
Abstract
Cell death processes in eukaryotes shape normal development and responses to the environment. For plant–microbe interactions, initiation of host cell death plays an important role in determining disease outcomes. Cell death pathways are frequently initiated following detection of pathogen-derived molecules which can lead to resistance or susceptibility to disease depending on pathogen lifestyle. We previously identified several small secreted proteins (SSPs) from the wheat-infecting fungus Zymoseptoria tritici that induce rapid cell death in Nicotiana benthamiana following Agrobacterium-mediated delivery and expression (agroinfiltration). Here we investigated whether the execution of host cells was mechanistically similar in response to different Z. tritici SSPs. Using RNA sequencing, we found that transient expression of four Z. tritici SSPs led to massive transcriptional reprogramming within 48 h of agroinfiltration. We observed that distinct host gene expression profiles were induced dependent on whether cell death occurs in a cell surface immune receptor-dependent or -independent manner. These gene expression profiles involved differential transcriptional networks mediated by WRKY, NAC and MYB transcription factors. In addition, differential expression of genes belonging to different classes of receptor-like proteins and receptor-like kinases was observed. These data suggest that different Z. tritici SSPs trigger differential transcriptional reprogramming in plant cells.
Similar content being viewed by others


Introduction
Multicellular organisms frequently sacrifice individual cells during specific developmental stages or in response to environmental cues. Forms of programmed cell death (PCD) such as apoptosis and autophagy are a normal part of growth and development and also contribute towards the recycling of nutrients. In flowering plants, initiation of PCD plays important roles in processes as diverse as temperature stress, hypoxia, organ development and response to biotic stimuli1,2,3.
In plant-pathogen interactions, cell death is an essential part of the plant immune system2. In interactions with biotrophic pathogens, the active triggering of host cell death termed the hypersensitive response (HR) is often associated with disease resistance4. HR is considered an orderly form of PCD, characterised by DNA laddering, organelle fragmentation and cell shrinkage5,6,7. Induction of HR is assumed to both deny a nutrient supply and spatially restrict invading pathogens8. Activation of HR is mediated by plant disease resistance (R) proteins directly recognising secreted pathogen virulence proteins (effectors) or through recognition of effector action on other host proteins9,10,11 In contrast to HR, necrosis or other uncontrolled forms of cell death are often beneficial to pathogens. Necrotic tissue is characterised by the rupture of plasma membrane and release of cytoplasm to the extracellular spaces. Necrotrophic pathogens in particular benefit from the release of nutrients during necrosis. The importance of control of cell death is illustrated by the variety of pathogen-produced molecules which interfere with these processes. Biotrophic or hemibiotrophic pathogens frequently secrete effectors that are able to suppress activation of immune stimulation that may lead to HR12. In contrast, necrotrophic pathogens produce necrotrophic effectors that actively trigger host cell death pathways13. The lifestyle of each pathogen determines whether induction of cell death has a beneficial or detrimental outcome to the host.
The ascomycete fungus Zymoseptoria tritici (Z. tritici) causes Septoria tritici blotch (STB) disease of wheat (Triticum aestivum) and is a major threat to wheat productivity globally14. Z. tritici is hemibiotrophic, with infection typically being symptomless for 10–14 days, before a rapid transition to necrotrophic phase of the life cycle15. This is initially characterised by leaf chlorosis, followed by the appearance of necrotic lesions in infected areas and sometimes even death of infected leaves. There is considerable transcriptional reprogramming both in host plants and in the fungal cells during infection16,17,18. Wheat responses are characterised by downregulation of defence-related genes during the early symptomless phase, followed by upregulation of many of the same genes during the transition to necrotrophy16,17. In the fungus, there is upregulation of numerous secreted proteins that are likely to function as effectors and in genes associated with production of secondary metabolites16,17.
In previous work, we identified > 100 Z. tritici small secreted proteins (SSPs) that were upregulated during the switch from symptomless to necrotrophic growth16. These were classed as candidate effectors that might be involved in the induction of cell death during this transition. We used the model plant Nicotiana benthamiana to identify a number of SSPs with ability to induce macroscopic cell death in leaves19,20. We found that 13 SSPs induced cell death, and that for 12 of these, initiation of cell death required protein localisation to the apoplastic space. Further, for a smaller group of SSPs (Zt9, Zt11, Zt12) we showed that cell death required the Brassinosteroid Insensitive 1 (BRI1)‐Associated Receptor Kinase 1 (BAK1) and Suppressor of BIR1‐1 (SOBIR1) receptor-like kinases (RLKs). Both BAK1 and SOBIR1 are important co-receptors for the initiation of intracellular signalling following perception of extracellular ligands. These ligands are frequently microbe-associated molecular patterns (MAMPs) or apoplastic effectors. This indicated that initiation of cell death in response to the Z. tritici SSPs occurs at the cell-surface and is likely dependent on recognition by currently unidentified cell-surface immune receptors. In contrast, Zt6 was identified as a ribonuclease toxin that initiates cell death independent of BAK1/SOBIR120. Moreover, Zt6 induced cell death irrespectively of whether it was secreted to the apoplast or localised to the cytoplasm. Zt6 was demonstrated to have RNase activity against rRNA and display a broad toxicity against monocot and dicot plants, yeast and bacteria, though not to Z. tritici itself20.
Based on previous results, we hypothesised that Z. tritici effectors may trigger different immune pathways in N. benthamiana that ultimately lead to macroscopically similar cell death phenotypes. To test this hypothesis, we used a transcriptomic approach (RNA sequencing, RNA-seq) to investigate early host responses to transient expression of a group of previously described Z. tritici SSPs that induce cell death in either a BAK1/SOBIR1-dependent or -independent manner.
Results
RNAseq overview
We aimed to determine the changes in the N. benthamiana transcriptome that occur preceding cell death driven by non-host recognition of three (Zt9, Zt11, and Zt12) Z. tritici SSPs, and a secreted phytotoxic RNase (Zt6) in comparison to a green fluorescent protein (GFP) control. GFP, Zt6 and SSPs were transiently expressed in N. benthamiana leaves using agroexpression and samples were collected at 24- and 48-h post-inoculation (hpi), i.e. prior to the HR becoming visible by eye. RNA was extracted from treated leaves to produce 30 RNA-seq libraries (five treatments x three biological replicates x two timepoints) for sequencing by paired-end sequencing on the Illumina Hiseq 2000 platform. The libraries contained 23.95–35.42 million raw reads. Subsequent quality filtering reduced the number of reads in each library by 37.4% to 51.15%. Of the remaining reads, 92.8% to 98.9% were successfully mapped to the reference N. benthamiana genome21 (Table 1).
Principle component analysis (PCA) of the overall gene-expression profile showed that replicates of each treatment clustered tightly, as well as revealing minimal difference between the GFP and SSP treatments at 24hpi (Fig. 1A). This pattern changed by 48hpi, with SSP treatments showing clear separation compared to the GFP control, although with minimal difference among themselves. In contrast, Zt6 expression induced a different gene-expression profile compared to both the GFP and SSP treatments at 24hpi. This was further exaggerated by the 48hpi timepoint (Fig. 1A).

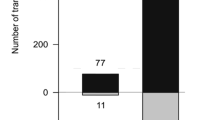
(A) PCA plot of RNA-seq data. SSP treatments and timepoints indicated. (B) Total numbers of upregulated (red) and downregulated (blue) DEGs.
To examine these different responses, differential expression analysis was conducted using DESeq2 Bioconductor package22, with FDR less than 0.05, to identify differentially expressed genes (DEGs) between the SSP and Zt6 treatments compared to the GFP control (Fig. 1B). At 24hpi across all treatments, more DEGs were upregulated than downregulated. At 48hpi, the ratio of upregulated and downregulated DEGs was similar, although the total number of DEGs greatly increased. The increase in number of DEGs was most noticeable for the Zt9 treatment, which had a much smaller number of DEGs at 24hpi than the other treatments (Fig. 1B).
The profound difference between the Zt6 and SSP treatments observed by PCA (Fig. 1A), was reflected in the number of DEGs shared between treatments (Fig. 2). For example, the number of upregulated genes common to all three SSP treatments at 48 h (1294) was far higher than that of any other group of three treatments at 48hpi that contained Zt6 (732, 131, 127 DEGs respectively, Fig. 2A). A similar pattern was observed for downregulated genes, although one particular Zt6-containing group of treatments (Zt11/Zt9/Zt6 vs GFP) at 48 hpi shared a much larger number of DEGs (1069) than other groupings, and almost as much as the SSP treatments group (Zt9/Zt11/Zt12 vs GFP, 1104 DEGs, Fig. 1B). Noticeably, at 48 hpi the Zt11 treatment shared far more DEGs with the Zt6 treatment (482 and 808 upregulated and downregulated respectively) than either of the other two SSP treatments, but at 24hpi was much more similar to the Zt12 treatment (Fig. 1).

Plots showing total numbers of significantly upregulated (A) and significantly downregulated (B) DEGs. Numbers of DEGs shared among one, two, and three treatment combinations are represented by bar size in each main plot, with the individual treatments that make up each combination represented in the bead and line chart below. Total numbers of DEGs in each individual treatment are represented by bar size on the lower left of each plot. Plots generated using UpsetR63.
Gene ontology enrichment analysis
In order to examine whether differentially expressed genes (DEGs) were involved in specific developmental processes, we performed a GO-enrichment analysis of up- and down-regulated genes for the Zt6 (Tables 2, 3) and SSP treatments (Tables 4, 5). For brevity, only the top 20 GO terms (i.e. those with the largest number of DEGs averaged across treatments) enriched in the SSP treatments are listed here.
Most significantly enriched GO terms were present across several treatments, and most were enriched only at the 48hpi time point. Among upregulated DEGs, only four GO terms were enriched in more than one treatment at 24hpi, whilst none were enriched among downregulated DEGs. Out of all significantly enriched GO terms (70 in downregulated DEGs and 113 in upregulated DEGs) 37 were enriched only at the 24hpi time point of only one treatment. Noticeably, 20 of these were enriched only in the Zt12 treatment, while eight, seven, and two were exclusive to the Zt11, Zt9, and Zt6 treatments respectively. Only one of the eight GO terms exclusive to Zt11 (sulfate reduction) was enriched in the downregulated DEGs.
The contrasting transcriptional response to SSP and Zt6 treatments revealed by PCA (Fig. 1A) was also observed in terms of enriched GO categories. Among the upregulated DEGs, only seven GO terms were enriched in all treatments at 48hpi (structural constituent of ribosome, translation initiation factor activity, intracellular, ribosome, translation, metabolic process, and ribosome biogenesis) (Tables 2, 4). Most other GO terms were enriched in either one or all SSP treatments, or they were exclusive of the Zt6 treatment. There were only two terms (hydrolase activity, and catalytic activity) that were common to Zt6 and at least one other treatment. This pattern was similar in downregulated DEGs, where only nine GO terms were enriched across all treatments (carbohydrate metabolic process, extrinsic component of membrane, fructose 1,6-bisphosphate 1-phosphatase activity, membrane, photosynthesis, light harvesting, photosystem I reaction centre, photosystem II, and photosystem II oxygen evolving complex) (Tables 3, 5) also in this case at 48hpi. Strikingly, these nine common GO terms included five (of a total six) photosynthesis-related GO terms. Of those remaining, only five were shared between the Zt6 treatment and at least one SSP treatment. Furthermore, whilst four photosynthesis related GO terms were also significantly enriched among upregulated DEGs, three were exclusive to the Zt11 treatment at 24hpi, and one was significantly enriched in both Zt11 and Zt12 treatments, also only at 24hpi.
The response to Zt6 expression was characterised by an overrepresentation of up-regulated genes involved in protein phosphorylation and kinase activity (Table 2). This included the GO terms transferase activity (of phosphorous-containing groups), protein phosphorylation, protein kinase activity, and protein serine/threonine kinase activity. By contrast, these signaling-related GO terms were enriched amongst downregulated genes for the SSP treatments (Table 5).
GO categories overrepresented among upregulated genes in the SSP treatments were those related to cellular protein catabolic processes. This included threonine type endopeptidase activity, endopeptidase activity, proteasome complex, and proteolysis involved in cellular catabolic process, as well as categories involved in other cellular protein metabolic processes such as protein folding (Table 4).
Over-represented among genes downregulated in the SSP treatments were those involved in diverse processes such as response to hormone and signal transduction, binding and activity of microtubules, lipid and fatty acid metabolism, DNA replication, and protein complex genes (e.g. MCM complex and kinesin complex) (Table 5). Closer investigation showed that downregulation of histone protein genes was the sole driver of enrichment of the GO term nucleosome. GTPase activity and GTP-catabolic process were the only GO terms enriched among both up and downregulated genes of the SSP treatments.
Differential expression of immune receptor-like genes
We previously demonstrated the requirement for the cell-surface co-receptors NbBAK1 and NbSOBIR1 for full induction of cell death by the SSP effector group19. These co-receptors are not required for Zt6-induced cell death20. It is therefore likely that cell death induced by the SSP treatments is a form of immune receptor-mediated programmed cell death. In contrast, Zt6-induced cell death is likely to be more similar to necrosis. Expression of receptors is often upregulated in response to the presence of their ligand23,24,25. We therefore assessed whether there were differential expression patterns of cell surface and cytoplasmic receptor gene families commonly associated with pathogen-associated molecular pattern (PAMP) and/ or effector recognition. Lists of these gene family members used in this assessment were obtained by filtering of the N. benthamiana genome annotation (GFF3) file according to their description in the note field (Supplementary material 1).
There were clear differences in transcriptional profiles across receptor families induced by Zt6 and SSP treatments. At 24hpi, transcriptional changes among wall-associated kinases (WAKs), receptor like kinases (RLKs), leucine-rich repeat receptor like-kinases (LRR-RLKs), and lectin-receptor kinases (LecRLKs) showed a clear bias toward upregulation in the Zt6 treatment (Fig. 3). For WAKs and RLKs this bias was reversed in the SSP treatments, and for LRR-RLKs there was a smaller number of genes upregulated in the SSP treatments in comparison to Zt6. For LecRLKs, a similar bias towards upregulation existed between the Zt6, Zt11, and Zt12 treatments, although this was less prominent for Zt11 and Zt12. At 48hpi, LRR-RLKs in the SSP treatments showed a bias towards downregulation, this was less prominent in the Zt6 treatment. Interestingly, at 48hpi cysteine-rich receptor-like kinases (CRKs) showed a strong bias toward downregulation across all treatments. Genes annotated as nucleotide-binding site leucine-rich repeats (NLRs) showed bias toward upregulation at 48hpi across all treatments, though this was a comparatively small number of genes relative to the total N. benthamiana NLR gene complement.

Total numbers of DEGs from each of five pathogen response associated gene families in each treatment. CRKs, LecRLKs, LRR-RLKs, NLRs, WAKs and RKs. Number of upregulated DEGs are represented by bar size above the x-axis, number of downregulated DEGs are represented by bar size below the x-axis.
Given the differences between the transcriptomes of the Zt6 and SSP treatments, we next identified and investigated specific genes within these five receptor gene families, based on whether they showed a marked difference in expression change between the Zt6 and SSP treatments. For the purposes of this investigation, we defined a “marked difference” as a log2 fold change of an absolute value of at least 1.0 in one or all of the SSP treatments which was either not present or reversed in the Zt6 treatment, or a log2 fold change of an absolute value of at least 1.0 in the Zt6 treatment which was either not present or reversed across all the SSP treatments. For all six receptor gene families, few expression changes were apparent at 24hpi in comparison to the GFP control. However, at 48hpi a clear difference in expression pattern is visible between the Zt6 and SSP treatments. Zt6 treatment specifically induced expression of 11 out of a total 149 LRR-RLKs, 4 out of a total 65 NLRs, 7 out of a total 107 WAKs/RLKs and 7 out of a total 54 LecRLKs in the current N. benthamiana genome annotation (Fig. 4). Zt6 also induced expression of 34 out of a total 795 RLPKs in the current N. benthamiana genome annotation (Fig. 5). Only two LRR-RLKs, two LecRLKs and 7 RLPKs were downregulated in response to Zt6 treatment (Fig. 4). Interestingly, no CRKs showed any transcriptional response to Zt6 (Fig. 4). Very few receptor genes were induced by the SSP treatments, although notable exceptions included one NLR gene induced by all three SSP treatments at 48hpi (Fig. 4), and two RLPK genes relatively strongly induced only by the Zt12 treatment at 48hpi (Fig. 5). Most receptor family genes that were transcriptionally responsive to the SSP treatments were downregulated. These included a notable over-abundance of WAKs and RLKs that were strongly downregulated in response only to the Zt11 treatment at 48hpi. Together, this suggests that there is reprogramming of receptor gene expression following exposure to SSPs, and that this differs between individual SSPs.

Expression profile of defence associated genes with marked difference in expression change between Zt6 and Zt9, Zt11, and Zt12.

Expression profile of RLPKs genes with marked difference in expression change between Zt6 and Zt9, Zt11, and Zt12.
Differential expression of transcription factors, senescence- and programmed cell death-associated genes
There are differences in how developmental, pathogen-associated, and stress-induced PCD is executed. These distinct but partially overlapping pathways share some common components, although no pathway is fully characterised26,27,28. Our GO enrichment analysis showed that Zt6 and SSP treatments appear to be inducing different types of transcriptional reprogramming, and that Zt6-induced cell death is distinct from ordered receptor-mediated PCD26. We therefore investigated whether gene families commonly associated with transcriptional reprogramming related to senescence and PCD displayed differential expression patterns between the Zt6 and SSP effector treatments. We first evaluated the expression of the NAC, WRKY, TCP and MYB transcription factor (TF) families (Fig. 6), which are widely reported as regulators of leaf senescence29,30,31,32. At 24hpi there were few differential expression changes across any of the treatments. However, by 48hpi there were clear differences in expression patterns between Zt6 and SSP treatments. Overall, Zt6 treatment induced expression of 29 out of a total 241 NACs, 20 out of a total 151 WRKYs, 5 out of a total 61 TCPs and 18 out of a total 242 MYBs in the current N. benthamiana genome annotation. Only four TFs (all NAC family) were downregulated in response to Zt6 at this timepoint. In contrast, few TFs of any family were induced by the SSP treatments. Indeed, for WRKY, MYB and TCP families the majority of differentially expressed TFs were downregulated in response to effector treatment. There was minimal overlap between the individual genes transcriptionally induced by Zt6, and those transcriptionally repressed by the SSP treatments.

Expression profile of senescence regulating transcription factor genes with marked difference in expression change between Zt6 and Zt9, Zt11, and Zt12.
Several other non-TF gene families have previously been linked to senescence and cell death in plants. Metacaspases, zinc-finger domain containing proteins and HR-inducing proteins have all been shown to regulate PCD or HR33,34. The senescence-associated genes SAG1 (Arabidopsis thaliana), SAG102 (Medicago truncatula), phytoalexin–deficient 4 (PAD4/SAG101) and harpin-induced gene 1 (HIN1), are markers of senescence and HR respectively35,36,37. In addition, STAY-GREEN (SGR) genes operate downstream of NAC TF regulation of senescence in the catabolism of chlorophyll38. We investigated expression of these genes across all treatments and found that expression of genes encoding metacaspases, SAG, HR-inducing and SGR proteins between Zt6 and SSP treatments was similar (Fig. S1). However, a zinc-finger containing protein encoding gene was strongly induced by Zt6 treatment at 24- and 48hpi but not by the SSP treatments.
Discussion
In this investigation, we aimed to understand the mechanisms of cell death induced by several SSPs of Z. tritici in a non-host plant N. benthamiana. To do this, we made use of the Agrobacterium-mediated transient expression system originally used to identify the cell death-inducing activity of these proteins. Our results indicate that expression of this group of proteins initiate massive transcriptional reprogramming of plant cells prior to onset of macroscopic cell death. Furthermore, the cytotoxic secreted ribonuclease Zt6 induces a transcriptional response distinct from other SSPs.
The transcriptional responses to SSP treatments were broadly similar, and clearly distinct from that induced by Zt6 expression. PCA (Fig. 1A) illustrates clustering of SSP treatments at 24hpi and obvious separation from both Zt6 and GFP control treatments. This separation is further exaggerated by 48hpi. The total number of genes that are differentially expressed is greater in Zt6 treated leaves at 24hpi in comparison to SSP treatments. This indicates that transcriptional reprogramming begins earlier for Zt6 compared to SSP treatments. This is consistent with the earlier onset of macroscopic cell death induced by Zt6 in comparison with the SSPs20. This may reflect that Zt6 induces cell death due to its enzymatic RNase activity targeting rRNA20, whereas the SSPs presumably induce immune receptor-mediated cell death19.
Given the dependency of SSP induced cell death on BAK1, broad similarity of transcriptional response to all three SSP treatments is not surprising. BAK1 functions as a co-receptor for various RLKs, including some whose ligands are PAMPs or secreted effectors; it is a convergence point of multiple pathogen-triggered physiological pathways that lead to PCD39. However, BAK1 dependent PCD has been shown to be highly ligand specific and could still proceed via a variety of mechanisms, dependent upon how its PAMP/effector co-receptor role disrupts its normal functioning and modifies how it subsequently interacts with other RLKs (including but not limited to BIR1 and SOBIR1), which also have important roles in regulation of PCD39.
A notable difference in response to the Zt6 and SSP treatments was differential regulation of genes involved in microtubule activity, movement and binding. Downregulation of genes in these categories was enriched in SSP treatments at 48hpi (Table 5), but not in Zt6 at either timepoint (Table 3). Depolymerisation of the microtubule network has been associated with PCD40, in particular in HR reactions in A. thaliana41 and soybean42. Microtubule reorganisation is also associated with developmental PCD processes such as self-incompatibility43. That genes facilitating maintenance of a normal microtubule network are downregulated in SSP treatments is a characteristic of an orderly form of PCD. The absence of this downregulation during Zt6 expression is consistent with a form of cell death relying less on cytoskeleton arrangement. In contrast, GO term analysis revealed enrichment of terms associated with ribosome, structural constituent of the ribosome, translation and translation initiation factor activity in genes upregulated by Zt6 at 48hpi (Table 2). This suggests Zt6 treatment induced a significant stress on ribosome function and on protein translation in general. These terms are not enriched in either up- or downregulated groups for the SSP treatments at either timepoint (Tables 4, 5). An upregulation of genes related to ribosome structure and function could be indicative of cells experiencing ribosomal stress and therefore perturbations in protein synthesis. This would be anticipated in cells expressing Zt6 which has previously been shown to cleave plant rRNA in a semi-specific manner20. These expression signatures are a likely response to compensate for reduced ribosome functionality in cells expressing Zt6.
The GO terms protein kinase activity and protein serine/threonine kinase activity were strongly induced by Zt6 treatment at the 24hpi timepoint (Table 2). In contrast, these groups were unchanged at 24hpi and subsequently downregulated at 48hpi in the SSP treatment group (Table 5). The activity of several serine/threonine kinase proteins is important for control of apoptosis and autophagy in animal systems44. However, the role of this protein class in direct activation of plant cell death is less clear. Many transmembrane receptor kinases and intracellular kinases play important roles in ligand perception and signal transduction during pathogen interaction. We were specifically interested in expression patterns of genes belonging to the RLP, RLK and WAK receptor families. These classes of receptors are well known to be involved in recognition of PAMPs or apoplastic effectors. Indeed, the only two cloned R genes against Z. tritici are Stb6 and Stb16q, encoding a WAK and a CRK respectively24,45. Expression of immune receptors is often upregulated in response to pathogens23,24,25. Here, we found a number of genes annotated as receptor-like kinases, WAKs, LRR-RLKs, CRKs and LecRLKs were transcriptionally responsive to SSP expression (Fig. 4). Expression patterns were similar between SSP effector treatments in comparison to Zt6. Most differentially expressed receptors in these classes were downregulated upon SSP expression, but upregulated in response to Zt6. A small number of genes annotated as NLRs were induced by Zt6 expression only. A larger number of genes annotated as RLPKs were differentially expressed upon SSP treatment. Again, there is clear differentiation between expression patterns induced by SSPs in comparison to Zt6. Nearly all RLPKs differentially expressed in the Zt6 group were upregulated, whereas nearly all RLPKs that changed in response to SSPs were downregulated. Our data therefore provides a small number of candidate cell surface immune receptors for recognition of Z. tritici SSPs for validation in a follow-on study.
The GO terms proteasome core complex and proteolysis involved in cellular protein catabolic process were enriched amongst upregulated genes for all three of the SSP group treatments at 48hpi. In contrast, proteolysis was enriched amongst downregulated genes at 48hpi for the Zt6 treatment. The contribution of proteasome function to PCD has previously been investigated. Silencing of components of the 26S proteasome leads to build-up of polyubiquitinated proteins and induction of PCD46, suggesting proteasome function negatively regulates PCD. In contrast, Hatsugai and colleagues identified a mechanism involving PBA1 linking proteasome function with promotion of PCD47. These contrasting results suggest that role of the proteasome in cell death may be highly complex.
Given the differences in expression profile of many receptor gene groups between Zt6 and SSP treatments, we focussed on TF family expression patterns. Overall, Zt6 expression led to induction of many NAC, MYB and WRKY TFs (Fig. 6). In contrast, the majority of these genes were either unresponsive or repressed by SSP expression. This pattern was also true for TCP TFs, although the number of genes that were differentially expressed were much lower. NAC TFs play important roles in senescence and in both biotic and abiotic stress responses. Expression of the NAC transcription factor gene Niben101Scf01498g04003 was striking as it was strongly upregulated in the Zt6 48 h treatment exclusively (Fig. 6A). This gene shows a high level of homology to A. thaliana ANAC032 (E-value = 8e−08). ANAC032 regulates senescence through modulation of AtNYE1, the so-called STAY-GREEN gene involved in ability to catabolize chlorophyl38. In N. benthamiana there are six STAY-GREEN genes, however, only two were responsive to treatment and this pattern was similar across Zt6 and SSP treatments. This suggests that while transcriptional promotion of senescence at the level of TF genes appears starkly different between Zt6 and SSP treatments, there may yet be some downstream regulatory convergence.
Other pathways by which upregulation of Niben101Scf01498g04003 may promote senescence are alluded to by its other closest homologs in A. thaliana. The second of these was ANAC047 (AKA. SPEEDY HYPONASTIC GROWTH) (E-value = 1e−07). ANAC047 is upregulated during and may promote leaf senescence via regulation of ACO5 (1-aminocyclopropane-1-carobxylic acid oxidase 5), an enzyme involved in ethylene biosynthesis48,49. In our results, a number of genes annotated as ACO5 and ACO5 homologs exhibit both up and downregulation in the Zt6 treatments. Alternatively, the 10th closest homolog of Niben101Scf01498g04003 is ANAC082 (E-value = 2e−05) which plays a role in the sensing of nucleolar stress50. This is due to the presence of an upstream open reading frame (uORF) in ANAC082 mRNA. Previously studied uORFs act as negative regulators of the main ORF due to ribosome stalling on the mRNA. Upregulation of ANAC082 expression could therefore be indicative of ribosome instability induced by Zt6 expression. Given the known interaction between Zt6 and rRNA, this could provide a mechanism for Zt6-specific patterns of transcriptional reprogramming and a role for this TF as a master regulator of downstream gene expression.
A number of MYB TF genes were also highly upregulated by Zt6 but not by other treatments (Fig. 6B). Of these, Niben101Scf04560g07009 and Niben101Scf01694g12010 were most strongly induced. These genes are orthologous to AtMYB36/MYB68 and AtMYB119 respectively. These TFs are known to have roles in root cell differentiation51, root development52 and cellular differentiation during female gametogenesis53. However, these TFs are not documented to be involved in induction of cell death or pathogen responses. Similarly, the N. benthamiana ortholog of AtTCP5 was induced by Zt6 treatment alone (Fig. 6C). This TF has roles in floral development and ethylene biosynthesis54 but not known to be involved in cell death pathways.
WRKY transcription factors have been identified as important regulators of biotic stress responses55. It is possible that increased WRKY activity accounts for the modified expression of defence genes such as RLPs and RLKs. Therefore, the complex interplay between these TF families might explain the transcriptional profile induced by the effector treatments. A number of WRKYs were transcriptionally induced by Zt6 but not by SSP expression (Fig. 6D). Of these, Niben101Scf01281g05001 and Niben101Scf12560g00018 were the most strongly upregulated. These TFs are orthologues to AtWRKY14/35 and WRKY22 respectively. Constitutively activated WRKY14 is known to promote cell death in N. benthamiana56. WRKY22 has previously been implicated in plant defence responses and loss of WRKY22 expression compromises effector-induced cell death in N. benthamiana57. This suggests that WRKY-dependent transcriptional reprogramming may contribute to cell death induced by Zt6.
Taken together our results show that Z. tritici secreted ribonuclease Zt6 and three SSPs, trigger cell death in non-host N. benthamiana at least partially via gene expression changes in clearly distinct cohorts of genes. Within these cohorts, only a small number of genes that could function as immune receptors were upregulated in response to SSP expression, and therefore provide a manageable set of candidates for further study as potential Z. tritici non-host R-genes. Among TF genes in these cohorts, those transcriptionally responsive to Zt6 expression suggest a potential pathway to ribosomal stress induced PCD. This work provides a detailed picture of transcriptional changes that occur in N. benthamiana prior to cell death induced by apoplastic recognition of non-host pathogen SSPs, and ribonuclease activity.
Methods
Plants and bacterial strains
All N. benthamiana plants were from a seed stock used in our previous investigation under the same growth conditions19,20. The Agrobacterium tumefaciens GV3101 (pMP90) strains expressing Zt6, Zt9, Zt11 and Zt12 from pEAQ-HT-DEST3 were described previously19,20.
Generation of RNA samples
Leaves of 5-week old N. benthamiana seedlings were syringe infiltrated with Agrobacterium suspensions at OD600 = 1.2 in Agroinfiltration buffer (10 mM MgCl2, 10 mM MES, 150 µM acetosyringone, pH 5.6). Six plants were infiltrated per treatment with 30 plants used in total. Leaf sampling was performed at 24 h and 48 h post infiltration. Three infiltrated leaf patches, one each from three individual plants were cut from leaves and pooled to produce each sample. Samples were snap frozen in liquid nitrogen and stored at − 80 °C until processing.
RNA extraction
Frozen leaf samples were ground in liquid nitrogen using a mortar and pestle. RNA was extracted using a Trizol/Chloroform procedure as described previously58. DNase digest was performed using RQ1 DNase (Promega) and RNA recovered by ethanol precipitation. RNA quality and purity was measured using Qubit and Nanodrop. RNA-seq was performed by BGI on the Illumina HiSeq2500 platform.
Quality control and alignment
Quality of raw reads was manually assessed using FastQC software v0.11959. Filtering of raw reads was then performed using PRINSEQ-lite software v0.20.4 to a minimum Phred-quality score of 2660. Version 1.0.1 of the N. benthamiana reference genome was downloaded from the Sol Genomics Network ftp site (ftp://ftp.solgenomics.net/genomes/Nicotiana_benthamiana). Index of the reference genome was built using the build function in HISAT2 v2.1.0, filtered paired-end reads were aligned to the reference genome using the same software61. Following alignment, resulting .BAM files for each treatment were checked for uniformity of gene body coverage and sufficient reads per kilo base per million mapped reads (RPKM) saturation using RSeQC v2.6.462.
Read counting and differential expression analysis
Reads mapped to each gene were counted using HTSeq v0.11.0. Differential expression analysis was conducted between each treatment and the control treatment (GFP) at the appropriate time point using the DESeq2 R package v1.32.0, with a Benjamini-Hochberg – false discovery rate (BH-FDR) corrected P-value of 0.0522.
Gene ontology enrichment analysis
Significantly differentially expressed genes (DEGs) were divided into those that were up-regulated and those that were down-regulated at each time point in each treatment. Gene ontology enrichment analysis was conducted on each of these two groups of DEGs using the GOseq R package with BH-FDR P-value adjustment and gene length bias correction, GO terms with adjusted P-values less than 0.05 were considered to be significantly enriched in that group22.
Ethics approval
All handling of plants, microorganisms and associated samples in this study was performed under biosafety regulations in place at Rothamsted Research. All work was carried out under the Department for Environment, Food and Rural Affairs (Defra) plant health licenses Nos. 101941/197343/8 and 101948/198285/4.
Data availability
All RNA-seq raw sequencing data used in this study were deposited into the NCBI SRA under BioProject accession number PRJNA858969.
References
Reape, T. J., Molony, E. M. & McCabe, P. F. Programmed cell death in plants: Distinguishing between different modes. J. Exp. Bot. 59(3), 435–444 (2008).
Dickman, M. B. & de Figueiredo, P. Death be not proud—Cell death control in plant fungal interactions. PLoS Pathog. 9(9), e1003542 (2013).
Daneva, A., Gao, Z., Van Durme, M. & Nowack, M. K. Functions and regulation of programmed cell death in plant development. Annu. Rev. Cell Dev. Biol. 32(1), 441–468. https://doi.org/10.1146/annurev-cellbio-111315-124915 (2016).
Kamoun, S., Huitema, E. & Vleeshouwers, V. G. Resistance to oomycetes: A general role for the hypersensitive response?. Trends Plant Sci. 4(5), 196–200 (1999).
Salguero-Linares, J. & Coll, N. S. Plant proteases in the control of the hypersensitive response. J. Exp. Bot. 70(7), 2087–2095 (2019).
Yao, N. et al. Apoptotic cell death is a common response to pathogen attack in oats. Mol. Plant Microbe Interact. 15(10), 1000–1007 (2002).
Kiba, A., Takata, O., Ohnishi, K. & Hikichi, Y. Comparative analysis of induction pattern of programmed cell death and defense-related responses during hypersensitive cell death and development of bacterial necrotic leaf spots in eggplant. Planta 224(5), 981–994 (2006).
Mur, L. A., Kenton, P., Lloyd, A. J., Ougham, H. & Prats, E. The hypersensitive response; The centenary is upon us but how much do we know?. J. Exp. Bot. 59(3), 501–520 (2008).
Heath, M. C. Apoptosis, programmed cell death and the hypersensitive response. Eur. J. Plant Pathol. 104(2), 117–124 (1998).
Van Doorn, W. G. et al. Morphological classification of plant cell deaths. Cell Death Differ. 18(8), 1241–1246 (2011).
Dalio, R. J. et al. Hypersensitive response: From NLR pathogen recognition to cell death response. Ann. Appl. Biol. 178(2), 268–280 (2021).
Dou, D. & Zhou, J. M. Phytopathogen effectors subverting host immunity: Different foes, similar battleground. Cell Host Microbe 12(4), 484–495 (2012).
Shao, D., Smith, D. L., Kabbage, M. & Roth, M. G. Effectors of plant necrotrophic fungi. Front. Plant Sci. 12, 995 (2021).
Kettles, G. J. & Kanyuka, K. Dissecting the molecular interactions between wheat and the fungal pathogen Zymoseptoria tritici. Front. Plant Sci. 7, 508 (2016).
Sánchez-Vallet, A., McDonald, M. C., Solomon, P. S. & McDonald, B. A. Is Zymoseptoria tritici a hemibiotroph?. Fungal Genet. Biol. 79, 29–32 (2015).
Rudd, J. J. et al. Transcriptome and metabolite profiling of the infection cycle of Zymoseptoria tritici on wheat reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 167(3), 1158–1185 (2015).
Ma, X., Keller, B., McDonald, B. A., Palma-Guerrero, J. & Wicker, T. Comparative transcriptomics reveals how wheat responds to infection by Zymoseptoria tritici. Mol. Plant Microbe Interact. 31(4), 420–431 (2018).
Kellner, R. et al. Expression profiling of the wheat pathogen Zymoseptoria tritici reveals genomic patterns of transcription and host-specific regulatory programs. Genome Biol. Evol. 6(6), 1353–1365 (2014).
Kettles, G. J., Bayon, C., Canning, G., Rudd, J. J. & Kanyuka, K. Apoplastic recognition of multiple candidate effectors from the wheat pathogen Zymoseptoria tritici in the nonhost plant Nicotiana benthamiana. New Phytol. 213(1), 338–350 (2017).
Kettles, G. J. et al. Characterization of an antimicrobial and phytotoxic ribonuclease secreted by the fungal wheat pathogen Zymoseptoria tritici. New Phytol. 217(1), 320–331 (2018).
Bombarely, A. et al. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol. Plant Microbe Interact. 25(12), 1523–1530 (2012).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 1–21 (2014).
Li, B., Meng, X., Shan, L. & He, P. Transcriptional regulation of pattern-triggered immunity in plants. Cell Host Microbe 19(5), 641–650 (2016).
Saintenac, C. et al. Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat. Genet. 50(3), 368–374 (2018).
Beck, M. et al. Expression patterns of flagellin sensing 2 map to bacterial entry sites in plant shoots and roots. J. Exp. Bot. 65(22), 6487–6498 (2014).
Zhao, Z. X. et al. RPW8. 1 enhances the ethylene-signaling pathway to feedback-attenuate its mediated cell death and disease resistance in Arabidopsis. New Phytol. 229(1), 516 (2021).
Cubría-Radío, M. & Nowack, M. K. Transcriptional networks orchestrating programmed cell death during plant development. Curr. Top. Dev. Biol. 131, 161–184 (2019).
Coll, N. S., Epple, P. & Dangl, J. L. Programmed cell death in the plant immune system. Cell Death Differ. 18(8), 1247–1256 (2011).
Buchanan-Wollaston, V. et al. Comparative transcriptome analysis reveals significant differences in gene expression and signalling pathways between developmental and dark/starvation-induced senescence in Arabidopsis. Plant J. 42(4), 567–585 (2005).
Guo, P. et al. A tripartite amplification loop involving the transcription factor WRKY75, salicylic acid, and reactive oxygen species accelerates leaf senescence. Plant Cell 29(11), 2854–2870 (2017).
Ren, T. et al. Involvement of NAC transcription factor SiNAC1 in a positive feedback loop via ABA biosynthesis and leaf senescence in foxtail millet. Planta 247(1), 53–68 (2018).
Zhang, Y., Wang, H. L., Li, Z. & Guo, H. Genetic network between leaf senescence and plant immunity: Crucial regulatory nodes and new insights. Plants 9(4), 495 (2020).
Coll, N. S. et al. Arabidopsis type I metacaspases control cell death. Science 330(6009), 1393–1397 (2010).
Dietrich, R. A., Richberg, M. H., Schmidt, R., Dean, C. & Dangl, J. L. A novel zinc finger protein is encoded by the Arabidopsis LSD1 gene and functions as a negative regulator of plant cell death. Cell 88(5), 685–694 (1997).
Lapin, D. et al. A coevolved EDS1-SAG101-NRG1 module mediates cell death signaling by TIR-domain immune receptors. Plant Cell 31(10), 2430–2455 (2019).
Oh, S. A., Lee, S. Y., Chung, I. K., Lee, C. H. & Nam, H. G. A senescence-associated gene of Arabidopsis thaliana is distinctively regulated during natural and artificially induced leaf senescence. Plant Mol. Biol. 30(4), 739–754 (1996).
Espinoza, C., Medina, C., Somerville, S. & Arce-Johnson, P. Senescence-associated genes induced during compatible viral interactions with grapevine and Arabidopsis. J. Exp. Bot. 58(12), 3197–3212 (2007).
Mahmood, K., Xu, Z., El-Kereamy, A., Casaretto, J. A. & Rothstein, S. J. The Arabidopsis transcription factor ANAC032 represses anthocyanin biosynthesis in response to high sucrose and oxidative and abiotic stresses. Front. Plant Sci. 7, 1548 (2016).
Gao, X., Ruan, X., Sun, Y., Wang, X. & Feng, B. BAKing up to survive a battle: Functional dynamics of BAK1 in plant programmed cell death. Front. Plant Sci. 9, 1913 (2019).
Smertenko, A. & Franklin-Tong, V. E. Organisation and regulation of the cytoskeleton in plant programmed cell death. Cell Death Differ. 18(8), 1263–1270 (2011).
Takemoto, D., Jones, D. A. & Hardham, A. R. Re-organization of the cytoskeleton and endoplasmic reticulum in the Arabidopsis pen1-1 mutant inoculated with the non-adapted powdery mildew pathogen, Blumeria graminis f. sp. hordei. Mol. Plant Pathol. 7(6), 553–563 (2006).
Cahill, D., Rookes, J., Michalczyk, A., McDonald, K. & Drake, A. Microtubule dynamics in compatible and incompatible interactions of soybean hypocotyl cells with Phytophthora sojae. Plant. Pathol. 51(5), 629–640 (2002).
Poulter, N. S., Vatovec, S. & Franklin-Tong, V. E. Microtubules are a target for self-incompatibility signaling in Papaver pollen. Plant Physiol. 146(3), 1358–1367 (2008).
Singh, P., Ravanan, P. & Talwar, P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front. Mol. Neurosci. 9, 46 (2016).
Saintenac, C. et al. A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch. Nat. Commun. 12(1), 1–10 (2021).
Kim, M. et al. Activation of the programmed cell death pathway by inhibition of proteasome function in plants. J. Biol. Chem. 278(21), 19406–19415 (2003).
Hatsugai, N. et al. A novel membrane fusion-mediated plant immunity against bacterial pathogens. Genes Dev. 23(21), 2496–2506 (2009).
Moschen, S. et al. Identification of candidate genes associated with leaf senescence in cultivated sunflower (Helianthus annuus L.). PLoS ONE 9(8), e104379 (2014).
Rauf, M. et al. NAC transcription factor speedy hyponastic growth regulates flooding-induced leaf movement in Arabidopsis. Plant Cell 25(12), 4941–4955 (2013).
Ohbayashi, I. & Sugiyama, M. Plant nucleolar stress response, a new face in the NAC-dependent cellular stress responses. Front. Plant Sci. 8, 2247 (2018).
Liberman, L. M., Sparks, E. E., Moreno-Risueno, M. A., Petricka, J. J. & Benfey, P. N. MYB36 regulates the transition from proliferation to differentiation in the Arabidopsis root. Proc. Natl. Acad. Sci. 112(39), 12099–12104 (2015).
Feng, C. et al. Arabidopsis MYB68 in development and responses to environmental cues. Plant Sci. 167(5), 1099–1107 (2004).
Rabiger, D. S. & Drews, G. N. MYB64 and MYB119 are required for cellularization and differentiation during female gametogenesis in Arabidopsis thaliana. PLoS Genet. 9(9), e1003783 (2013).
van Es, S. W. et al. Novel functions of the Arabidopsis transcription factor TCP 5 in petal development and ethylene biosynthesis. Plant J. 94(5), 867–879 (2018).
Wani, S. H., Anand, S., Singh, B., Bohra, A. & Joshi, R. WRKY transcription factors and plant defense responses: Latest discoveries and future prospects. Plant Cell Rep. 40(7), 1071–1085 (2021).
Adachi, H. et al. WRKY transcription factors phosphorylated by MAPK regulate a plant immune NADPH oxidase in Nicotiana benthamiana. Plant Cell 27(9), 2645–2663 (2015).
Ramos, R. N., Martin, G. B., Pombo, M. A. & Rosli, H. G. WRKY22 and WRKY25 transcription factors are positive regulators of defense responses in Nicotiana benthamiana. Plant Mol. Biol. 105(1), 65–82 (2021).
Kettles, G. J. et al. sRNA profiling combined with gene function analysis reveals a lack of evidence for cross-kingdom RNAi in the wheat–Zymoseptoria tritici pathosystem. Front. Plant Sci. 10, 892 (2019).
Andrews, S. FastQC: A quality control tool for high throughput sequence data. Available online at http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27(6), 863–864 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37(8), 907–915 (2019).
Wang, L., Wang, S. & Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 28(16), 2184–2185 (2012).
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940 (2017).
Acknowledgements
The authors would like to thank Rothamsted Research horticultural services for plant care and maintenance. High-performance computing resources were provided by the University of Birmingham BLUEbear cluster.
Author information
Authors and Affiliations
Contributions
Agroinfiltrations were performed by G.K. and RNA extracted and purified by C.B. Computational analysis of RNAseq data was done by T.W., G.K., J.R. and K.K. conceived the project and T.W., G.K., J.R. and K.K. wrote the manuscript. This work was funded by Rothamsted Research Fellowship Grant (to J.R. and K.K.) and a JABBS Foundation startup package to G.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Welch, T., Bayon, C., Rudd, J.J. et al. Induction of distinct plant cell death programs by secreted proteins from the wheat pathogen Zymoseptoria tritici. Sci Rep 12, 17880 (2022). https://doi.org/10.1038/s41598-022-22660-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-22660-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
