
Abstract
Petrocodon and Primulina are two characteristic genera of Gesneriaceae that exhibit remarkable species and floral diversity, and high endemism across the Sino-Vietnamese Limestone Karsts. To better understand the evolution of limestone gesneriad plastomes, we report nine complete plastomes of seven Primulina and two Petrocodon which have never been assembled before. The newly generated plastomes range from 152,323 to 153,786 bp in size and display a typical quadripartite structure. To further explore the plastome evolution across Gesneriaceae, we assembled five additional plastomes from public reads data and incorporated 38 complete Gesneriaceae plastomes available online into comparative and phylogenomic analyses. The comparison of 52 Gesneriaceae plastomes reveals that not only Primulina and Petrocodon but all gesneriad genera analyzed are highly conserved in genome size, genome structure, gene contents, IR boundary configurations, and codon usage bias. Additionally, sliding window analyses were implemented across alignments of Primulina and Petrocodon for identifying highly variable regions, providing informative markers for future studies. Meanwhile, the SSRs and long repeats of Gesneriaceae plastomes were characterized, serving as useful data in studying population and repetitive sequence evolutions. The results of plastome phylogenetics represent a preliminary but highly resolved maternal backbone genealogy of Primulina and the Old World subtribes of Gesneriaceae.
Similar content being viewed by others


Gesneriaceae is a medium-sized plant family comprising more than 3400 species in ca. 150 genera distributed primarily in tropics and subtropics around the world1,2. Species of Gesneriaceae inhabit a wide range of habitats and exhibit multifarious evolutionary adaptations2,3, resulting in numerous aesthetic plants with high horticultural values4. One of the most notable habitats that gesneriads thrive is the limestone karst5,6, a distinctive landscape and internal drainage system formed by the dissolution of the highly soluble and porous carbonate sedimentary rock7,8. Because of the high salinity and porosity, poorly developed soils, and fragmented landscapes, limestone karsts generally impose hardship on the survival of plants8, allowing only those that have evolved to adapt to the limestone bedrock7,9. Striding across the bordering areas of southern China and northern Vietnam lies an immense and spectacular landscape10 noted as the Sino-Vietnamese Limestone Karsts (SVLK)7,9. As one of the largest continuous karsts in the world10, SVLK are known for many celebrating tourist destinations and UNESCO World Heritages Sites11 of international geomorphological significance. Additionally, SVLK are also major biodiversity hotspots9,11 abounding with many species-rich and narrowly distributed endemic plants9 such as the two sister gesneriad genera3 Primulina Hance6 and Petrocodon Hance12.
Prior to the molecular recircumscription, Primulina (Pr.) was known as a monotypic genus composed solely of Pr. tabacum Hance, a prioritized key protected species distributed in southern China13. Molecular phylogenetic studies, however, showed that Pr. tabacum was nested within a strongly supported clade composed of Chirita sect. Gibbosaccus Clarke (ca. 120 spp.), Chiritopsis W.T.Wang (17 taxa), and two of the three described species of Wentsaiboea D.Fang & D.H.Qin that are all mainly distributed in the SVLK6,14,15. Based on taxonomic priority, Primulina was expanded to include the ca. 140 species of these three genera14,15. With more than 70 new species added to the genus after its recircumscription, Primulina now contains more than 200 species, representing one of the largest Old World gesneriad genera5,6. The high species diversity and restricted distributions on limestone habitats have made the calciphilous Primulina an ideal study subject for understanding plant radiation on SVLK6. Several studies have employed Sanger sequencing data to reconstruct phylogenetic relationships of Primulina for understanding its underlying speciation mechanisms6,16,17. Additionally, the genome-scale data of Primulina have gradually increased with the advancement of next generation sequencing (NGS) technology. For instance, Ai et al.18 sequenced 11 Primulina transcriptomes, and Feng et al.19 assembled the complete genome of Pr. huaijiensis Z.L.Ning & Jing Wang. Furthermore, there are eight plastomes of seven Primulina species, i.e., Pr. brachytricha var. magnibracteata (W.T.Wang & D.Y.Chen) Mich.Möller & A.Weber, Pr. eburnea (Hance) Yin Z.Wang, Pr. huaijiensis, Pr. liboensis (W.T.Wang & D.Y.Chen) Mich.Möller & A.Weber, Pr. linearifolia (W.T.Wang) Yin Z.Wang, Pr. ophiopogoides (D.Fang & W.T.Wang) Yin Z.Wang, and Pr. tenuituba (W.T.Wang) Yin Z.Wang, available on GenBank up to date (Table S1).
Similar to Primulina, the circumscription of Petrocodon (Pe.) also experienced drastic changes as molecular phylogenetic analyses revealed that the originally defined Petrocodon is polyphyletic, nested within a strongly supported clade composed of the mono- or oligotypic genera (i.e., Calcareoboea C.Y.Wu ex H.W.Li, Dolicholoma D.Fang & W.T.Wang, Lagarosolen W.T.Wang, Paralagarosolen Y.G.Wei, and Tengia Chun), and some species of Didymocarpus Wall. and Wentsaiboea D.Fang & D.H.Qin that are also restricted to SVLK20. Comparing to the species-rich Primulina, however, Petrocodon contains only ca. 50 taxa5,12 or a quarter of the species diversity of its sister genus. However, flower morphology of Petrocodon is even more variable than Primulina in terms of corolla color, symmetry, shapes, and even stamen number12,20,21, likely resulting from adaptive evolution triggered by diverse pollination syndromes21. However, since all previous phylogenetic studies of Petrocodon relied solely on two DNA markers, the nuclear ITS and chloroplast trnLUAA-trnFGAA, its interspecific relationships have yet to be robustly resolved12,20. Additionally, in comparison with Primulina, the dearth of genomic data in Petrocodon is apparent since the only available data to date is the plastome of Pe. jinxiensis (Yan Liu, H.S.Gao & W.B.Xu) A.Weber & Mich.Möller (Table S1). As a result, it is necessary to generate more genomic data in order to provide greater insight into the evolution of the extraordinary floral diversity of Petrocodon.
Despite thousand copies per organelle, plastid genome (plastome) is overall highly conserved across various land plant lineages22. The typical structure of a plastome is represented as a circular molecule consisting of two large rRNA-encoding inverted repeats (IRs) separated by a large single copy (LSC) region and a small single copy (SSC) region, forming a quadripartite structure22,23. Most of the land plant plastomes contain about 110–130 genes including eight rRNA genes (four duplicated genes in IRs), 30–35 tRNA genes, and 70–88 protein coding genes, and the genome sizes are usually in the range around 150–160 kb22. Though evolving about half slower than nuclear DNAs22, plastid DNAs have been essential in plant molecular phylogenetics24 for their high copy number, uniparental inheritance, relatively small genome size, and haploidy that are easier to work with and more likely to obviate problems caused by frequent recombination, polyploidy, or paralogues in nuclear phylogenetic analyses25. Currently, 8887 plastome sequences of land plants (https://www.ncbi.nlm.nih.gov/genome/browse/#!/organelles/; accessed 25 July 2022) have been deposited in GenBank up to date, mostly assembled using NGS technologies in the recent decade. The boom of data allows more plastomes to be compared and analyzed, facilitating our understanding of plastome evolution and greatly improving phylogenetic resolutions23,26.
In this study, we report nine complete plastomes of seven Primulina and two Petrocodon species, all of which have never been sequenced before (Table 1). In addition, all available Gesneriaceae plastomes on the GenBank were downloaded (accessed in February 1st, 2022) and five whole-genome shotgun (WGS) sequencing data from the Sequence Read Archive (SRA) were retrieved to generate additional plastomes (Table S1). Based on these data, we intend to (1) conduct the comparative analyses of plastome structure, gene content, repeat content, and codon usage bias of Gesneriaceae plastomes to improve our understanding of plastome evolution in the family; (2) identify the variable regions in Primulina and Petrocodon plastomes for further phylogeographic and phylogenetic studies; (3) reconstruct the phylogenetic relationships using plastome sequences to provide fundamental insights into the phylogenetic relationships of Primulina and other Gesneriaceae taxa. Our data and results could serve as valuable resources for both the evolutionary and conservation biology of Primulina and Petrocodon.
Results
Features of the newly sequenced Petrocodon and Primulina plastomes
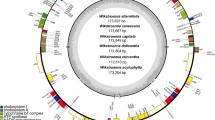
Sizes of the nine newly sequenced plastomes of Petrocodon and Primulina range from 152,323 bp in Pr. medica (D.Fang ex W.T.Wang) Yin Z.Wang to 153,786 bp in Pr. cordata Mich.Möller & A.Weber (Tables 1, S1). These nine plastomes display the typical quadripartite structure of most angiosperms (Figs. 1, S1–S8), with one LSC (83,218 in Pr. pengii W.B.Xu & K.F.Chung to 84,890 bp in Pr. medica), one SSC (16,489 in Pr. medica to 18,279 bp in Pe. multiflorus F.Wen & Y.S.Jiang), and a pair of IRs (25,415 in Pe. multiflorus to 25,495 bp in Pr. pengii). GC contents of the nine plastomes also show limited variation (37.5–37.7%). For assembly quality, there were 35,440 (Pr. cordata) to 188,042 (Pe. multiflorus) reads mapped to the nine plastomes whose mean coverages range from 60.9 ± 16.5 × in Pr. cordata to 318.3 ± 61.9 × in Pe. multiflorus (Table 1). The gene content and order are identical in the nine plastomes (Fig. 1, S1–8). These plastomes contain 114 unique genes, including 80 protein coding genes, 30 tRNA genes, and four rRNA genes, of which 15 genes have one intron (including rps12), two genes have two introns, and 17 genes are completely duplicated in IRs. Similar to a majority of angiosperm plastomes27, the rps12 in the nine plastomes is a trans-splicing gene with three exons of which two are duplicated in IRs (Table 2; Fig. 1). In addition, ndhF, rps19, and ycf1 are partially duplicated in IRs since these genes are located at IR/SC boundaries (Figs. S9, S10).

The plastome map of Primulina lutea. Genes drawn on the inner side of the outer circle are transcribed clockwise, and those on the outer side are transcribed counterclockwise. IRs are shown in bold line in the outer circle. The inner circle indicates GC contents across the genome with lighter gray indicating AT contents. Genes belonging to different functional groups were shown in different colors. Gene name ends with asterisk (*) indicates the intron containing gene.
Comparisons of plastome structure and IR boundaries
The comparison of sequence similarity performed by mVISTA28 showed that both the structure and sequence content are highly conserved across different gesneriad species and genera (Fig. 2). The IR/SC boundaries and the positions of the genes flanking these boundaries among the Gesneriaceae plastomes analyzed here are highly conserved with no significant boundary shift (Figs. S9, S10). Among all the examined gesneriad plastomes, except for Haberlea rhodopensis Friv. and several Paraboea (C.B.Clarke) Ridl. species, the boundaries of LSC/IRB, IRB/SSC, SSC/IRA, and IRA/LSC are located within the reading frames of rps19, ndhF, ycf1, and between the trnH-GUG and the partially duplicated rps19, respectively (Figs. S9, S10). The IRB/SSC boundary of H. rhodopensis differs from others since the coding region of its ndhF is shorter (2241 bp) than other species (2262–2307 bp), and thus does not extend into IRB (Fig. S9). The LSC/IRB boundary of the eight Paraboea species located outside rps19, resulting in no partial duplication of rps19 at the IRA/LSC boundary in those plastomes (Fig. S9).

The plastome similarity comparison of Gesneriaceae plastomes using mVISTA. Primulina lutea was set as the reference. The filled boxes inside each row indicate the sequence similarities. Similarities lower than 50% are not shown. Purple boxes represent the protein-coding regions, cyan blue boxes represent the RNA genes, and red boxes are intergenic (non-coding) regions.
SSR and long repeat analyses of Gesneriaceae plastomes
The simple sequence repeats (SSRs) detected by MISA29 are listed in Table S2 and their distributions in each species are plotted in Fig. 3. A total of 3551 SSRs were found in the 52 Gesneriaceae plastomes, with the number of SSRs in each sequence ranging from 53 in Dorcoceras hygrometricum Bunge to 100 in Corallodiscus lanuginosus (Wall. ex R.Br.) B.L.Burtt. Dinucleotide repeats are the most abundant type across all species (totaling 1860 SSRs), followed by mono- (1097), tetra- (424), tri- (136), penta- (23), and hexanucleotide repeats (11). Among 31 different kinds of repeat units identified, AT-rich units are more abundant in terms of both unit types and SSR counts than GC-rich units (Fig. 3). For example, the A/T mononucleotide SSRs are greater in number than G/C mononucleotide SSRs in all samples, while many of the pent- and hexanucleotide repeat units consist exclusively of A/T bases (Fig. 3). Additionally, all of the hexa- and some of the pentanucleotide SSRs are unique to specific species or genera. For instance, AATTC/AATTG is unique to Streptocarpus Lindl., and AATATT/AATATT was only found in Pr. ophiopogoides (D.Fang & W.T.Wang) Yin Z.Wang (Fig. 3).

The balloon plot of detailed numbers of each SSR repeat pattern (unit pattern) of 52 Gesneriaceae plastomes with one IR excluded. Total number of SSRs in each plastome is shown in the parentheses after the scientific name. 1: Pr. eburnea MF177038. 2: Pr. eburnea MF472011. 3: Pr. eburnea SRR9711276.
The long repeats detected by REPuter30 are summarized in Table S3 and Fig. 4. The total number of long repeats in each plastome varies from 27 in Paraboea filipes (Hance) B.L.Burtt, Pr. sclerophylla (W.T.Wang) Yan Liu, and Pr. liboensis to 36 in Pr. hezhouensis (W.H.Wu & W.B.Xu) W.B.Xu & K.F.Chung. Among the four repeat types, palindromic repeat is the most abundant type (totaling 664 repeats across all plastomes), followed by forward (540), reverse (41), and complement (19) repeats (Fig. 4B). Most of the repeats fall into the range class of 30–39 bp in length (867 repeats across all plastomes), followed by the range class 40–49 bp (177), ≥ 70 bp (55), 50–59 bp (47), and 60–69 bp (18) (Fig. 4A), while the longest repeat reaches 200 bp found in Lysionotus pauciflorus Maxim. (Table S3).

The statistics of long repeat contents in 52 Gesneriaceae plastomes with one IR excluded. (A) The length distribution of long repeats in five length classes. (B) The number of four types of long repeats in each plastome. 1Pr. eburnea MF177038. 2Pr. eburnea MF472011. 3Pr. eburnea SRR9711276.
Codon usage bias in Gesneriaceae
The observed codon usage frequencies and relative synonymous codon usage (RSCU) values of 80 protein-coding genes for each of the 52 Gesneriaceae plastomes evaluated by DAMBE v.7.3.231 are provided in Table S4 and the average RSCU values were summarized in Fig. 5. Of the observed codon usage frequency, the codon AUU encoding isoleucine is the most frequent codon (a total of 50,164 counts), while the stop codon UGA is the least frequent (892) counts (Table S4). The comparison of RSCU values across each sample demonstrates that the codon usage pattern is highly conserved across Gesneriaceae (Fig. 5A) and is biased toward A or T, especially at the third codon position of synonymous codons (Fig. 5). For instance, every sample has the same set of 30 highly preferred codons (RSCU > 1), two codons without preferential bias (since there is no synonymous codon for UGG and AUG; RSCU = 1), and 32 less preferred codons (RSCU < 1), while there is no codon terminated in C or G among the 30 highly preferred codons (Table S4; Fig. 5).

The RSCU of 80 protein-coding genes of 52 Gesneriaceae Plastomes. (A) The heat map of RSCU of 63 codons across 52 plastomes. Red boxes indicate highly preferred synonymous codon (RSCU > 1), while the blue boxes imply the less preferred synonymous codon (RSCU < 1). (B) The average RSCU of 52 Gesneriaceae plastomes colored by the content of the first (1st C) and the third codon positions (3rd C). 1Pr. eburnea MF177038. 2Pr. eburnea MF472011. 3Pr. eburnea SRR9711276.
Sequence divergence analyses
To detect variable regions in Primulina and Petrocodon, we conducted sliding window analyses for three alignments, Primulina (19 sequences, 132,094 bp), Petrocodon (three sequences, 128,477 bp), and Primulina + Petrocodon (22 sequences, 132,546 bp), and identified variable regions for each alignment based on the windows with 95th percentile pi values using DnaSP v.6.12.0332. In the alignment of Primulina + Petrocodon, six intergenic spacers (trnHGUG-psbA, trnKUUU-rps16, petN-psbM, ndhF-rpl32, rpl32-trnLUAG, and ndhD-psaC), petD intron, and two coding regions in the ycf1, i.e., ycf1(3′) closer to the 3′ end of the reading frame and ycf1(5′) closer to the 5′ end of the gene, were detected as highly variable regions (Fig. 6A). In the alignment containing only Primulina, five intergenic spacers (trnHGUG-psbA, trnKUUU-rps16, petN-psbM, ndhF-rpl32, and ndhD-psaC), flanking regions around trnT-GGU, petD intron, and CDS of ycf1(3′) and ycf1(5′) were considered as highly variable regions (Fig. 6B). In the alignment of three Petrocodon plastomes, we identified five intergenic spacers (trnHGUG-psbA, trnKUUU-rps16, psbK-psbI, ndhC-trnVUAC, and ndhF-rpl32), petD intron, and ndhF, ycf3(3′), and ycf1(5′) CDS as potential phylogenetic markers for the genus (Fig. 6C). Six regions, i.e., trnHGUG-psbA, trnKUUU-rps16, petD intron, ndhF-rpl32, ycf1(3′), and ycf1(5′), were detected as most variable in all of the three analyses, while trnT-GGU flanking regions was recognized exclusively in Primulina, psbK-psbI, trnC-trnV and ndhF CDS exclusively in Petrocodon, and petN-psbM and ndhD-psaC in both Primulina and Primulina + Petrocodon alignments (Fig. 6). In addition, there is no variable region detected in IR since its pi values are generally lower than the SC regions (Fig. 6).

The nucleotide diversity among 19 Primulina and three Petrocodon plastomes. (A) The sliding window analysis across the alignment of 19 Primulina + three Petrocodon accessions. (B) The sliding window analysis across the alignment of 19 Primulina plastomes. (C) The sliding window analysis across the alignment of three Petrocodon plastomes. The red dashed line indicates the lowest pi value of 95th percentile windows in the specific alignment. Regions whose pi values are higher than the red dashed line were marked as variable regions. Names of the variable regions colored in black indicate that they are universal to three alignments, while those colored in red indicate they were identified only in one or two of the alignment(s). *The flanking regions of trnT-GGU.
Phylogenetic analyses
The partitioned alignment of 61 plastomes with one IR excluded is 153,963 bp in length, of which 41,572 (30.5% of the total length) were variable and 20,830 (15.3%) were parsimoniously informative. The substitution models and best-fit partition schemes estimated by ModelFinder33 for IQ-TREE34 and MrBayes35 are summarized in Table S5. Phylogenetic relationships reconstructed by the three phylogenetic programs RAxML36, IQ-TREE34, and MrBayes35 are all highly supported and congruent except for the position of Pr. huaijiensis that varied slightly among different analyses (Figs. 7, S11, S12). Specifically, Pr. huaijiensis is sister to Pr. mollifolia (D.Fang & W.T.Wang) J.M.Li & Yin Z.Wang + Pr. renifolia (D.Fang & D.H.Qin) J.M.Li & Yin Z.Wang with poor support value (ultrafast bootstrap support = 66) in the IQ-TREE phylogram (Fig. 7), while the species is sister to Pr. tenuituba + Pr. eburnea (MF177038) with low supports (bootstrap value/posterior probability = 48/0.9994) in the trees computed by RAxML (Fig. S11) and MrBayes (Fig. S12), respectively.

The ML phylogram of 54 Gesneriaceae plastomes and seven Lamiales outgroups reconstructed by IQ-TREE. Nodes with full ultrafast bootstrap support values were not labeled with any number, while those values which are not equal to 100 were labeled out. Each clade within Gesneriaceae was colored by subtribes following the classification in Weber et al.1. The three major clades of Primulina were labeled corresponding to the Clades B, C, and D in Kong et al.16. Species name in bold indicates that the plastome is generated or assembled in this study.
As depicted in Fig. 7, monophylies of tribes, subtribe, and genera circumscribed by Weber et al.1 are fully supported and relationships among these taxa are all well resolved (Figs. 7, S11, S12). Specifically, the predominantly New World subfamily Gesnerioideae is monophyletic and sister to the monophyletic and mainly Old World subfamily Didymocarpoideae. Within Gesnerioideae, Achimenes Pers. (subtribe Gloxiniinae, tribe Gesnerieae) is sister to Rhabdothamnus A.Cunn. + Coronanthera Vieill. ex C.B.Clarke of the subtribe Coronantherinae, tribe Coronanthereae. Within the subfamily Didymocarpoideae (only tribe Trichiosporeae sampled), subtribes Loxocarpinae and Didymocarpinae form a clade, with subtribes Streptocarpinae (Streptocarpus Lindl.), Ramondiinae (Haberlea), and Corallodiscinae (Corallodiscus) forming successive sister groups to Loxocarpinae + Didymocarpinae. Within subtribe Didymocarpinae, Lysionotus and Henckelia form successive sister groups to a poorly supported clade (ultrafast bootstrap value of IQ-TREE/bootstrap value of RAxML/posterior probability of MrBayes = 66/77/1.00) composed of two clades, Petrocodon + Primulina and Oreocharis Benth. + Hemiboea C.B.Clarke (Figs. 7, S11, S12).
Within Primulina, three major clades corresponding to Clades B, C, and D designated in Kong et al.16 are also consistently resolved in all three analyses with full support values (Figs. 7, S11, S12). Notably, the three accessions of Pr. eburnea are not clustered together. Instead, MF177038, MF472011, and SRR9711276 are sister to Pr. tenuituba, Pr. pengii, and Pr. lutea (Yan Liu & Y.G.Wei) Mich.Möller & A.Weber, respectively (Figs. 7, S11, S12). In Petrocodon, Pe. jingxiensis is sister to the remaining two species. In Oreocharis, O. cotinifolia (W.T.Wang) Mich.Möller & A.Weber and O. mileensis (W.T.Wang) Mich.Möller & A.Weber, which previously belonged to two different genera5, are clustered with O. esquirolii H.Lév. (Fig. 7). In Paraboea, the species sampled here form two major clades (Figs. 7, S11, S12), consistent with Xu et al.37.
Discussion
Highly conserved Gesneriaceae plastomes
Our analyses indicate that the genome size, structure, gene content, and IR boundaries of all 52 Gesneriaceae plastomes investigated are highly conserved. Except for a slightly larger outlier of 156,614 bp in Corallodiscus lanuginosus (Wall. ex R.Br.) B.L.Burtt, the size of our sampled Gesneriaceae plastomes all falls within the range between 152,323 in Pr. medica to 154,245 bp in Paraboea wenshanensis Xin Hong & F.Wen (Table 1, S1), close to the median plastome size (154,953 bp) of angiosperms38. Gene contents of all 52 gesneriad plastomes are also highly conserved, with only an 83-bp-deletion detected near the 5′-terminal of the reading frame of rps14 in Oreocharis mileensis (W.T.Wang) Mich.Möller & A.Weber (≡ Paraisometrum mileense W.T.Wang) that might affect the function of the gene, though this likely case of gene deletion was neglected in the original publication39. Compared to the dynamic IR/SC boundaries in plastomes of other Lamiales taxa such as Amphilophium Kunth of Bignoniaceae40, the configurations around IR/SC boundaries of the 52 gesneriad plastomes are highly conserved (Figs. 2, S9, S10), with the minor variation only detected in Haberlea rhodopensis (ndhF is not included in IR) and some species of Paraboea species (LSC/IRB is located at rps19-rpl2 spacer) (Fig. S9). No conspicuous boundary shift was detected in the plastomes examined here (Figs. S9, S10), and no genome rearrangement was identified either (Fig. 2).
Comparative plastome analyses across diverse plant lineages such as cupressophytes41, Fabaceae42, Geraniaceae43, Asarum L. (Aristolochiaceae)44, and Trachelium L. (Camapnulaceae)45 revealed a positive correlation between the number of repeats and the extent of plastome rearrangement, suggesting a general trend of the repeat-mediated recombination that could have resulted in plastome structural variation42. Although plastomes of some species such as the hot pepper Capsicum annuum L. possess a considerable number of repetitive sequences and yet are structurally highly conserved46, the relatively low number of repeats (Fig. 4) and highly conserved nature of plastomes of Gesneriaceae reported in current study appear to conform well to the expectation of the repeat-mediate recombination hypothesis.
Codon usage bias in Gesneriaceae plastomes
The redundancy of 64 codons encoding only 20 amino acid and one stop signal, together with factors such as selections and mutational biases, inevitably leads to codon usage bias, and this phenomenon is ubiquitous and variable among different organisms or even within genomes47. In angiosperm plastomes, the usage of codons usually biases toward codons with high AT content, especially those synonymous codons with the third position in A or T48. This biased tendency is consistent with our results of RSCU analysis (Table S4; Fig. 5). Furthermore, we found that all of the Gesneriaceae plastomes analyzed exhibit highly conserved codon usage patterns (Fig. 5A). Since synonymous codon usage might be associated with various genetic events, e.g., natural selection, mutation, genetic drift49, our results of RSCU analysis and the discovery of conserved usage pattern provide preliminary information for better understanding of plastid gene expressions and plastome evolution of Gesneriaceae.
Potential phylogenetic markers for Primulina, Petrocodon, and gesneriads
Despite recent advancements in high throughput sequencing technologies, generating and analyzing large-scale plastome data for species-rich genera with more than 200 species such as Primulina or population samplings remains a costly and time- and labor-consuming task50,51. For efficient species identifications, taxonomic revisions, and inter- and intraspecific studies, it is still crucial to develop phylogenetic informative makers23. Since the hypervariable regions are often lineage-specific even among close-related groups51, we performed sliding window analyses on three alignments, i.e., Primulina, Petrocodon, and Primulina + Petrocodon. Thirteen different variable regions were identified across the three datasets, and six of them are constantly identified in all three alignments (Fig. 6). Amongst the 13 hypervariable regions, four (i.e., trnHGUG-psbA, rpl32-trnLUAG, ycf1_1, and ycf1_2) have been employed for phylogenetic studies of Primulina16,17,52,53, suggesting the potential of the other nine markers in improving the phylogenetic resolution of Primulina. However, compared to the top-9-ranked intrageneric variable regions identified in Feng et al.50, only three regions (i.e., trnHGUG-psbA, ndhF-rpl32, rpl32-trnLUAG) are common in the former and current studies, indicating that variable markers are indeed lineage specific. Nevertheless, the hypervariable regions identified by current study should greatly facilitate systematic and evolutionary research of Primulina and especially Petrocodon considering all previous studies of the latter genus12,20 employed only trnLUAA-trnFGAA that is slow-evolving and phylogenetically less informative24.
Additionally, the plastid SSRs identified in current study (Table S2) provides further molecular markers for population genetic studies of Gesneriaceae. Although contents of SSRs vary across different plastomes in our results, the dominance of A/T mononucleotide SSRs and the strongly AT-biased unit types (Fig. 3) are consistent with the general trend in angiosperm plastomes54. Of the 52 gesneriad species investigated, the plastome of Corallodiscus lanuginosus possesses the highest number of SSRs (100 repeats) and the second highest number of long repeats (34 repeats, Fig. 4), and these features might have contributed to its larger plastome size (156,614 bp). Interestingly, C. lanuginosus is known as a highly variable species with ca. 30 synonyms and distributed widely across remarkable altitudinal, latitudinal, and longitudinal gradients55. Zhou et al.55 proposed that the wide niche and morphological diversity of C. lanuginosus complex might be the consequence of its old divergence in Himalayan-Hengduan Mountains and hybridization/introgression from secondary contacts among populations. Comparing to the low numbers of repetitive sequences and SSRs in plastomes and relatively narrow distribution ranges of a majority of gesneriad species, the abundance of plastid repetitive elements in C. lanuginosus might have also been crucial in contributing its intraspecific variation and adaptation to diverse habitats55.
Plastome phylogenomics of Gesneriaceae
In this study, highly supported phylogenies of 54 Gesneriaceae plastomes rooted by seven Lamiales outgroups (Fig. 7) were reconstructed. Of the 19 Primulina accessions analyzed, our results are generally congruent with the grouping in Kong et al.16 as well as intrageneric relationships inferred by several studies with larger sampling using ITS and plastid markers6,16,17,53 with a few exceptions. One notable difference is the position of Pr. brachytricha var. magnibracteata. In Sanger sequencing-based phylogenetic analyses, Pr. brachytricha (W.T.Wang & D.Y.Chen) R.B.Mao & Yin Z.Wang var. brachytricha was placed in Clade D16,17 (equivalent to Clade A in Guo et al.53), while Pr. brachytricha var. magnibracteata was grouped in Clade B (Fig. 7) in current study, indicating that Pr. brachytricha var. brachytricha and Pr. brachytricha var. magnibracteata are distantly related. Recent phylogenetic studies have shown that several intraspecific varieties of Primulina, e.g., Pr. repanda var. guilinensis (W.T.Wang) Mich.Möller & A.Weber and Pr. glandulosa var. yangshuoensis (F.Wen, Yue Wang & Q.X.Zhang) Mich.Möller & A.Weber, were polyphyletic, necessitating nomenclatural changes to restore the monophyly of these taxa6. The likely polyphyletic nature of Pr. brachytricha urges further taxonomic study. On the other hand, while all three sampled Pr. eburnea are all placed in Clade D, they do not form a monophyletic clade (Fig. 7). Primulina eburnea is a species complex with a wide geographic range and morphological variation52,56 and the non-monophyly of the species has been noted in a previous study6. Zhang et al.57 reported hybrid individuals between Pr. eburnea and the sympatrically distributed Pr. mabaensis K.F.Chung & W.B.Xu, suggesting that hybridization could have contributed to the non-monophyly of the three plastomes of Pr. eburnea in current study, though further investigation is needed to test alternative explanation such as incomplete lineage sorting52,56.
Although our sampling of Gesneriaceae is far from comprehensive, phylogenomic relationships based on plastome sequences are highly congruent with current infrafamilial classification1 and relationships among subfamilies, tribes, and subtribes are also largely congruent with phylogenies inferred from the supermatrix of 26 PCR markers of 768 gesneriad species3 and a recent study using target enrichment2 capturing 418 genes of 78 taxa except for relationships among a few subtribes in tribe Trichosporeae. Specifically, our plastome trees placed Loxocarpinae and Didymocarpinae in one clade sister to Streptocarpinae (Figs. 7, S11, S12), while in Roalson and Roberts3 the clade of Streptocarpinae + Loxocarpinae is successively sister to Didissandrinae and Didymocarpinae, and in Ogutcen et al.2 the clade of Didymocarpinae + Streptocarpinae is sister to the clade of Loxocarpinae + Didissandrinae. These discordances could have resulted from sampling issues, hybridization/introgression, chloroplast capture, or incomplete lineage sorting26, and thus more studies leveraging informative genomic data with comprehensive sampling are needed to explore the extent and causations of the discordance across Gesneriaceae.
Material and methods
Taxon sampling
Two species of Petrocodon and six species of Primulina were collected from Guangxi and Hunan, China, and Pr. fimbrisepala was sampled from the botanical garden of Guangxi Institute of Botany, Guangxi, China (Tables 1 and S1). Voucher specimens were deposited in the Herbarium of Biodiversity Research Center, Academia Sinica, Taipei (HAST) and the Herbarium of Guangxi Institute of Botany (IBK). All species were identified by the second author (W.-B.X.), a leading authority in the taxonomy of Chinese Gesneriaceae5,6,12,21,37,53. Since all sampled species are not included in the “Threatened Species List of China’s Higher Plants”13 and were not collected from protected areas, no permission was required for sampling. All experiments, including the collection of plant materials, were complying with the relevant institutional, national, and international guidelines and legislation. In addition to the newly sequenced species, we downloaded all the 38 complete and two incomplete Gesneriaceae plastomes available on the GenBank, and assembled five additional plastomes from the WGS raw read data of four Primulina and one Henckelia species from the SRA database (accessed on February 1st, 2022). Consequently, a total of 52 plastomes from 13 Gesneriaceae genera, i.e., Achimenes, Corallodiscus, Damrongia, Dorcoceras, Haberlea, Hemiboea, Henckelia, Lysionotus, Oreocharis, Paraboea, Petrocodon, Primulina, and Streptocarpus, were included in our plastome comparative analyses, and 54 sequences of 15 Gesneriaceae genera (with Coronanthera and Rhabdothamnus included) were sampled for phylogenetic analyses. Overall, 19 Primulina (17 species) and three Petrocodon (three species) plastomes were analyzed in this study. Following the names used in Xu et al.5 and Plants of the World Online (http://plantsoftheworldonline.org/), the species names and accession numbers of the plastomes and raw reads sampled in this study are summarized in Table S1, regardless the names used in the original publication.
DNA extraction and next generation sequencing
Total genomic DNAs were extracted from 0.01 g of silica gel dried leaves using the modified CTAB method53. The quantity and quality of extracted DNAs were assessed by 1% gel electrophoresis, Nanodrop 2000 (Thermo Scientific Inc., Carlsbad, CA, USA), and Qubit Fluorometer (Thermo Scientific). The DNAs passing were sent to High Throughput Genomics Core at Biodiversity Research Center, Academia Sinica, Taipei, Taiwan to conduct library preparation using TruSeq Nano DNA Library Prep kit (Illumina Inc., San Diego, CA, USA) following manufacturer’s instructions with insert size 550 bp in length. The DNA libraries were sequenced using Illumina MiSeq System (Illumina) in pair-end mode with read length of 300 bp.
Plastome assembly and annotation
The WGS raw reads data of four Primulina and one Henckelia species were retrieved via NCBI SRA Toolkits v.2.1.11 for plastome assembly. The sequencing quality of both the newly sequenced and downloaded raw reads were evaluated via FastQC v.0.11.558. Low quality portions of the reads were trimmed and filtered by Trimmomatic v.0.3659 with the setting of “LEADING:25 TRAILING:25 SLIDINGWINDOW:4:20 MINLEN:200” for the nine newly sequenced samples and customized settings for the five downloaded samples of different read lengths. The trimmed reads were de novo assembled by GetOrganelle v.1.7.5.160 with the settings of “-R 15 -w 0.9 -k 55,85,105,127,131,143,151,171 -F embplant_pt --reduce-reads-for-coverage inf”. The resulting complete sequences or incomplete contigs were checked and finalized by mapping reads to respective sequences using “Map to Reference” function with “Medium–Low Sensitivity/Fast” settings in Geneious Prime v.2021.1.161.
All newly assembled plastomes were annotated based on the annotations of Arabidopsis thaliana (L.) Heynh. (GenBank accession: AP000423) using the “Map to Reference” and “Transfer Annotation” function under Geneious. The start and stop codons of each protein-coding gene were manually checked and adjusted. Additionally, length and anti-codon name of each tRNA gene were further verified by searching the gene sequence against the database of tRNAscan-SE web server62. Annotated genomes were visualized by OGDRAW v.1.3.163.
Comparative analyses of Gesneriaceae plastomes and evaluation of sequence divergence in Primulina and Petrocodon
To compare genome structures and sequence similarities among the 52 complete Gesneriaceae plastomes, sequences were aligned by Shuffle-LAGAN64, which could detect rearrangement, and analyzed using mVISTA web-interface28. To compare sequence variations across Primulina and Petrocodon, three alignments (19 Primulina, three Petrocodon, and 19 Primulina + three Petrocodon) were analyzed by sliding window computation of nucleotide diversity (pi) with the window size of 600 bp and step size of 200 bp in DnaSP v.6.12.0332. The regions with the 95th percentile windows were considered highly variable.
Simple sequence repeats and long repeats characterization of Gesneriaceae plastomes
To characterize SSR contents in plastomes, we utilized MISA29 to identify SSRs in all Gesneriaceae plastomes with one IR excluded and analyzed with the threshold of ten repeat units for mono-, five for di-, four for tri-, and three repeat units for tetra-, penta-, and hexanucleotide SSRs. Compound SSRs were detected by setting the interval up to 100 bp in length. The results were visualized in balloon plot generated in R v.4.0.1 using ‘reshape’ and ‘ggplot2’ packages65. For the detection of long repeats, REPuter30 was employed to search four repeat categories, i.e., forward (direct), reverse, complement, and palindromic (reverse complement) repeats with the minimum repeat size of 30 bp and ≥ 90% repeat identity by setting Hamming distance of 3.
Codon usage analysis of Gesneriaceae plastomes
For each Gesneriaceae plastome, the protein coding sequences (CDS) of all the 80 unique protein coding genes (Table 2) were extracted under Geneious. The codon usage frequencies and RSCU values of CDS in each plastome were evaluated in DAMBE v.7.3.231 with the first codon excluded in the calculation. The results of RSCU were visualized in heatmap using TBtools v.1.0985466.
Phylogenetic analyses of Gesneriaceae plastomes
In addition to the nine newly sequenced plastomes, we incorporated all the 38 complete and two incomplete Gesneriaceae plastomes available on Genbank and five plastomes assembled from released WGS reads data of Gesneriaceae species into our phylogenetic analyses. For the outgroup, due to the lack of available plastomes from the two most close-related families, Calceolariaceae and Peltantheraceae, plastomes from seven other Lamiales families were sampled (Table S1). A total of 61 plastomes with one IR excluded were aligned by MAFFT v.745067 under Geneious with default settings and were further partitioned into six parts, i.e., CDS codon position 1 (CDS1), CDS codon position 2 (CDS2), CDS codon position 3 (CDS3), introns, tRNA + rRNA genes (RNA), and intergenic spacers. The phylogenetic relationships were reconstructed by both maximum likelihood (ML) and Bayesian inference (BI) methods implemented in RAxML v.8.2.1236 and IQ-TREE v.2.0.634, and MrBayes v.3.2.735, respectively. For ML in RAxML, we executed the rapid bootstrap (BS) analysis with “autoFC” bootstrapping criterion and searched for the best-scoring ML tree based on the GTR + GAMMA nucleotide substitution model on the partitioned dataset. In IQ-TREE, the automatic model selection and the best-fit partitioning scheme search were conducted by ModelFinder33, and the ML tree reconstruction was carried out using “-m MFP + MERGE” option with 5000 replicates of ultrafast bootstrap (UFBS) approximation68. For BI, prior to MrBayes analyses, the best models fitting into MrBayes and the best-fit partition scheme of the partitioned dataset were evaluated by ModelFinder in IQ-TREE using “-m TESTMERGEONLY -mset mrbayes” options. In MrBayes, the priors were set according to the results of IQ-TREE with models unlinked among each partition, and the Marko chain Monte Carlo (MCMC) simulation of 2,500,000 generations was conducted in two independent runs with four chains each and sampling frequency of every 1000 generations. The first 25% of sampled trees were discarded as burn-in, and the remaining trees were summarized into a phylogram with clade credibility (posterior probability) values. The final phylogram from each analysis was visualized by FigTree v.1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Data availability
The newly assembled plastome sequences of Petrocodon and Primulina have been submitted to NCBI (https://www.ncbi.nlm.nih.gov/) under the accession numbers MZ675547–MZ675555.
References
Weber, A., Clark, J. L. & Möller, M. A new formal classification of Gesneriaceae. Selbyana 31, 68–94 (2013).
Ogutcen, E. et al. Phylogenomics of Gesneriaceae using targeted capture of nuclear genes. Mol. Phylogenet. Evol. 157, e107068. https://doi.org/10.1016/j.ympev.2021.107068 (2021).
Roalson, E. H. & Roberts, W. R. Distinct processes drive diversification in different clades of Gesneriaceae. Syst. Biol. 65, 662–684. https://doi.org/10.1093/sysbio/syw012 (2016).
Mason, J., Cole, G., Beermann, M. & Fraser, A. (eds) Gesneriads—African Violets, Gloxinias, Streptocarpus and Others (ACS Distance Education, 2007).
Xu, W.-B., Guo, J., Pan, B., Zhang, Q. & Liu, Y. Diversity and distribution of Gesneriaceae in China. Guihaia 37, 1219–1226. https://doi.org/10.11931/guihaia.gxzw201707004 (2017).
Xu, W.-B., Chang, H., Huang, J. & Chung, K.-F. Molecular systematics of Chiritopsis-like Primulina (Gesneriaceae): One new species, one new name, two new combinations, and new synonyms. Bot. Stud. 60, e18. https://doi.org/10.1186/s40529-019-0266-x (2019).
Tseng, Y.-H. et al. Phylogeography of Begonia luzhaiensis suggests both natural and anthropogenic causes for the marked population genetic structure. Bot. Stud. 60, e20. https://doi.org/10.1186/s40529-019-0267-9 (2019).
Liu, C.-N. et al. Plant adaptability in karst regions. J. Plant Res. 134, 889–906. https://doi.org/10.1007/s10265-021-01330-3 (2021).
Chung, K.-F. et al. Phylogenetic analyses of Begonia sect. Coelocentrum and allied limestone species of China shed light on the evolution of Sino-Vietnamese karst flora. Bot. Stud. 55, e1, https://doi.org/10.1186/1999-3110-55-1 (2014).
Sweeting, M. M. Karst in China: Its Geomorphology and Environment (Springer, 1995).
Clements, R., Sodhi, N. S., Schilthuizen, M. & Ng, P. K. L. Limestone karsts of Southeast Asia: Imperiled arks of biodiversity. Bioscience 56, 733–742. https://doi.org/10.1641/0006-3568(2006)56[733:LKOSAI]2.0.CO;2 (2006).
Xu, W.-B. et al. Petrocodon (Gesneriaceae) in the limestone karsts of Guangxi, China: Three new species and a new combination based on morphological and molecular evidence. Syst. Bot. 39, 965–974. https://doi.org/10.1600/036364414X681437 (2014).
Qin, H. et al. Threatened species list of China’s higher plants. Biodiv. Sci. 25, 696–744. https://doi.org/10.17520/biods.2017144 (2017).
Wang, Y.-Z. et al. Phylogenetic reconstruction of Chirita and allies (Gesneriaceae) with taxonomic treatments. J. Syst. Evol. 49, 50–64. https://doi.org/10.1111/j.1759-6831.2010.00113.x (2011).
Weber, A. et al. Molecular systematics and remodelling of Chirita and associated genera (Gesneriaceae). Taxon 60, 767–790. https://doi.org/10.1002/tax.603012 (2011).
Kong, H.-H. et al. Both temperature fluctuations and East Asian monsoons have driven plant diversification in the karst ecosystems from southern China. Mol. Ecol. 26, 6414–6429. https://doi.org/10.1111/mec.14367 (2017).
Xu, M.-Z., Yang, L.-H., Kong, H.-H., Wen, F. & Kang, M. Congruent spatial patterns of species richness and phylogenetic diversity in karst flora: Case study of Primulina (Gesnariaceae). J. Syst. Evol. 59, 251–261. https://doi.org/10.1111/jse.12558 (2021).
Ai, B. et al. Comparative transcriptome resources of eleven Primulina species, a group of ‘stone plants’ from a biodiversity hot spot. Mol. Ecol. Resour. 15, 619–632. https://doi.org/10.1111/1755-0998.12333 (2014).
Feng, C. et al. The genome of a cave plant, Primulina huaijiensis, provides insights into adaptation to limestone karst habitats. New Phytol. 227, 1249–1263. https://doi.org/10.1111/nph.16588 (2020).
Weber, A. et al. A new definition of the genus Petrocodon (Gesneriaceae). Phytotaxa 23, 49–67. https://doi.org/10.11646/PHYTOTAXA.23.1.3 (2011).
Lu, Y.-B. et al. Floral evolutionary trend of Petrocodon (Gesneriaceae) and its significance in classification. Guihaia 37, 1227–1239. https://doi.org/10.11931/guihaia.gxzw201611029 (2017).
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F. & Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. https://doi.org/10.1007/s11103-011-9762-4 (2011).
Kuo, W.-H. et al. Plastome phylogenomics of Allaeanthus, Broussonetia and Malaisia (Dorstenieae, Moraceae) and the origin of B. × kazinoki. J. Plant Res. 135, 203–220. https://doi.org/10.1007/s10265-022-01369-w (2022).
Shaw, J. et al. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: the Tortoise and the Hare IV. Am. J. Bot. 101, 1987–2004. https://doi.org/10.3732/ajb.1400398 (2014).
Doyle, J. J. Defining coalescent genes: Theory meets practice in organelle phylogenomics. Syst. Biol. 71, 476–489. https://doi.org/10.1093/sysbio/syab053 (2022).
Hsieh, C.-L., Yu, C.-C., Huang, Y.-L. & Chung, K.-F. Mahonia vs. Berberis unloaded: Generic delimitation and infrafamilial classification of Berberidaceae based on plastid phylogenomics. Front. Plant Sci. 12, 720–171. https://doi.org/10.3389/fpls.2021.720171 (2022).
Xie, C.-Z. et al. Comparative genomic study on the complete plastomes of four officinal Ardisia species in China. Sci. Rep. 11, e22239. https://doi.org/10.1038/s41598-021-01561-3 (2021).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. https://doi.org/10.1093/nar/gkh458 (2004).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 33, 2583–2585. https://doi.org/10.1093/bioinformatics/btx198 (2017).
Kurtz, S. et al. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. https://doi.org/10.1093/nar/29.22.4633888 (2001).
Xia, X.-H. DAMBE7: New and improved tools for data analysis in molecular biology and evolution. Mol. Biol. Evol. 35, 1550–1552. https://doi.org/10.1093/molbev/msy073 (2018).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. https://doi.org/10.1038/nmeth.4285 (2017).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–11534. https://doi.org/10.1093/molbev/msaa015 (2020).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Xu, W.-B. et al. Three new species of Paraboea (Gesneriaceae) from limestone karsts of China based on morphological and molecular evidence. Bot. Stud. 58, e56. https://doi.org/10.1186/s40529-017-0207-5 (2017).
Park, S., An, B. & Park, S. Reconfiguration of the plastid genome in Lamprocapnos spectabilis: IR boundary shifting, inversion, and intraspecific variation. Sci. Rep. 8, e13568. https://doi.org/10.1038/s41598-018-31938-w (2018).
Meng, J., Zhang, L.-N. & He, J. Complete plastid genome of the endangered species Paraisometrum mileense (Gesneriaceae) endemic to China. Mitochondrial DNA B 4, 3585–3586. https://doi.org/10.1080/23802359.2019.1677186 (2019).
Thode, V. A. & Lohmann, L. G. Comparative chloroplast genomics at low taxonomic levels: A case study using Amphilophium (Bignonieae, Bignoniaceae). Front. Plant Sci. 10, e796. https://doi.org/10.3389/fpls.2019.00796 (2019).
Hao, Z.-D. et al. The complete chloroplast genome sequence of a relict conifer Glyptostrobus pensilis: Comparative analysis and insights into dynamics of chloroplast genome rearrangement in cupressophytes and Pinaceae. PLoS ONE 11, e0161809. https://doi.org/10.1371/journal.pone.0161809 (2016).
Wu, S. et al. Extensive genomic rearrangements mediated by repetitive sequences in plastomes of Medicago and its relatives. BMC Plant Biol. 21, e421. https://doi.org/10.1186/s12870-021-03202-3 (2021).
Weng, M.-L., Blazier, J. C., Govindu, M. & Jansen, R. K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 31, 645–659. https://doi.org/10.1093/molbev/mst257 (2014).
Sinn, B. T., Sedmak, D. D., Kelly, L. M. & Freudenstein, J. V. Total duplication of the small single copy region in the angiosperm plastome: Rearrangement and inverted repeat instability in Asarum. Am. J. Bot. 105, 71–84. https://doi.org/10.1002/ajb2.1001 (2018).
Haberle, R. C., Fourcade, H. M., Boore, J. L. & Jansen, R. K. Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 66, 350–361. https://doi.org/10.1007/s00239-008-9086-4 (2008).
Jo, Y. D. et al. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep. 30, 217–229. https://doi.org/10.1007/s00299-010-0929-2 (2011).
Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 2, 13–34. https://doi.org/10.1093/oxfordjournals.molbev.a040335 (1985).
Morton, B. R. Selection on the codon bias of chloroplast and cyanelle genes in different plant and algal lineages. J. Mol. Evol. 46, 449–459. https://doi.org/10.1007/PL00006325 (1998).
Akashi, H. Gene expression and molecular evolution. Curr. Opin. Genet. Dev. 11, 660–666. https://doi.org/10.1016/S0959-437X(00)00250-1 (2001).
Feng, C., Xu, M.-Z., Feng, C., von Wettberg, E. J. B. & Kang, M. The complete chloroplast genome of Primulina and two novel strategies for development of high polymorphic loci for population genetic and phylogenetic studies. BMC Evol. Biol. 17, e224. https://doi.org/10.1186/s12862-017-1067-z (2017).
Särkinen, T. & George, M. Predicting plastid marker variation: Can complete plastid genomes from closely related species help?. PLoS ONE 8, e82266. https://doi.org/10.1371/journal.pone.0082266 (2013).
Gao, Y., Ai, B., Kong, H.-H., Kang, M. & Huang, H.-W. Geographical pattern of isolation and diversification in karst habitat islands: A case study in the Primulina eburnea complex. J. Biogeogr. 42, 2131–2144. https://doi.org/10.1111/jbi.12576 (2015).
Guo, J., Pan, B., Liu, J., Xu, W.-B. & Chung, K.-F. Three new species of Primulina (Gesneriaceae) from limestone karsts of China based on morphological and molecular evidence. Bot. Stud. 56, 1–13. https://doi.org/10.1186/s40529-015-0115-5 (2015).
George, B., Bhatt, B. S., Awasthi, M., George, B. & Singh, A. K. Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr. Genet. 61, 665–677. https://doi.org/10.1007/s00294-015-0495-9 (2015).
Zhou, P., Li, J. & Möller, M. Secondary contact, hybridization and polyploidization add to the biodiversity in the Hengduan Mountains, exemplified by the widespread Corallodiscus lanuginosus (Gesneriaceae). Plant Syst. Evol. 303, 587–602. https://doi.org/10.1007/s00606-017-1392-0 (2017).
Wang, J., Ai, B., Kong, H.-H. & Kang, M. Speciation history of a species complex of Primulina eburnea (Gesneriaceae) from limestone karsts of southern China, a biodiversity hot spot. Evol. Appl. 10, 919–934. https://doi.org/10.1111/eva.12495 (2017).
Zhang, X.-L., Yang, L.-H. & Kang, M. Post-pollination reproductive isolation of sympatric populations of Primulina eburnea and P. mabaensis (Gesneriaceae). Biodiv. Sci. 25, 615–620. https://doi.org/10.17520/biods.2017029 (2017).
Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. https://doi.org/10.1093/bioinformatics/btu170 (2014).
Jin, J.-J. et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome. Biol. 21, e241. https://doi.org/10.1186/s13059-020-02154-5 (2020).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. https://doi.org/10.1093/bioinformatics/bts199 (2012).
Lowe, T. M. & Chan, P. P. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res 44, W54–W57. https://doi.org/10.1093/nar/gkw413 (2016).
Greiner, S., Lehwark, P. & Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. https://doi.org/10.1093/nar/gkz238 (2019).
Brudno, M. et al. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 19, i54–i62. https://doi.org/10.1093/bioinformatics/btg1005 (2003).
R Core Team. R: A Language and Environment for Statistical Computing. https://www.R-project.org/ (2020).
Chen, C.-J. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Minh, B. Q., Nguyen, M. A. T. & von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. https://doi.org/10.1093/molbev/mst024 (2013).
Acknowledgements
This study was supported by the Minister of Science and Technology, Taiwan (MOST 103-2621-B-002-004-MY3) to K.-F.C. and in part by the National Natural Science Foundation of China (Grant No. 31860043) to W.-B.X.
Author information
Authors and Affiliations
Contributions
W.-B.X. and K.-F.C. designed the study. C.-L.H., W.-B.X., and K.-F.C. collected the plant materials. C.-L.H. carried out the experiments and data analyses. C.-L.H. wrote the draft and W.-B.X. and K.-F.C. revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hsieh, CL., Xu, WB. & Chung, KF. Plastomes of limestone karst gesneriad genera Petrocodon and Primulina, and the comparative plastid phylogenomics of Gesneriaceae. Sci Rep 12, 15800 (2022). https://doi.org/10.1038/s41598-022-19812-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19812-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
