbering is shown in Fig. 6). All spectra are available in the supplementary information S1.

Atom numberings for compound 14 for NMR assignment.
Biological evaluation
Compound 14 was tested on eight Gram-positive and four Gram-negative strains using teicoplanin and vancomycin as reference antibiotics (Table 3).
The new polymyxin-like compound showed a surprising profile of activity against Gram-positive strains, with a remarkable effect against resistant strains and only a moderate effect against non-resistant strains. The VanB type (vancomycin-resistant, teicoplanin-sensitive) E. faecalis was more susceptible to compound 14 than to vancomycin (32 vs. 128 μg/mL). More curiously, the vancomycin and teicoplanin resistant VanA type E. faecalis was even more susceptible with a MIC of 8 μg/mL. VanA phenotype resistance is the most frequently encountered glycopeptide antibiotic resistance in enterococci, which causes cell wall reprogramming, resulting in incorporation of d-alanine-d-lactate or d-alanine-d-serine into the peptidoglycan precursors instead of the natural d-alanine-d-alanine (d-Ala-d-Ala)8. This modification in the dipeptide unit greatly reduces the affinity of glycopeptide antibiotics teicoplanin and vancomycin to peptidoglycan. We assume that, on the one hand, compound 14 can bind strongly to the cell membrane through its lipophilic decanoyl side chain, and on the other hand, due to synthetic modifications of the peptide core, its binding affinity towards the modified peptidoglycan precursors might be higher than that of the native glycopeptide antibiotics. Together, these effects may result in the high activity of compound 14 against vanA E. faecalis.
The S. epidermidis strain carrying the mecA gene, which is also teicoplanin resistant, was also more susceptible to the new derivative than to vancomycin or teicoplanin.
At the same time, bacteria that are not glycopeptide resistant, such as MSSA, MRSA and E. faecalis (ATCC 29212) were found to be generally less (or equally) susceptible to compound 14 than to the reference antibiotics. The most striking example is probably the comparison of MIC values obtained for vancomycin sensitive E. faecalis ATCC 29212 and vancomycin/teicoplanin resistant VanA E. faecalis. Compound 14 is 16 times less active against the vancomycin sensitive strain, than vancomycin, but it is 32 times more potent against the resistant VanA strain. We assume that the reduced activity against vancomycin-susceptible strains can be explained by the highly modified aglycon part, as a result of which the binding affinity/accessibility of 14 to the d-Ala-d-Ala peptidoglycan precursors decreases.
Unfortunately, compound 14 did not exert activity against any of the Gram-negative strains tested. Despite this inactivity, we hypothesized that due to its polycationic structure it should have membrane activity, so it may potentiate glycopeptide antibiotics against Gram negative bacteria. It is worth noting here that the synergistic activity of glycopeptide antibiotics vancomycin, teicoplanin and telavancin in combination with colistin (polymyxin E) against Gram-negative bacteria has been previously reported12,34,35,36,37. However, only Acinetobacter baumanniii was included in the synergistic studies with teicopanin34 and vancomycin35, and there are no results in the literature on the activity of these combinations against other Gram-negative strains.
To determine the activity profile of teicoplanin and vancomycin in combination with compound 14, combined susceptibility tests were perfomed against five Gram negative strains including Escherichia coli, Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae bacteria. The combined effects of compound 14 and the glycopeptide antibiotics were determined by conventional checkerboard assays (images of the chequerboard assay for synergistic combinations are shown in Supplementary Information S1).
Upon combining compound 14 with teicoplanin against Escherichia coli, Pseudomonas aeruginosa and Acinetobacter baumannii marked synergy was observed with ΣFIC = 0.2578, 0.375 and 0.2656, respectively. When vancomycin was combined with 14, additive interactions were detected against these strains (ΣFIC = 0.5078, 0.5625 and 0.75, respectively). Combining 14 with teicoplanin or vancomycin against Klebsiella pneumoniae resulted in neither synergy nor additive activity (Table 4).
The additive or synergistic effects of the compound mixtures are visualized by isobologram graphs of the combinations, generated from the FIC values (Fig. 7).

Isobolograms for combinations of compound 14 with teicoplanin and vancomycin tested at various ratios against Acinetobacter baumannii ATCC BAA1605, Escherichia coli ATCC 25218 and Pseudomonas aeruginosa ATCC 27853.
The cationic modification performed on vancomycin could significantly alter the toxicity profile of the modified derivative. The potential toxicity of compound 14, was tested against Caco-2 and hCMEC/D3 cell lines. Caco-2 cells are human intestinal epithelial cells, while hCMEC/D3 cell line is a human brain capillary endothelial cell line. The two cell types were chosen to test the biocompatibility of antibiotics on different barriers in the body after the administration through the two major application routes, the oral or intravenous application. The cationic vancomycin derivative 14 showed a dose-dependent cytotoxicity both on Caco-2 and hCMEC/D3 cells, but fortunately low toxicity was observed in both cases. The IC50 value was 56.22 ± 7.74 µM (116.29 ± 16 µg/mL) for hCMEC/D3 cells, and > 100 µM (> 206.86 µg/mL) for Caco-2 cells (Fig. 8).

Dose-dependent cytotoxicity of compound 14 towards Caco-2 and hCMEC/D3 cells.
For comparison, the effect of vancomycin and teicoplanin on the viability of the two cell types was also tested (Fig. 9). Teicoplanin did not show cytotoxic effect on Caco-2 or hCMEC/D3 cell lines between 0.082 and 200 µM (0.155–375.94 µg/mL). Interestingly Caco-2 cells were more sensitive to vancomycin than hCMEC/D3 cells. The IC50 value of vancomycin on Caco-2 cells was 54.24 µM (78.65 µg/mL).

Cytotoxicity of glycopeptide antibiotics vancomycin and teicoplanin towards Caco-2 and hCMEC/D3 cells.
Finally, to ascertain the biocompatibility of the compound mixtures, cytotoxicity assays were performed with the combinations in checkerboard layout. The combinations of derivative 14 with teicoplanin (Fig. 10), and with vancomycin (Fig. 11) showed cytotoxicity in a lower concentration range, compared to the effect of antibiotics alone, however, they inhibited only partially the cell viability between 2 and 64 µg/mL. Importantly, there are combinations, which did not show or only minor toxicity on the cells, but had synergistic effects on bacteria. The combination teicoplanin : compound 14—32:2 µg/mL was not toxic on either cell line, and the combination teicoplanin : compound 14—2:32 µg/mL only slightly decreased the viability of hCMEC/D3 cells to 81.4%. These combinations had synergistic effects on Acinetobacter baumannii and Escherichia coli, respectively. However, it is important to note, that the possibility of clinical use of the combination against A baumannii is questionable, since concentration of teicoplanin effective in synergism would probably result in toxic side effects in vivo.

Cytotoxic effect of teicoplanin in combination with compound 14 towards Caco-2 and hCMEC/D3 cells, measured by checkerboard method.

Cytotoxic effect of vancomycin in combination with compound 14 towards Caco-2 and hCMEC/D3 cells, measured by checkerboard method.
Conclusions
The new polymyxin-like vancomycin aglycone derivative 14 showed remarkable activity against resistant Gram-positive bacteria but exerted only moderate activity against non-resistant strains. The reason for this seemingly controversial activities against Gram-positive strains might be a considerably different mode of action of the new derivative compared to teicoplanin or vancomycin. The diminished activity against the vancomycin sensitive strains could be due to the heavily modified aglycone part. Plausible explanations could be e.g. the binding pocket becoming less accessible for the peptidoglycan precursors due to steric hindrance and/or the slightly altered orientation of the binding pocket due to intramolecular interactions. Concerning the enhanced activity against vancomycin and teicoplanin resistant bacteria, we assume that the sufficiently strong binding affinity of compound 14 to both modified peptidoglycan precursors and the cell membrane may play a role in the mechanism of action. At the same time, a polymyxin-like effect, based on interaction with the cell wall teichoic acids of Gram-positive bacteria, cannot be ruled out either25,26.
Despite a thoughtful and reasonable design, compound 14 alone did not show antibacterial activity against the tested Gram-negative strains. Uhl and co-workers recently reported similar results when modifying vancomycin with a polycationic peptide; the modification was found to be effective against resistant Gram-positive bacteria but did not lead to activity against Gram-negative strains38.
The main finding of our study is that compound 14 was able to synergize with teicoplanin and strongly potentiated vancomycin against Gram-negative bacteria including Escherichia coli, Pseudomonas aeruginosa and Acinetobacter baumannii, demonstrating the ability of compound 14 to compromise the integrity of the outer membrane of Gram-negative strains.
Although earlier reports of combination of colistin with teicoplanin and vancomycin exist, these studies were limited to determination of the activity against A. baumanii34,35. Herein, we performed an extended combined susceptibility study by incorporating five Gram negative strains.
We observed relevant differences in potentiation efficacy against different bacterial strains with the same antibiotic. Although this phenomenon seems surprising, similar results have been reported by the Wareham group in the study of the synergistic effect of telavancin and colistin36. The membrane perturbation ability of compound 14, which plays a key role in the potentiating effect, is associated with its binding to bacterial lipopolysaccharides. The composition of the lipopolysaccharide is different in the bacteria involved the present study, and the phosphorylation pattern of the lipopolysaccharides may also be different. We hypothesize that compound 14 interacts with different affinities with different lipopolysaccharides, leading to different membrane perturbation ability and thus different antibacterial activity.
Our results provides further evidence that using glycopeptide antibiotics in combination with membrane-active agents is a viable and resourceful strategy to repurpose them against Gram-negative bacteria.
Experimental
General information
Vancomycin hydrochloride was a gift from TEVA Pharmaceutical Industries Ltd. (Debrecen, Hungary). TLC was performed on Kieselgel 60 F254 (Merck) with detection either by immersing into ammonium molybdate-sulfuric acid solution followed by heating or by using Pauly’s reagent for detection. Flash column chromatography was performed using Silica gel 60 (Merck 0.040–0.063 mm) and Silica gel 60 silanized (0.063–0.200 mm). The 1H NMR (360, 400 and 500 MHz), 13C NMR (90, 100 and 125 MHz) and 2D NMR spectra were recorded with a Bruker DRX-360, a Bruker DRX-400 and a Bruker Avance II 500 spectrometers at 300 K. Chemical shifts are referenced to Me4Si and to the solvent residual signals. MALDI-TOF MS measurements were carried out with a BIFLEX III mass spectrometer (Bruker, Bremen, Germany) or a Bruker Autoflex Speed mass spectrometer equipped with time-of-flight (TOF) mass analyzer. 2,5-Dihydroxybenzoic acid (DHB) was used as matrix and CF3COONa as cationizing agent in DMF. For analytical RP-HPLC a Waters 2695 Separations Module (Waters Corp., Milford, USA) was used. The separations were carried out on a VDSphere PUR 100 C18-M-SE, 5 μm, 150 × 4.6 mm column at an injection volume of 10 μL, using a flow rate of 1.0 mL/min with a Waters 2996 DAD set at 254 nm and a Bruker MicroTOF-Q type Qq-TOF MS instrument (Bruker Daltonik, Bremen, Germany) as detectors. The following system was used for the elution: Solvent A: water:MeCN 9:1 + 0.0025%v/v TFA and Solvent B: MeCN. Gradient elution: from 0% of B to 20% from 0 to 10 min then from 20% of B to 80% from 10 to 30 min and 80% of B from 30 to 35 min. The MicroTOF-Q mass spectrometer was equipped with an electrospray ion source. The mass spectrometer was operated in positive ion mode with a capillary voltage of 3.5 kV, an endplate offset of − 500 V, nebulizer pressure of 1.8 bar, and N2 as drying gas with a flow rate of 9.0 l/min at 200 °C. The mass spectra were recorded by means of a digitizer at a sampling rate of 2 GHz. The mass spectra were calibrated externally using the exact masses of clusters [(NaTFA)n+TFA]+ from the solution of sodium trifluoroacetate (NaTFA). The spectra were evaluated with the DataAnalysis 3.4 software from Bruker.
The routine antibacterial evaluations were carried out as it was described in our previous publication39. Antimicrobial synergy was determined by evaluating the fractional inhibitory concentration (FIC) index and was characterized by a conventional checkerboard assay40. Bacteria grown to mid-logarithmic phase in MHB were pre-incubated with serially diluted concentrations of teicoplanin or vancomycin and compound 14. The sum of the FICs (ΣFIC) was calculated with the equation ΣFIC = FICA + FICB = (CA/MICA) + (CB/MICB), where MICA and MICB are the MICs of antimicrobial A and B alone, respectively, and CA and CB are the inhibitory concentrations of the drugs when combined, respectively. Synergy was defined as a ΣFICs ≤ 0.5 and additive activity was defined as a ΣFICs > 0.5 ≤ 1.0.
The human immortalized hCMEC/D3 cells (Merck KGaA, Darmstadt, Germany, Cat. # SCC066) were cultured in EndoGRO-MV Complete Culture Media (supplemented with the components of the kit and fibroblast growth factor 2 (FGF-2) at 1 ng/mL final concentration). Human Caco-2 intestinal epithelial cells were obtained from European Collection of Cell Cultures (ECACC, UK) and grown routinely in Dulbecco’s Minimum Essential Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid and 1% penicillin–streptomycin solution.
Vancomycin aglycone hexapeptide (2)
The preparation of the title compound was based on literature procedures29,30,33. Vancomycin hydrochloride (1) (1.0 g, 0.673 mmol) was added to 30 mL of pyridine:water 1:1 mixture. Phenyl isothiocyanate (242 μL, 2.019 mmol, 3 equiv.) was added and the reaction mixture was stirred for 3 h at room temperature. Then, 200 mL of acetone was added, and the white precipitate was filtered off and washed with an additional 100 mL of acetone, 100 mL of diethyl ether, then dried. The crude material was dissolved in 10 mL of dry TFA and stirred for one hour at room temperature. Then, 1 mL of water was added and the reaction mixture was heated at 50 °C for 3 h. The mixture was concentrated to a smaller volume and the product was precipitated by the addition of diethyl ether (100 mL), filtered off and washed several times with diethyl ether. The crude product was dissolved in water (75 mL) and the pH was set to ~ 5–6 by adding 1 N sodium hydroxide solution. A yellowish green suspension formed which was filtered, washed with additional water, then after drying, with acetonitrile (50 mL) and ether (50 mL). TLC (Normal Phase Silica, MeCN:H2O = 87:13) indicated, that the cleaved carbohydrates were washed away, there was one glycopeptide derivative and an unknown yellow colored impurity. The yield of the crude product was 600 mg (70%) yellowish solid. MALDI-TOF m/z: 1038.261 [M+Na]+ (calcd. 1038.172 for C46H39Cl2N7O16Na); Rf: 0.35 (Normal Phase Silica, MeCN:H2O = 87:13).
N-decanoyl-D-Leu (6)
Decanoic acid 4 (2 g, 11.6 mmol) was dissolved in thionyl chloride (3 mL) and stirred while heating at 80 °C for two hours. After cooling to room temperature, the reaction mixture was co-evaporated with toluene in vacuo 3 times to remove the excess thionyl chloride. D-leucine 3 (787 mg, 6 mmol) was dissolved in a mixture of 1 M NaOH (6 mL) and THF (6 mL). The freshly prepared decanoyl chloride 5 (1.6 mL, about 1.3 equiv.) was dissolved in 3 mL of THF, and slowly added to the reaction mixture. After stirring for 5 h at room temperature, the solution was diluted with water (100 mL), acidified by cc. HCl solution (750 μL) and extracted with ethyl acetate (3 × 60 mL). The combined organic phase was dried on Na2SO4, filtered and evaporated to dryness at reduced pressure. The white residue was suspended in 50 mL of i-hexane and stirred for half an hour, after which it was filtered off and washed with an additional 50 mL of i-hexane, and dried in vacuum. Yield: 1.40 g (81%), white powder. MALDI-TOF m/z: 308.642 [M+Na]+ (calcd. 308.220 for C16H31NO3Na); 1H NMR (360 MHz, CDCl3) δ 11.22 (s, 1H), 6.32 (d, J = 8.1 Hz, 1H), 4.63 (td, J = 8.4, 4.5 Hz, 1H), 2.25 (t, J = 7.5 Hz, 2H), 1.77–1.66 (m, 2H), 1.66–1.53 (m, 3H), 1.33–1.24 (m, 10H), 0.95 (d, J = 4.6 Hz, 6H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (91 MHz, CDCl3) δ 176.55, 174.44, 77.16, 50.95, 41.39, 36.57, 31.96, 29.56, 29.42, 29.36, 29.30, 25.78, 24.99, 22.94, 22.76, 21.99, 14.20. Rf: 0.62 (Normal Phase Silica, MeCN:H2O = 85:15).
N-decanoyl norvancomycin aglycone (7)
N-Decanoyl-D-Leu (6) (285 mg, 1 mmol) was dissolved in dichloromethane (3 mL) and a suspension of EDCI.HCl (211 mg, 1.1 mmol) in DCM (2 mL) was added dropwise to the solution. After stirring for 30 min at room temperature, N-hydroxysuccinimide (127 mg, 1.1 mmol) was added, and the stirring was continued for 18 h, after which TLC (i-hexane:ethyl acetate = 7:3) indicated almost total consumption of the starting material. The solvent was evaporated.
Crude vancomycin aglycone hexapeptide (2) (500 mg, ~ 0.5 mmol) was dissolved in DMF (4 mL). The above prepared crude NHS ester was dissolved in 1 mL of DMF and added to the reaction mixture. After 2 h of stirring at room temperature, TLC (Normal Phase Silica, toluene:methanol = 1:1) indicated the total consumption of 2. Ethyl acetate (75 mL) and diethyl ether (75 mL) was added and the resulting precipitate was filtered off and washed with additional ether, then dried. The product (753 mg, yellow powder) was used in the next step in its crude form. MALDI-TOF m/z: 1305.361 [M+Na]+ (calcd. 1305.392 for C62H68Cl2N8O18Na); Rf: 0.28 (Normal Phase Silica, toluene:MeOH = 1:1).
N-Boc-ethylenediamine (9)
Ethylenediamine 8 (1.12 mL, 16.7 mmol, 6 equiv.) was added to DCM (5 mL). Boc2O (610 mg, 2.8 mmol) was dissolved in DCM (40 mL) in a dropping funnel and added to the reaction mixture in about 3 h at room temperature. Stirring was continued overnight, then the mixture was concentrated at reduced pressure. The crude material was dissolved in 2 M sodium carbonate solution (100 mL), and extracted with ethyl acetate (3 × 40 mL). The organic phase was dried on Na2SO4, filtered, concentrated then co-evaporated with toluene two times. Yield: 380 mg (85%) (colorless syrup). 1H NMR (400 MHz, CDCl3) δ 5.14 (s, 1H), 3.17 (q, J = 5.5 Hz, 2H), 2.79 (t, J = 5.9 Hz, 2H), 1.45 (s, 9H), 1.39 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 156.30, 79.14, 43.43, 41.90, 28.45. Rf: 0.27 (Normal Phase Silica, i-hexane:acetone = 6:4 + 1% v/v Et3N).
N-decanoyl norvancomycin aglycone N-Boc-aminoethyl amide (10)
Compound 7 (500 mg, 0.39 mmol) was dissolved in 5 mL of DMF:DMSO 1:1 mixture, then Et3N (55 μL, 0.39 mmol) and 9 (125 mg, 0.78 mmol) were added, followed by PyBOP (203 mg, 0.39 mmol). After stirring for 3 h at room temperature, the starting material was consumed as indicated by TLC (Normal Phase Silica, toluene:methanol = 6:4). Diethyl ether (100 mL) was added, and the precipitate was filtered off and washed several times with ether. The resulting yellow material was dissolved in methanol, flash silica gel was added (Kieselgel 60) and the solvents were removed under reduced pressure. The product was purified by flash column chromatography using gradient elution (toluene:MeOH = 90:10, 85:15, 80:20, 75:25). Yield: 199 mg, yellow powder (25% from vancomycin·HCl) MALDI-TOF m/z: 1447.45 [M+Na]+ (calcd. 1447.50 for C69H82Cl2N10O19Na); Rf: 0.63 (Normal Phase Silica, toluene:MeOH = 6:4).
N-Boc-bromoethylamine (12)
Boc2O (2.18 g, 10 mmol) was dissolved in DCM (50 mL) and cooled to 0 °C. 2-bromoethylamine hydrobromide 11 (2 g, 10 mmol) was added, followed by the dropwise addition of Et3N (2.1 mL, 15 mmol). The reaction mixture was allowed to warm up to room temperature, and was stirred overnight. Then, it was diluted with DCM (400 mL) and washed with sat. aq. NH4Cl (2 × 50 mL), sat. aq. NaHCO3 (2 × 50 mL) and brine (2 × 50 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The resulting syrup was chromatographed by flash column chromatography using gradient elution (i-hexane:acetone = 99:1, 97:3, 94:6) yielding 1.78 g (84%) of 12 as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.02 (br s, 1H, NH), 3.57–3.49 (m, 2H, CH2), 3.45 (tr, J = 5.5 Hz, 2H, CH2), 1.45 (s, 9H, 3 × CH3). 13C NMR: δ 155.70 (CO), 79.85 (Cq), 42.45 (NH-CH2), 32.83 (Br-CH2), 28.43 (3C, 3 × CH3). Rf: 0.59 (Normal Phase Silica, i-hexane:EtOAc = 8:2).
Tetraalkyl N-decanoyl norvancomycin aglycone N-Boc-aminoethyl amide (13)
Compound 10 (200 mg, 0.14 mmol) was dissolved in DMF (2 mL). Cs2CO3 (684 mg, 2.1 mmol, 15 equiv.) was added and the reaction mixture was stirred for 30 min at room temperature followed by the addition of N-Boc-bromoethylamine (12) (1.255 g, 5.6 mmol, 40 equiv.) in DMF (1 mL). After stirring for 16 h, the reaction mixture was diluted with methanol (10 mL), silica gel was added, and the mixture was evaporated to dryness at reduced pressure. Purification started with flash chromatography using gradient elution (toluene:MeOH = 99:1, 95:5, 90:10, 85:15, 80:20). Further purification was carried out by dissolving the product in a DCM:MeOH 1:1 mixture, then applying it to a preparative TLC plate. (Normal Phase Silica, layer thickness: 1 mm, EtOAc:toluene:MeOH = 6:3:1 was used as eluent.) Yield: 88 mg (32%) white powder. MALDI-TOF m/z: 2019.78 [M+Na]+ (calcd. 2019.88 for C97H134Cl2N14O27Na);
Tetraaminoethyl N-decanoyl norvancomycin aglycone aminoethyl amide trifluoroacetate salt (14)
Compound 13 was dissolved in dry TFA (1 mL) and stirred at room temperature for 2 h. Then, diethyl ether (50 mL) was added, the white precipitate was filtered off, and washed with additional 50 mL of ether, then dried. The solid was dissolved in H2O:MeCN = 9:1 + 0.2%v/v TFA, silanized silica was added and the slurry was applied to a silanized silica gel column in the same solvent mixture. A step gradient was used (H2O:MeCN = 85:15, 8:2, 7:3, 6:4 + 0.2%v/v TFA) for elution, clean fractions were collected, n-BuOH was added and the solvents were evaporated under reduced pressure (co-evaporated with toluene to remove TFA). The solid material was dissolved in water and lyophilized overnight to obtain 14 as a white foam. Yield: 64 mg (78%, HPLC purity 96%); MALDI-TOF m/z: 1519.60 (calcd. 1519.62 for C72H94Cl2N14O17Na); Rf: 0.28 (Silanized Silica, H2O:MeCN = 6:4 + 0.5%v/v TFA).
Cytotoxicity experiments
The human immortalized hCMEC/D3 cells (Merck KGaA, Darmstadt, Germany, Cat. # SCC066) were cultured in EndoGRO-MV Complete Culture Media (supplemented with the components of the kit and fibroblast growth factor 2 (FGF-2) at 1 ng/mL final concentration).
Human Caco-2 intestinal epithelial cells were obtained from European Collection of Cell Cultures (ECACC, UK) and grown routinely in Dulbecco’s Minimum Essential Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid and 1% penicillin–streptomycin solution.
In cell viability experiments, 1 × 104 hCMEC/D3 or Caco-2 cells/well were seeded on 96-weel plates. After 24 h of incubation, cells were treated with test solutions in different concentrations. The stock solution of the test molecule was dissolved in DMSO and further diluted with cell culture medium to prepare test solutions. The final concentration of DMSO in the test solutions did not exceed 0.5 V/V%. The control group received culture medium. Cells were incubated with the test solutions for 72 h at 37 °C in an atmosphere of 5% CO2, and test solutions were replaced with 0.05 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solutions prepared in PBS. Cells were incubated with MTT solutions for 4 h at 37 °C and the formed dark blue formazan crystals were dissolved in acidic isopropanol (isopropanol: 1.0 N hydrochloric acid = 25:1). The absorbance was measured at 570 nm against a 690 nm reference wavelength with a Thermo Fisher Multiskan Go microplate reader (Thermo Fisher, Waltham, MA, USA). Cell viability was expressed as the percentage of the untreated control and IC50 values were calculated by GraphPad Prism 7 software (GraphPad Software Inc., San Diego, CA, USA).
Cells were also treated with the combinations of derivative 14 and teicoplanin or vancomycin between 2 and 64 µg/mL in checkerboard configuration by the above detailed MTT test.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author (A.B.) on reasonable request.
References
Nicolaou, K. C., Boddy, C. N. C., Brase, S. & Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem.-Int. Ed. 38, 2096–2152 (1999).
Butler, M. S., Hansford, K. A., Blaskovich, M. A., Halai, R. & Cooper, M. A. Glycopeptide antibiotics: back to the future. J. Antibiot. 67(9), 631–644 (2014).
McDonald, L. C. et al. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 66, e1–e48 (2018).
Stevens, V. W. et al. Use of oral vancomycin for Clostridioides difficile infection and the risk of vancomycin-resistant enterococci. Clin. Infect. Dis. 71(3), 645–651 (2020).
Zeng, D. et al. Approved glycopeptide antibacterial drugs: Mechanism of action and resistance. Cold Spring Harb. Perspect. Med. 6, a026989 (2016).
Biondi, S., Chugunova, E. & Panunzio, M. From Natural products to drugs: Glyco- and lipoglycopeptides, a new generation of potent cell wall biosynthesis inhibitors. Stud. Nat. Prod. Chem. 50, 249–297 (2016).
McComas, C. C., Crowley, B. M. & Boger, D. L. Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J. Am. Chem. Soc. 125(31), 9314–9315 (2003).
Sarkar, P., Yarlagadda, V., Ghosh, C. & Haldar, J. A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Med. Chem. Comm. 8(3), 516–533 (2017).
CDC’s antibiotic resistance threats in the United States, 2019. Centers for Disease Control and Prevention, 2019, Atlanta, GA.
Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nature 543(7643), 15–15. https://doi.org/10.1038/nature.2017.21550 (2017).
Peri, A. M. et al. Antimicrobial treatment challenges in the era of carbapenem resistance. Diagn. Microbiol. Infect. Dis. 94(4), 413–425 (2019).
Acharya, Y., Bhattacharyya, S., Dhanda, G. & Haldar, J. Emerging roles of glycopeptide antibiotics: Moving beyond gram-positive bacteria. ACS Infect. Dis. 8(1), 1–28 (2022).
Blaskovich, M. A. T. et al. Developments in glycopeptide antibiotics. ACS Infect. Dis. 4(5), 715–735 (2018).
Dhanda, G., Sarkar, P., Samaddar, S. & Haldar, J. Battle against vancomycin-resistant bacteria: Recent developments in chemical strategies. J. Med. Chem. 62(7), 3184–3205 (2019).
Malabarba, A. et al. Synthesis and antibacterial activity of a series of basic amides of teicoplanin and deglucoteicoplanin with polyamines. J. Med. Chem. 35, 4054–4060 (1992).
Yarlagadda, V., Akkapeddi, P., Manjunath, G. B. & Haldar, J. Membrane active vancomycin analogues: A strategy to combat bacterial resistance. J. Med. Chem. 57(11), 4558–4568 (2014).
Yarlagadda, V. et al. Glycopeptide antibiotic to overcome the intrinsic resistance of gram-negative bacteria. ACS Infect. Dis. 2(2), 132–139 (2016).
Antonoplis, A., Zang, X., Wegner, T., Wender, P. A. & Cegelski, L. Vancomycin-arginine conjugate inhibits growth of carbapenem-resistant E. coli and targets cell-wall synthesis. ACS Chem. Biol. 14(9), 2065–2070 (2019).
Neville, L. F. et al. In vivo targeting of Escherichia coli with vancomycin-arginine. Antimicrob. Agents Chemother. 65(4), E02416–E02420 (2021).
Shi, W. et al. Design, synthesis, and antibacterial evaluation of vancomycin-LPS binding peptide conjugates. Bioorg. Med. Chem. Lett. 45, 128122 (2021).
Cochrane, S. A. & Vederas, J. C. Lipopeptides from bacillus and paenibacillus spp.: A gold mine of antibiotic candidates. Med. Res. Rev. 36, 4–31 (2014).
Velkov, T., Roberts, K. D., Nation, R. L., Thompson, P. E. & Li, J. Pharmacology of polymyxins: New insights into an “old” class of antibiotics. Future Microbiol. 8, 711–724 (2013).
Velkov, T., Thompson, P. E., Nation, R. L. & Li, J. Structure–activity relationships of polymyxin antibiotics. J. Med. Chem. 53(5), 1898–1916 (2010).
Blanchet, M., Borselli, D. & Brunel, J. M. Polyamine derivatives: A revival of an old neglected scaffold to fight resistant Gram-negative bacteria?. Future Med. Chem. 8, 963–973 (2016).
Yin, J. et al. Mechanisms of bactericidal action and resistance of polymyxins for Gram-positive bacteria. Appl. Microbiol. Biotechnol. 104, 3771–3780 (2020).
Rudilla, H. et al. (2018) Novel synthetic polymyxins kill Gram-positive bacteria. J. Antimicrob. Chemother. 73, 3385–3390 (2018).
Szűcs, Z. et al. N-Terminal guanidine derivatives of teicoplanin antibiotics strongly active against glycopeptide resistant Enterococcus faecium. J. Antibiotics 73, 603–614 (2020).
Bereczki, I., Vimberg, V., Lőrincz, E., Papp, H., Nagy, L., Kéki, S., Batta, G., Mitrović, A., Kos, J., Zsigmond, Á., Hajdú, I., Lőrincz, Z., Bajusz, D., Petri, L., Hodek, J., F., Keserű, G. M., Weber, J., Naesens, L., Herczegh, P., & Borbás, A. Semisynthetic teicoplanin derivatives with dual antimicrobial activity against SARS-CoV-2 and multiresistant bacteria, Sci Rep. 12, 16001 (2022).
Booth, P. M., Stone, D. J. M., & Williams, D. H. The Edman degradation of vancomycin: Preparation of vancomycin hexapeptide. J. Chem. Soc. Chem. Commun. 1694–1695 (1987).
Szűcs, Z. et al. Reprogramming of the antibacterial drug vancomycin results in potent antiviral agents devoid of antibacterial activity. Pharmaceuticals 13(7), 139 (2020).
Kam, Y., Rubinstein, A., Nissan, A., Halle, D. & Yavin, E. Detection of endogenous K-ras mRNA in living cells at a single base resolution by a PNA molecular beacon. Mol. Pharmaceut. 9, 685–693 (2012).
Luescher, M. U., Vo, C. V. & Bode, J. W. SnAP reagents for the synthesis of piperazines and morpholines. Org. Lett. 16, 1236–1239 (2014).
Crane, C. M., Pierce, J. G., Leung, S. S. F., Tirado-Rives, J., Jorgensen, W. L., & Boger. D. L. Synthesis and evaluation of selected key methyl ether derivatives of vancomycin aglycon. J. Med. Chem. 53, 7229–7235 (2010).
Wareham, D. W., Gordon, N. C. & Hornsey, M. In vitro activity of teicoplanin combined with colistin versus multidrug-resistant strains of Acinetobacter baumannii. J. Antimicrob. Chemother. 66(5), 1047–1051 (2011).
Gordon, N. C., Png, K. & Wareham, D. W. Potent synergy and sustained bactericidal activity of a vancomycin-colistin combination versus multidrug-resistant strains of Acinetobacter baumannii. Antimicrob. Agents Chemother. 54(12), 5316–5322 (2010).
Hornsey, M., Longshaw, C., Phee, L. & Wareham, D. W. In vitro activity of telavancin in combination with colistin versus Gram-negative bacterial pathogens. Antimicrob. Agents Chemother. 56(6), 3080–3085 (2012).
van Groesen, E. et al. Vancomyxins: Vancomycin-polymyxin nonapeptide conjugates that retain anti-Gram-positive activity with enhanced potency against Gram-negative strains. ACS Infect. Dis. 7(9), 2746–2754 (2021).
Umstätter, F. et al. Vancomycin resistance is overcome by conjugation of polycationic peptides. Angew. Chem. Int. Ed. 59, 8823–8827 (2020).
Szűcs, Z. et al. Lipophilic teicoplanin pseudoaglycon derivatives are active against vancomycin- and teicoplanin-resistant enterococci. J. Antibiot. 70, 664–670 (2017).
Fernandez-Cuenca, F., Martinez-Martinez, L., Pascual, A. & Perea, E. J. In vitro activity of azithromycin in combination with amikacin, ceftazidime, ciprofloxacin or imipenem against clinical isolates of Acinobacter baumannii. Chemotherapy 49, 24–26 (2003).
Acknowledgements
This work was supported by the European Regional Development Fund under projects GINOP-2.3.2-15-2016-00044 and GINOP-2.3.4-15-2020-00008. This research was also funded by the National Research, Development and Innovation Office of Hungary (K132870 and FK 142315). The MS and NMR instrumental background was provided by the GINOP-2.3.3-15-2016-00021 and GINOP-2.3.3-15-2016-00004 projects. The authors are grateful to Gyula Batta for NMR experiments and to Lajos Nagy for HPLC measurements.
Funding
Open access funding provided by University of Debrecen.
Author information
Authors and Affiliations
Contributions
P.H., and A.B. supervised this project. Z.S. and I.B. performed the chemical synthesis and NMR and MALDI-MS measurements. E.O. performed the antibacterial tests. F.F. performed the cytotoxicity study. Z.S., I.B., E.O. and A.B. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szűcs, Z., Bereczki, I., Fenyvesi, F. et al. Synthesis of an amphiphilic vancomycin aglycone derivative inspired by polymyxins: overcoming glycopeptide resistance in Gram-positive and Gram-negative bacteria in synergy with teicoplanin in vitro. Sci Rep 12, 20921 (2022). https://doi.org/10.1038/s41598-022-24807-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-24807-0
This article is cited by
-
In vitro and in vivo antiplasmodial evaluation of sugar-modified nucleoside analogues
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
