
Abstract
Growing evidence suggests that the origins of the panzootic amphibian pathogens Batrachochytrium dendrobatidis (Bd) and Batrachochytrium salamandrivorans (Bsal) are in Asia. In Taiwan, an island hotspot of high amphibian diversity, no amphibian mass mortality events linked to Bd or Bsal have been reported. We conducted a multi-year study across this subtropical island, sampling 2517 individuals from 30 species at 34 field sites, between 2010 and 2017, and including 171 museum samples collected between 1981 and 2009. We analyzed the skin microbiome of 153 samples (6 species) from 2017 in order to assess any association between the amphibian skin microbiome and the probability of infection amongst different host species. We did not detect Bsal in our samples, but found widespread infection by Bd across central and northern Taiwan, both taxonomically and spatially. Museum samples show that Bd has been present in Taiwan since at least 1990. Host species, geography (elevation), climatic conditions and microbial richness were all associated with the prevalence of infection. Host life-history traits, skin microbiome composition and phylogeny were associated with lower prevalence of infection for high altitude species. Overall, we observed low prevalence and burden of infection in host populations, suggesting that Bd is enzootic in Taiwan where it causes subclinical infections. While amphibian species in Taiwan are currently threatened by habitat loss, our study indicates that Bd is in an endemic equilibrium with the populations and species we investigated. However, ongoing surveillance of the infection is warranted, as changing environmental conditions may disturb the currently stable equilibrium.
Similar content being viewed by others

Introduction
Globalization and environmental change has led to the emergence of infectious diseases that threaten biodiversity and contribute to the ongoing 6th mass extinction1,2. Prominent examples are white-nose-syndrome in bats3, ash tree dieback4 and chytridiomycosis in amphibians5. Diseases caused by fungal pathogens such as these are emerging worldwide, leading to attrition in biodiversity, ecosystems and food security6,7. Declines due to epizootics that affect multiple host species simultaneously are leading to cascading effects across food webs through changes in species interactions and resulting ecosystem-level changes8,9.
Two emerging fungal pathogens, Batrachochytrium dendrobatidis (Bd) and Batrachochytrium salamandrivorans (Bsal), which cause the disease chytridiomycosis, have impacted amphibian populations around the world10,11,12. The geographic origin of Bd has been contested and the sources were variously suggested to be South Africa13, North America14, South America15, Japan16, and East Asia17. However, there is now compelling evidence that Bd may have originated from the Korean Peninsula18; this region is a global center of Bd genetic diversity19 and places the ancestral population of Bd within East Asia, corroborating results from China20, Korea21,22, Japan16, and Indonesia23. An out-of-Asia origin for Bsal has also been proposed based on the co-occurrence of these species in the region, and the lack of epizootics11. While Bsal has been found in East Asian countries11, predominantly in Vietnamese salamanders24, a widespread occurrence of Bsal has not been confirmed for China20,25 (but see26). Generally, no clear evidence for disease and declines associated with Bd or Bsal exist for Central Asia despite its potential role as source region of the most devastating infections ever recorded26.
The global trade in amphibians has led to ample opportunities for vectoring these pathogens worldwide27. While mass die-offs of amphibians have not been reported in Asia, our current knowledge about host range, environmental preferences, and distributions, as well as the potential impact of fungal pathogens in Asia is limited. In order to fill this knowledge gap, there is a pressing need to undertake surveillance of Bd and Bsal across their putative native range to more fully understand the factors that account for their distribution, epidemiology, and susceptibility of hosts26,28.
Identifying the biotic and abiotic factors that modify the prevalence of infection is necessary to better understand the interaction between hosts, pathogens, environment, and co-occurring microorganisms29,30. A large body of evidence supports the importance of symbiotic bacteria and skin defense peptides as defense mechanisms against pathogens including skin-infecting chytridiomycetes31,32. It has been shown that higher microbial richness and diversity can reduce the risk of Bd infection30. For example, laboratory infections have shown that bacterial diversity may influence whether Bd can infect an individual and to what intensity33. Under natural conditions, the skin microbiome of wild Bd-infected amphibians usually differs from uninfected individuals34. The differences were attributed to Bd-induced microbial dysbiosis, or a disruption of bacteria abundances35. We are also only at the beginning of our understanding of the factors that determine the structure of microbial communities on amphibian skin36,37,38,39. For instance, it is known that the amphibian skin microbiome is not wholly determined by the environmental microbial community, but also shows specificity to the host species30,40,41,42,43.
We surveyed wild amphibian populations in the tropical mountainous island of Taiwan, an important amphibian diversity hotspot in Asia. We sampled for the presence of Bd and Bsal and complemented our survey with formalin-preserved museum specimens collected between 1981 and 2009 to determine the historical presence of Bd in Taiwan. We analyzed species susceptibility, geography (elevation), microbial richness, and climatic conditions to determine chytrid infection dynamics in a potential source region of Bd and Bsal.
Results
We did not find any evidence of Bsal infections in any of our samples (field and museum) and we found relatively low infection prevalence and infection intensities of Bd, with 182 positive individuals (7%) in the field across all years and species. From the 30 species sampled in the field, we found that 9 species (N = 426) did not show evidence of Bd infection across all years sampled, 12 species had a low infection prevalence (< 10%) and five species had a Bd prevalence equal to or above 10% (Supplementary Table 1). Excluding specimen with no evidence of infection, mean infection intensities were low with only three species showing mean infection intensities above 100 zoospore equivalents (ZE; Supplementary Table 1) and four species showing maximum infection intensities above 1000 zoospore equivalents (ZE; Buergeria choui = 1125 ZE; Nidirana okinavana = 4239 ZE; Hylarana latouchii = 90,000 ZE; Limnonectes fujianensis = 16,500 ZE). Across samples from 2010 to 2017, differences of Bd prevalence and infection intensities between Anurans (Prevalence = 0.090 ± 0.286; Bd ZE = 69.470 ± 2075.703) and Caudata (Prevalence = 0.045 ± 0.210; Bd ZE = 3.784 ± 37.998) were not significant (Prevalence: U2090 = 134,029; p = 0.084; Bd load: U2090 = 134,186; p = 0.076). However, when tested at the family level, prevalence and infection intensity significantly differed between the seven different amphibian families (Prevalence: Χ24 = 57.85; Bd ZE: Χ24 = 60.610; p < 0.001). This difference was attributed mostly to Rhacophoridae in comparison to other families (Z2090 = 3.829; p < 0.001) and the difference between Ranidae and Bufonidae (Z1456 = − 3.134; p = 0.015). Comparisons between years of sampling showed that in 2016 Bd prevalence was the highest (Supplementary Fig. 1), while a peak in Bd load was observed in 2013 (ZE2013 = 1070 ± 976; ZE2010 = 19 ± 7; ZE2012 = 0; ZE2016 = 895 ± 743; ZE2017 = 623 ± 499).
We found that the best model to explain Bd prevalence and Bd load included genus, year, elevation, Palmer Drought Severity Index (PDSI) and the interaction of elevation or PDSI and genus (Table 1). Models including any of the temperature variables had lower AICs (Akaike’s Information Criterion; Table 1). Overall, our results suggest that PDSI and the interaction of PDSI and elevation are important drivers of Bd prevalence and load, with infected sites characterized by drier conditions and located in intermediate elevations. Elevation also contributes to explaining Bd load, but not Bd prevalence (Table 1). Results of the models including temperatures show that yearly mean temperature (Tmean) poorly explains Bd prevalence and load (in terms of AICs and p-values), compared to temperature extremes (yearly maximum temperature Tmax; and yearly minimum temperature Tmin) and their interactions with PDSI (Table 1). Drier habitats, and generally higher temperatures led to higher infection probability (Fig. 1). The different taxa show differences in their altitudinal distribution (Fig. 2), and the interaction between genus and elevation was a good predictor for Bd prevalence (F14, 2043 = 3.49; p < 0.001) and Bd load (F14, 2043 = 3.55, p < 0.001).

Bd-prevalence (%, bubble size, continuous) as a function of elevation, Palmers Drought Severity Index (PDSI), and yearly maximum temperature (Tmax; bubble color).

Bd occurrence by species and elevation. The grey bars represent the elevational range of a species. Horizontal lines show at which elevation the species was sampled and whether Bd was found in that species at that elevation (blue = 0, Bd does not occur; red = 1, Bd occurs).
In our museum samples (N = 171), we found five Bd positive individuals (prevalence = 3%), four of which were salamanders (2 specimens each of Hynobius ariasanensis (2001) and Hynobius formosanus (1994)) and only one was an anuran (Rana sauteri) from 1990 (Supplementary Table 2).
We observed a marked difference of the skin microbiome of adult frogs as compared to salamanders (F1,90 = 27.149, p < 0.001) and between species of frogs (F3,54 = 9.286, p < 0.001; Fig. 3), as well as an effect of life stage (tadpole vs. adult) on the skin microbiome of N. adenopleura (F1,67 = 13.946, p < 0.001). In N. adenopleura we observed that adults and tadpoles possessed a unique set of Amplicon Sequence Variants (ASVs), with a richer microbiome in tadpoles (NASVs = 4575) as compared to adults (NASVs = 1645). However, many of the ASVs unique to the two life stages had low abundance, while the most common ASVs from Gammaprotobacteria, Burkholderiales and Bacteroidota were shared between adults and tadpoles at the same time and site (Fig. 4). On salamanders, the most common genera (occurrence in 75% of samples) were Flavobacterium, Pseudomonas, and Cutibacterium, with only 5 core ASVs (4 of Flavobacterium and one of Cutibacterium). On adult frog skin, we observed very few Flavobacteria. The most common genera across all four species of frogs included Cutibacterium, Pseudomonas, Delftia, Blastococcus, Rheinheimera, and Lysinibacillus (Fig. 3).

The skin microbial communities of the core bacterial genera occurring in at least 75% of individuals of four species of frogs and two species of salamanders.

Skin microbiome composition of N. adenopleura at the tadpole (n = 18) and adult (n = 13) life stages caught at the same time in the same location and habitat. Only ASVs occurring in at least 3 different samples are visualized in the network. The size of each node corresponds to the mean relative abundance of an ASV in the considered samples, while the colour indicates the ASV’s taxonomic affiliation. The node size corresponds to ~ 4% for the largest circles to ~ 0.001% of the smallest ones (i.e. larger nodes are shared and smaller are unique but also highly variable among individuals). The Venn diagram shows the size of the unique and shared node sets.
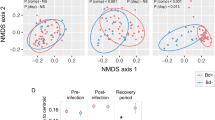
Our data does not support an effect of isolation-by-distance on the skin microbiome of both adult frogs (r418 = 0.242; p = 0.053) and tadpoles (r259 = 0.024; p = 0.386), but site was an important determinant of adult frog skin microbiome composition (permanova F4,53 = 27.785, p < 0.001).
For N. adenopleura and H. latouchii, we compared alpha diversity indices and observed a significantly higher richness and evenness in uninfected individuals (Fig. 5). The permanova analysis confirms a significant difference of the skin microbiome in infected vs. uninfected adults for N. adenopleura (F1,15 = 4.385, p = 0.003) and H. latouchii (F1,23 = 3.752, p = 0.009) with higher richness and evenness leading to lower infection probability (Fig. 5). The indicator analysis on the ASV level reveals that both frog species have in common Pseudomonas ASV 4, and Stenotrophomonas ASV 7 in infected specimens, and Limnobacter ASV 31 and Cetobacterium ASV 1 in uninfected specimens (Supplementary Fig. 2).

Comparison of species richness (number of observed ASVs) and evenness (inverse Simpson index) of infected and uninfected specimens of N. adenopleura and H. latouchii. We observed a significantly higher richness (NASVs, N. adenopleura: U16 = 50.5, p = 0.003, H. latouchii: U24 = 116, p = 0.031) and evenness (Inverse Simpson, N. adenopleura: U16 = 47.0, p = 0.015; H. latouchii: U24 = 124, p = 0.009) in uninfected individuals as compared to infected individuals.
Across all species and life-stages, species evenness, but not richness, is explained by climatic variables and elevation (Inverse Simpson, Tmax: F1,149 = 7.59, p = 0.007; PDSI: F1,149 = 4.93, p = 0.028) and their interaction (F1,149 = 6.79, p = 0.010). Considering adult frogs only, species richness and evenness were both significantly linked to temperature, PDSI and their interaction (Table 2). The relationship is not driven by Bd-induced dysbiosis, as it holds true for uninfected frog adults only and the relationship of evenness with Tmax (F1,39 = 6.70, p = 0.013), PDSI (F1,39 = 6.54, p = 0.015) and the interaction of Tmax and PDSI (F1,39 = 6.84, p = 0.013). Generally, microbial species richness and evenness were higher at sites with colder and drier climate conditions.
Discussion
Here, we investigated the presence of the fungal pathogens Bd and Bsal in amphibians of Taiwan, an island of high amphibian diversity close to the putative region of endemism for both Bd18 and Bsal26. We did not detect Bsal in any of our samples, however, it is likely that the global panzootic lineage of Bd (Bd-GPL) is potentially widespread in Taiwan, both taxonomically and spatially, based on whole genome sequencing of representative isolates from the island18. Analysis of museum samples suggest that Bd has been present in Taiwan since at least 1990. The pattern of Bd infection appears to be influenced by host species susceptibility, geography (elevation), climatic conditions (temperature extremes and dryness) and host skin microbiome composition. Our study suggests that alpha-diversity on amphibian skin was driven by climatic conditions and landscape context and not by Bd-driven dysbiosis.
Our data suggest that environmental conditions, such as elevation, dryness and temperature are important factors determining the prevalence of infection, but the interaction with host-specific properties also appear to be important. Bd infection dynamics are complex and multiple factors may affect the outcome44. Our study suggests that Bd was more commonly found on hosts that live at medium elevation (1000–1600 m a.s.l), at temperatures up to 25 °C and drier sites. In such habitats, anurans in the families Ranidae and Rhacophoridae are particularly numerous, suggesting that life-history traits, i.e. habitat preferences and aquatic larval stages of these host species may modulate Bd-dynamics. Generally, the relationship between Bd infection, amphibian hosts and temperature is not yet clearly understood45. In culture, Bd develops particularly well between 17 and 23 °C and dies at temperature above 29 °C and below freezing46,47. Under natural conditions Bd growth and infection is also related to the temperature dependent amphibian immune system48 and, as our study suggests, to the level of environmental humidity. In the warm temperate and mesic environmental zone, we found a high number of different host species as well as the most highly infected individuals and the highest Bd prevalence. In the lowland regions of Taiwan, where we found the least Bd infected hosts, temperatures can easily exceed the maximum temperature of Bd (29 °C), and even if individuals get infected, they might be able to clear Bd infections due to those high temperatures49. At higher elevations, host diversity and density are strongly reduced, and most of the species (most of them salamanders) are less aquatic. The adults are mainly terrestrial and although they lay eggs in nearby streams, they typically do not aggregate in breeding habitats for long periods of time. These life-history traits may lead to lower infection probabilities50 and may explain why we found the interaction between genus and elevation can help predict Bd prevalence and load on hosts. Temperatures continue to rise in Taiwan and elsewhere, and thus an upward elevational upward shift of Bd prevalence is predicted.
The already complex interactions of Bd, host, and environment might additionally be modulated by the host skin microbiome, as suggested by the recently proposed disease pyramid30. The climatic conditions defined by temperature and humidity are correlated with the richness and diversity of amphibian skin microbiomes38. We observed higher diversity and evenness of the skin microbiome in uninfected vs. infected individuals, and we suggest that temperature may not only have a direct impact on the host’s inner immune system and Bd growth, but also on the exterior host immune system formed by the skin microbiome30. Given that, in the Olive frog, the most common ASVs are shared between adults and tadpoles, the interactions between temperature and skin microbiome may be very similar in the different life stages. To understand the functional causes for differences in infected vs. uninfected individuals, and to predict the outcome for Taiwan’s amphibian diversity from the interaction of Bd, host, host microbiome, and climate change more data and more microbial research are needed.
One of the prevailing questions that remains regarding Bd is whether this pathogen is emerging, persisting, or endemic in Taiwan. Bd was found in a specimen of Adrianus japonicus collected in 1902 in Japan, and thus suggests that Bd is endemic in Japan16. Unlike other parts of the world that have experienced Bd-associated declines, there have not been any documented reports of enigmatic declines in Asian amphibian populations. In Taiwan, there has been no documentation of enigmatic declines in native amphibians, barring one report of a mass mortality event in R. sauteri that occurred in 1995 at the Sitou Forest Recreation Area (Chun-Fu Lin, personal comm.). During that event, around thirty individuals were found dead on the edge of a stream. Individuals exhibited red legs and skin lesions, symptoms associated with multiple pathogens, such as ranaviruses51 or infection by Aeromonas hydrophila, but not with Bd. Despite the presence of Bd in wild amphibian populations, we observed few individuals exhibiting disease symptoms characteristic of chytridiomycosis (tadpoles with missing mouthparts, lethargy, excessive skin sloughing, leg-locking, loss of righting reflex, etc.), with only one highly lethargic H. latouchii male exhibiting clinical signs associated with chytridiomycosis (ZE = 3.29). Generally, Bd was usually found at low levels of infection (less than 100 ZE) and with low prevalence in populations, which indicates that Bd is likely enzootic in Taiwan. Our method, however, does not allow to know if the global pandemic lineage of Bd (Bd-GPL) has been present in Taiwan before 1990 or if the less virulent lineage of Bd-Asia or unknown Bd lineages were present before, enabling the Taiwanese amphibian fauna to preadapt to Bd-GPL.
Even though Bd likely is enzootic in Taiwan, it may still drive population declines at low levels of disease prevalence52, likely also due to ongoing environmental changes such as climate change or pollution. Of greatest concern for conservation and Bd monitoring are those species that are endemic and endangered in Taiwan. Amphibians in this category that tested positive for Bd are salamanders within the family Hynobiidae (H. sonani, H. arisanensis, H. formosanus), usually limited to a specific mountain region of the Zhong Yang mountain range. However, only very few individuals tested positive for Bd and prevalence was well below 10%, indicating enzootic infection levels. Other threatened species found to be infected were Rana sauteri, and the highly endemic species of N. okinavana known only from three locations globally, of which two sites (one location) are found in Taiwan. Despite small sample sizes for these rare species, Bd was detected, but prevalence was low. However, the impact that Bd is having on these species is largely unclear, due to very limited data on population dynamics, currently not allowing population viability analyses. Low infection levels found in these individuals may indicate that they are persisting with low levels of infection, and may have achieved natural immune defenses against the fungus from a longer co-evolutionary interaction, but more intensive disease and population monitoring is needed.
Our study investigated multiple dimensions of disease dynamics in a poorly-studied yet diverse system that harbors Bd in an area of the world that has not experienced Bd epizootics. This information should help us better understand how the Bd pathogen is maintained in an enzootic state in multi-host systems. Our study further stresses that the Bd-amphibian disease system is highly complex44 and only a holistic research approach is able to unravel all the factors modulating infection outcome and disease expression, including micropredators53, pollution29, climate44, and microbial communities36,38.
Materials and methods
The island of Taiwan
The main island of Taiwan has a humid tropical climate and is characterized by 6 environmental zones54 (Fig. 6). The northern part around the city of Taipei, the low mountain range and the western plain receive variable precipitations based on season. Temperatures in the lowlands usually do not get below 5 °C, and the weather in summer is tropical and very humid, especially in mid to lower elevations. Lower temperatures and humidity can be observed between November and April and maximum temperatures around 38 °C can be observed during June to August. In some cases, there is frost or snow on the high mountains between January and March. The high relief of Taiwan and the climatic conditions lead to a high diversity of different habitats that span from lowlands to high elevations. These diverse environmental conditions gave opportunity for amphibians to fill a large range of different ecological niches, making Taiwan an important amphibian diversity hotspot with at least 41 currently known amphibian species, of which many have very restricted distribution areas (Amphibiaweb.org). The warm temperate and mesic zone, which contains mountains of low to medium elevation (500–1500 m above sea level), is the most amphibian rich zone of Taiwan. Thus, Taiwan is an ideal region to investigate climatic variables influencing the occurrence and prevalence of Bd and Bsal on amphibians.

Distribution of Bd in the different bioclimatic zones of Taiwan55. The circle size corresponds to the prevalence class.
Study sites and field sampling
We sampled 26 of the 41 currently known Taiwanese amphibian species (total N = 2517 individuals) across northern and central Taiwan in July/August 2010 (N = 328 individuals), February 2012 (N = 440), September 2013 (N = 373), April 2016 (N = 672) and May 2017 (N = 704). We obtained samples from different environmental zones (Fig. 6).
When possible, we sampled the same location over different years and aimed for 30 samples per species, life stage and location to ensure a 95% chance of detecting the pathogen at a prevalence of 5%56. All individuals were hand-caught and sampled by swabbing the ventral surface of adults and mouthparts of tadpoles with a synthetic cotton swab (Medical Wire MW-100) for Bd and Bsal analyses, and released at site of capture. Bd and Bsal swabs were stored in a cool box with cool packs and transferred to a refrigerator until the DNA was extracted.
In addition, in 2017 we took skin microbiome samples from up to 10 tadpoles and 10 adults per site by rubbing sterile dry swabs across the whole body surface. The microbiome swabs were immediately frozen at − 79 °C using dry ice and transferred to a freezer at earliest convenience (− 30 °C) until DNA extraction. We analysed skin microbiomes in a total of 153 samples: 34 salamander specimens (22 H. sonani, 12 H. formosanus), 58 adult frogs (17 N. adenopleura, 9 N. okinavana, 7 F. limnocharis, 25 H. latouchii), and 61 tadpoles (51 N. adenopleura, 10 H. latouchii).
We also sampled formalin-preserved specimens from the National Museum of Natural Science in Taichung, Taiwan, that were collected between 1986 and 2009, and formalin-preserved hynobiid salamanders from the lab of Dr. June-Shiang Lai at National Taiwan Normal University, Taipei, collected between 1981 and 2009 (N species = 7; N individuals = 171, Supplementary Table 2). Museum samples were first rinsed thoroughly with ethanol and swabbed with synthetic cotton swabs (Medical Wire MW-100) following established protocols57,58.
We did not conduct in-vivo experiments and our sampling protocol was not invasive, following national Taiwanese standards of animal wellbeing in force (Taiwan Animal Protection Act, Taiwan Wildlife Conservation Act) at the time of capture (amendments have been done in May 2021). As we did not use drugs, traps, firearms, poison or corrosive substances to capture animals, the Animal Protection Act of Taiwan does not oblige the calling on an ethics committee at the time of sampling. Further, the Taiwan Endemic Species Research Center (Chun-Fu Lin) is an officially authorized and commissioned organization within the Taiwan Wildlife Conservation Act to conduct all necessary work to protect wild species, including disease monitoring. All animals were released at their original site unharmed.
Genetic analysis and qPCR assays
DNA was extracted from swabs with commercially available kits (Nucleospin, Prepman). A cost-effective Prepman extraction was used for the chytrid pathogen extractions59 and Nucleospin kits were used for extraction of the microbiome as this captures the fungal part of the microbiome better.
For determination of Bd and Bsal prevalence and infection loads, extracts were diluted 1:10 in 0.25xTE Buffer and run using the Taqman real-time PCR protocol following previously published protocols60,61. In order to achieve a final infection intensity level measured in terms of zoospore equivalents (ZE), qPCR genomic equivalent (GE) values were multiplied by 40 to account for dilution during extraction. We report Bd loads as mean ZEswab ± SE, considering all ZE values above zero as positive. We define prevalence of infection as [(no. infected/no. sampled) * 100].
Skin bacterial communities were characterized using 16S amplicon sequencing. DNA was extracted from swabs using the PowerSoil DNA Isolation Kit (MoBio Laboratories, Carlsbad, CA, USA), and the hypervariable V3–V4 region of the bacterial 16S rRNA gene was amplified in triplicate using primers with overhang adaptors. Each 25-μL reaction consisted of 12.5 μL KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA), 5 μL forward and reverse primers (1 μM), and 2.5 μL template. PCR conditions were 95 °C for 180 s, followed by 25 cycles of 95 °C for 30, 60 °C for 15 s, 72 °C for 45 s, and a final extension of 72 °C for 120 s. The PCR products for triplicate reactions were pooled and purified using solid phase reversible immobilization (SPRI) beads (Agencourt AMPure XT, Agencourt Bioscience Corporation, Beverly, MA). All samples and negative control were visualized using gel electrophoresis. Dual indices, from the Illumina Nextera Index Kit, were attached to the purified amplicons using PCR. Each 25-µL reaction consisted of 12.5 µL Kapa HiFi HotStart ReadyMix, 2.5 µL forward and reverse primers (1 µM), 5 µL PCR-grade water, and 5 µL template. PCR conditions were 95 °C for 180 s, followed by 10 cycles of 95 °C for 30 s, 55 °C for 30 s, 72 °C for 30 s, and a final extension of 72 °C for 300 s. The PCR products were purified and visualized as described above. DNA concentrations were quantified using Qubit Fluorometric Quantification and samples were diluted and pooled at equimolar concentrations. Sequencing was performed on an Illumina MiSeq using a MiSeq Reagent Kit v3 (600 cycle) (Illumina, Inc., San Diego, CA).
Base calling and demultiplexing were performed using MiSeq Reporter (Illumina, Inc.), while removal of primer and adapter sequences was performed using Cutadapt62. Additional quality filtering and trimming (truncLen = c(260, 200), maxEE = c(5, 5)), formation of contiguous sequences (minOverlap = 12), identification of unique amplicon sequence variants (ASVs; pool = "pseudo") and chimera removal were performed using the DADA2 pipeline in R using default settings unless specified63. Taxonomy was assigned to ASVs using a local installation of SINA v1.7.264 and SILVA 138.165 release taxonomy. Samples were filtered out if they contained less than 100 ASVs. ASVs that were present in less than 0.001% of all reads and fewer than 2 samples were considered low quality and removed66. Samples were not rarefied, since rarefaction implies in loss of information and precision when defining a common read depth67. Data was normalized before the microbiome analysis.
Climatic and environmental variables
We sourced data from TerraClimate of Climatology Lab68, spanning our full sampling period from 1980 to 2017. We obtained yearly mean temperature (Tmean), yearly maximum temperature (Tmax) and yearly minimum temperature (Tmin). From the TerraClimate data set, we also obtained Palmers drought severity index (PDSI) data69, which uses readily available temperature and precipitation data to estimate relative dryness. It is a standardized index that spans from − 10 (dry) to + 10 (wet)69.
PDSI across our sampling sites has not changed over time, while the temperature has increased steadily over the years. The lowest maximum temperature observed was 20.07 °C in 1984, while the highest was observed in 2016 (21.45 °C). The year 1998 marks a first-time maximum temperature above 21 °C (21.43 °C), while the year 2012 marks the last time of a maximum temperature below 21 °C (20.55 °C). The lowest minimum temperature of 12.88 °C across our sites was observed in the year 1986, while the highest minimum temperature of 14.44 °C was observed in 1998. The driest years (PDSI < − 3.0) were 1993 (− 3.066), 1996 (− 3.272) and 2004 (− 3.824). The wettest years (PDSI > 3.0) were 1983 (4.690), 1998 (4.312) and 2001 (3.187). PDSI was not correlated with elevation (R42 = − 0.163, p > 0.05). All temperature variables and the elevation were highly intercorrelated (all R ≥ 0.611; all p < 0.01).
Statistical analyses
We performed statistical analyses using SAS v.9.4. First, we used the full dataset of field samples (N = 2518) to compare pathogen prevalence and infection load between the two orders (Anura vs. Caudata), seven families (Table 2) and five years of sampling, using non-parametric tests including an U-test for the pairwise comparison of Orders, and a Kruskal–Wallis-ANOVA for k variable comparisons for the comparisons between families and between years of sampling (2010–2017).
We examined pathogen prevalence and infection intensity in our field samples with GLMMs (SAS proc glimmix). We discarded all data of species with no Bd detection over the years, assuming that they might not be susceptible to Bd (N = 13). This selection left us with data from 17 species (11 genera), and a sample size of 2090 specimens (Table 2). We built a first set of GLMMs with Bd prevalence (Prev) as the dependent variable (with a binomial distribution of error terms and a logit link function), and a second set of models with log-transformed infection loads (log(ZE)) as the dependent variable (with negative binomial distribution of error terms and a logit link function). The independent variables included in the models were genus, year of sampling, PDSI, and a temperature/elevation parameter. We also included the interactions of PDSI or genus with temperature/elevation. Due to collinearity between elevation and temperature, we constructed separate models with Tmean, Tmax, Tmin or elevation, and then used the AIC to rank two sets of four models.
We used permutational multivariate analyses of variance (permanova) to compare the bacterial communities between the same groups of samples (order, species, infection status). For the permanova analysis, the ASV abundance was transformed to compositional data and the Bray–Curtis dissimilarity distance was calculated with the vegan R-package. We performed LDA Effect Size (LEfSe) analysis (LDA score ≥ 2, p ≤ 0.05)70 using the LEfSe online tool (http://huttenhower.sph.harvard.edu/galaxy/) to compare indicator species (ASVs) between two groups (infected vs. uninfected specimens). We performed a Mantel test (vegan R-package) between the matrix of the geographic distance between the sites (Haversine distance based on latitude and longitude) and the matrix of Bray–Curtis dissimilarity based on the mean relative abundance of the bacterial genera.
We computed species richness as number of observed ASVs, and species evenness using the inverse Simpson index. We z-transformed ASVs and log-transformed the inverse Simpson index to yield a normal distribution. We investigated the impact of the climate variables Tmax, PDSI, and their interaction on species richness and evenness using GLMMs (SAS proc Glimmix), assuming normal distribution and using the identity link function.
To compare alpha diversity indices (observed ASVs, inverse Simpson) between infected and uninfected individuals we used the non-parametric Mann–Whitney U-test, due to the low number of infected individuals in our dataset. We compared skin microbiomes of the tadpole and adult stages of N. adenopleura using a network analysis. We picture the distribution of unique and shared prokaryotic ASVs among the two life stages. The network analysis was done using the R-packages network, ggnetwork and ggVennDiagram.
References
McCallum, M. L. Vertebrate biodiversity losses point to a sixth mass extinction. Biodivers. Conserv. 24, 2497–2519 (2015).
Wake, D. B. & Vredenburg, V. T. Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. 105, 11466–11473. https://doi.org/10.1073/pnas.0801921105 (2008).
Blehert, D. S. et al. Bat white-nose syndrome: An emerging fungal pathogen?. Science 323, 227. https://doi.org/10.1126/science.1163874 (2009).
Pautasso, M., Aas, G., Queloz, V. & Holdenrieder, O. European ash (Fraxinus excelsior) dieback—A conservation biology challenge. Biol. Cons. 158, 37–49 (2013).
Daszak, P., Cunningham, A. A. & Hyatt, A. D. Infectious disease and amphibian population declines. Divers. Distrib. 9, 141–150 (2003).
Fisher, M. C., Gow, N. A. R. & Gurr, S. J. Tackling emerging fungal threats to animal health, food security and ecosystem resilience. Philos. Trans. R. Soc. B Biol. Sci. https://doi.org/10.1098/rstb.2016.0332 (2016).
Fisher, M. C. et al. Emerging fungal threats to animal, plant and ecosystem health. Nature 484, 186–194 (2012).
Lips, K. R., Reeve, J. D. & Witters, L. R. Ecological traits predicting amphibian population declines in Central America. Conserv. Biol. 17, 1078–1088 (2003).
Zipkin, E. F., DiRenzo, G. V., Ray, J. M., Rossman, S. & Lips, K. R. Tropical snake diversity collapses after widespread amphibian loss. Science 367, 814–816. https://doi.org/10.1126/science.aay5733 (2020).
Berger, L. et al. Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc. Natl. Acad. Sci. 95, 9031–9036 (1998).
Martel, A. et al. Recent introduction of a chytrid fungus endangers Western Palearctic salamanders. Science 346, 630–631. https://doi.org/10.1126/science.1258268 (2014).
Yap, T. A., Koo, M. S., Ambrose, R. F., Wake, D. B. & Vredenburg, V. T. Averting a North American biodiversity crisis. Science 349, 481–482 (2015).
Weldon, C., du Preez, L. H., Hyatt, A. D., Muller, R. & Speare, R. Origin of the amphibian chytrid fungus. Emerg. Infect. Dis. 10, 2100–2105 (2004).
Talley, B. L., Muletz, C. R., Vredenburg, V. T., Fleischer, R. C. & Lips, K. R. A century of Batrachochytrium dendrobatidis in Illinois amphibians (1888–1989). Biol. Cons. 182, 254–261 (2015).
Rodriguez, D., Becker, C., Pupin, N., Haddad, C. & Zamudio, K. Long-term endemism of two highly divergent lineages of the amphibian-killing fungus in the Atlantic Forest of Brazil. Mol. Ecol. 23, 774–787 (2014).
Goka, K. et al. Amphibian chytridiomycosis in Japan: Distribution, haplotypes and possible route of entry into Japan. Mol. Ecol. 18, 4757–4774 (2009).
Bataille, A. et al. Genetic evidence for a high diversity and wide distribution of endemic strains of the pathogenic chytrid fungus Batrachochytrium dendrobatidis in wild Asian amphibians. Mol. Ecol. 23, 4196–4209. https://doi.org/10.1111/mec.12385 (2013).
O’Hanlon, S. J. et al. Recent Asian origin of chytrid fungi causing global amphibian declines. Science 360, 621–627. https://doi.org/10.1126/science.aar1965 (2018).
Swei, A. et al. Is chytridiomycosis an emerging infectious disease in Asia?. PLoS ONE 6, e23179 (2011).
Bai, C. M., Garner, T. W. J. & Li, Y. M. First evidence of Batrachochytrium dendrobatidis in China: Discovery of chytridiomycosis in introduced American bullfrogs and native amphibians in the Yunnan Province, China. EcoHealth 7, 127–134. https://doi.org/10.1007/s10393-010-0307-0 (2010).
Yang, H. et al. First detection of the amphibian chytrid fungus Batrachochytrium dendrobatidis in free-ranging populations of amphibians on mainland Asia: Survey in South Korea. Dis. Aquat. Org. 86, 9–13 (2009).
Fong, J. J. et al. Early 1900s detection of Batrachochytrium dendrobatidis in Korean amphibians. PLoS ONE 10, e0115656 (2015).
Kusrini, M., Skerratt, L., Garland, S., Berger, L. & Endarwin, W. Chytridiomycosis in frogs of Mount Gede Pangrango, Indonesia. Diseases Aquat. Organ. 82, 187–194 (2008).
Laking, A. E., Ngo, H. N., Pasmans, F., Martel, A. & Nguyen, T. T. Batrachochytrium salamandrivorans is the predominant chytrid fungus in Vietnamese salamanders. Sci. Rep. 7, 44443. https://doi.org/10.1038/srep44443 (2017).
Zhu, W. et al. A survey for Batrachochytrium salamandrivorans in Chinese amphibians. Curr. Zool. 60, 729–735 (2014).
Beukema, W. et al. Environmental context and differences between native and invasive observed niches of Batrachochytrium salamandrivorans affect invasion risk assessments in the Western Palaearctic. Divers. Distrib. 24, 1788–1801. https://doi.org/10.1111/ddi.12795 (2018).
Auliya, M. et al. The global amphibian trade flows through Europe: The need for enforcing and improving legislation. Biodivers. Conserv. https://doi.org/10.1007/s10531-016-1193-8 (2016).
Scheffers, B. R., Edwards, D. P., Diesmos, A., Williams, S. E. & Evans, T. A. Microhabitats reduce animal’s exposure to climate extremes. Glob. Change Biol. 20, 495–503 (2014).
Schmeller, D. S. et al. People, pollution and pathogens—Global change impacts in mountain freshwater ecosystems. Sci. Total Environ. 622–623, 756–763. https://doi.org/10.1016/j.scitotenv.2017.12.006 (2018).
Bernardo-Cravo, A., Schmeller, D. S., Chatzinotas, A., Vredenburg, V. T. & Loyau, A. Environmental factors and host microbiomes shape host-pathogen dynamics. Trends Parasitol. 36, 29–36 (2020).
Harris, R. N. et al. Skin microbes on frogs prevent morbidity and mortality caused by a lethal skin fungus. ISME J. 3, 818–824. https://doi.org/10.1038/ismej.2009.27 (2009).
Harris, R. N., James, T. Y., Lauer, A., Simon, M. A. & Patel, A. Amphibian pathogen Batrachochytrium dendrobatidis is inhibited by the cutaneous bacteria of amphibian species. EcoHealth 3, 53–56. https://doi.org/10.1007/s10393-10005-10009-10391 (2006).
Piovia-Scott, J. et al. Greater species richness of bacterial skin symbionts better suppresses the amphibian fungal pathogen Batrachochytrium dendrobatidis. Microb. Ecol. 74, 217–226 (2017).
Ellison, S., Knapp, R. A., Sparagon, W., Swei, A. & Vredenburg, V. T. Reduced skin bacterial diversity correlates with increased pathogen infection intensity in an endangered amphibian host. Mol. Ecol. 28, 127–140 (2019).
Jani, A. J. & Briggs, C. J. The pathogen Batrachochytrium dendrobatidis disturbs the frog skin microbiome during a natural epidemic and experimental infection. Proc. Natl. Acad. Sci. USA 111, E5049-5058. https://doi.org/10.1073/pnas.1412752111 (2014).
Kueneman, J. G. et al. The amphibian skin-associated microbiome across species, space and life history stages. Mol. Ecol. 23, 1238–1250 (2014).
Kueneman, J. G. Ecology of the Amphibian Skin-Associated Microbiome and Its Role in Pathogen Defense (University of Colorado at Boulder, 2015).
Kueneman, J. G. et al. Community richness of amphibian skin bacteria correlates with bioclimate at the global scale. Nat. Ecol. Evolut. 3, 381–389. https://doi.org/10.1038/s41559-019-0798-1 (2019).
Jiménez, R. R. & Sommer, S. The amphibian microbiome: Natural range of variation, pathogenic dysbiosis, and role in conservation. Biodivers. Conserv. 26, 763–786. https://doi.org/10.1007/s10531-016-1272-x (2017).
Walke, J. B. et al. Amphibian skin may select for rare environmental microbes. ISME J 8, 2207–2217. https://doi.org/10.1038/ismej.2014.77 (2014).
McKenzie, V. J., Bowers, R. M., Fierer, N., Knight, R. & Lauber, C. L. Co-habiting amphibian species harbor unique skin bacterial communities in wild populations. ISME J 6, 588–596. https://doi.org/10.1038/ismej.2011.129 (2012).
Bates, K. A. et al. Amphibian chytridiomycosis outbreak dynamics are linked with host skin bacterial community structure. Nat. Commun. 9, 693. https://doi.org/10.1038/s41467-018-02967-w (2018).
Ellison, S. et al. The influence of habitat and phylogeny on the skin microbiome of amphibians in Guatemala and Mexico. Microb. Ecol. 78, 257–267 (2019).
Fisher, M. C., Pasmans, F. & Martel, A. Virulence and pathogenicity of chytrid fungi causing amphibian extinctions. Annu. Rev. Microbiol. https://doi.org/10.1146/annurev-micro-052621-124212 (2021).
Haver, M. et al. The role of abiotic variables in an emerging global amphibian fungal disease in mountains. Sci. Total Environ. 815, 152735 (2021).
Turner, A., Wassens, S., Heard, G. & Peters, A. Temperature as a driver of the pathogenicity and virulence of amphibian chytrid fungus Batrachochytrium dendrobatidis: A systematic review. J. Wildl. Dis. 57, 477–494 (2021).
Woodhams, D., Alford, R., Briggs, C., Johnson, M. & Rollins-Smith, L. Life history trade-offs influence disease in changing climates: Strategies of an amphibian pathogen. Ecology 89, 1627–1639 (2008).
Sonn, J. M., Berman, S. & Richards-Zawacki, C. L. The influence of temperature on chytridiomycosis in vivo. EcoHealth 14, 762–770. https://doi.org/10.1007/s10393-017-1269-2 (2017).
Schmidt, B., Küpfer, E., Geiger, C., Wolf, S. & Schär, S. Elevated temperature clears chytrid fungus infections from tadpoles of the midwife toad, Alytes obstetricans. Amphibia-Reptilia 32, 276–280 (2011).
Bielby, J., Cooper, N., Cunningham, A. A., Garner, T. W. J. & Purvis, A. Predicting susceptibility to future declines in the world’s frogs. Conserv. Lett. 1, 82–90 (2008).
Gray, M. J., Miller, D. L. & Hoverman, J. T. Ecology and pathology of amphibian ranaviruses. Dis. Aquat. Org. 87, 243–266 (2009).
Murray, K., Skerratt, L., Speare, R. & McCallum, H. Impact and dynamics of disease in species threatened by the amphibian chytrid fungus, Batrachochytrium dendrobatidis. Conserv. Biol. 23, 1242–1252 (2009).
Schmeller, D. S. et al. Microscopic aquatic predators strongly affect infection dynamics of a globally emerged pathogen. Curr. Biol. 24, 176–180. https://doi.org/10.1016/j.cub.2013.11.032 (2014).
Metzger, M. J. et al. Environmental stratifications as the basis for national, European and global ecological monitoring. Ecol. Ind. 33, 26–35. https://doi.org/10.1016/j.ecolind.2012.11.009 (2013).
Metzger, M. J. et al. A high-resolution bioclimate map of the world: A unifying framework for global biodiversity research and monitoring. Glob. Ecol. Biogeogr. 22, 630–638. https://doi.org/10.1111/geb.12022 (2013).
Clare, F., Daniel, O., Garner, T. & Fisher, M. Assessing the ability of swab data to determine the true burden of infection for the amphibian pathogen Batrachochytrium dendrobatidis. EcoHealth 13, 360–367. https://doi.org/10.1007/s10393-016-1114-z (2016).
Cheng, T. L., Rovito, S. M., Wake, D. B. & Vredenburg, V. T. Coincident mass extirpation of neotropical amphibians with the emergence of the infectious fungal pathogen Batrachochytrium dendrobatidis. Proc. Natl. Acad. Sci. 108, 9502–9507 (2011).
Vredenburg, V. T. et al. Pathogen invasion history elucidates contemporary host pathogen dynamics. PLoS ONE 14, e0219981. https://doi.org/10.1371/journal.pone.0219981 (2019).
Hyatt, A. D. et al. Diagnostic assays and sampling protocols for the detection of Batrachochytrium dendrobatidis. Dis. Aquat. Org. 73, 175–192 (2007).
Blooi, M. et al. Duplex real-time PCR for rapid simultaneous detection of Batrachochytrium dendrobatidis and B. salamandrivorans in amphibian samples. J. Clin. Microbiol. 51, 4173–4177 (2013).
Boyle, D. G., Boyle, D. B., Olsen, V., Morgan, J. A. T. & Hyatt, A. D. Rapid quantitative detection of chytridiomycosis (Batrachochytrium dendrobatidis) in amphibian samples using real-time Taqman PCR assay. Dis. Aquat. Org. 60, 141–148 (2004).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2012).
Bokulich, N. A. & Mills, D. A. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. https://doi.org/10.1128/aem.03870-12 (2013).
McMurdie, P. J. & Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10, e1003531 (2014).
Abatzoglou, J. T., Dobrowski, S. Z., Parks, S. A. & Hegewisch, K. C. TerraClimate, a high-resolution global dataset of monthly climate and climatic water balance from 1958–2015. Sci. Data 5, 170191. https://doi.org/10.1038/sdata.2017.191 (2018).
Wells, N., Goddard, S. & Hayes, M. J. A self-calibrating Palmer Drought Severity Index. J. Clim. 17, 2335–2351 (2004).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Fisher, M. C. et al. RACE: Risk assessment of chytridiomycosis to European Amphibian Biodiversity. Froglog 101, 45–47 (2012).
Acknowledgements
We thank T. C. Yeh and Y. L. Lin for their assistance during the fieldwork in Taiwan. We are much indebted to the late Dr. J. S. Lai for providing salamander specimens. This work received funding through the projects RACE (BiodivERsA, Risk Assessment of Chytridiomycosis to European amphibian biodiversity71), and P3 (People, Pollution, and Pathogens) financed through the call “Mountains as Sentinels of Change” by the Belmont-Forum (ANR-15-MASC-0001-P3, DFG-SCHM3059/6-1, NERC-163394829). D.S.S. currently holds the AXA Chair for Functional Mountain Ecology funded by the AXA Research Fund through the project GloMEc. MCF is a fellow in the CIFAR ‘Fungal Kingdom” program and is supported by UK-NERC. Information on ongoing activities can be found on p3mountains.org.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.L., D.S.S., and T.C. conceived the idea of the project. A.L., C.-F.L., D.S.S., J.S., T.C., and T.-S.D. conducted the sampling, and species identification. A.C.-A., A.L., D.S.S., and T.C. performed the laboratory work. A.B.-C., L.Z., Y.-P.L. helped D.S.S., A.L., and T.C. to analyze different parts of the dataset. A.S., V.T.V., M.C.F. contributed to the microbiome analysis. All authors discussed the results. A.L., D.S.S., T.C. and V.T.V. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schmeller, D.S., Cheng, T., Shelton, J. et al. Environment is associated with chytrid infection and skin microbiome richness on an amphibian rich island (Taiwan). Sci Rep 12, 16456 (2022). https://doi.org/10.1038/s41598-022-20547-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20547-3
This article is cited by
-
Associations of Batrachochytrium dendrobatidis with skin bacteria and fungi on Asian amphibian hosts
ISME Communications (2023)
-
High relative humidity and temperature limit disease development and mortality in golden frogs of Panama, Atelopus zeteki, infected with Batrachochytrium dendrobatidis
Evolutionary Ecology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
