
Abstract
Crohn’s disease (CD) is characterised by chronic inflammation. We aimed to identify a relationship between plasma inflammatory metabolomic signature and genomic data in CD using blood plasma metabolic profiles. Proton NMR spectroscopy were achieved for 228 paediatric CD patients. Regression (OPLS) modelling and machine learning (ML) approaches were independently applied to establish the metabolic inflammatory signature, which was correlated against gene-level pathogenicity scores generated for all patients and functional enrichment was analysed. OPLS modelling of metabolomic spectra from unfasted patients revealed distinctive shifts in plasma metabolites corresponding to regions of the spectrum assigned to N-acetyl glycoprotein, glycerol and phenylalanine that were highly correlated (R2 = 0.62) with C-reactive protein levels. The same metabolomic signature was independently identified using ML to predict patient inflammation status. Correlation of the individual peaks comprising this metabolomic signature of inflammation with pathogenic burden across 15,854 unselected genes identified significant enrichment for genes functioning within ‘intrinsic component of membrane’ (p = 0.003) and ‘inflammatory bowel disease (IBD)’ (p = 0.003). The seven genes contributing IBD enrichment are critical regulators of pro-inflammatory signaling. Overall, a metabolomic signature of inflammation can be detected from blood plasma in CD. This signal is correlated with pathogenic mutation in pro-inflammatory immune response genes.
Similar content being viewed by others
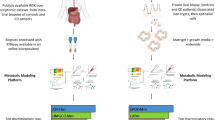


Introduction
Crohn’s disease (CD), one of the major subtypes of inflammatory bowel disease (IBD), is a heterogenous, relapsing, remitting condition characterised by transmural inflammation across the gastrointestinal tract. Disease aetiology centres on complex interaction between genetic predispostion and intestinal microbial exposure. Over 240 genes associated with IBD are enriched for proteins linked with bacterial recognition and response pathways, epithelial barrier integrity and downstream inflammatory signalling1,2. Whilst effective therapies exist, there is a clear need to stratify patients into risk groups for disease severity, complications and medication response. Reliable genetic and plasma biomarkers provide an attractive mechanism to stratify patients at diagnosis and during follow-up, whilst promoting novel drug discovery3.
Nuclear magnetic resonance (NMR) spectroscopy identifies precise constituents of biological samples, whereby molecules present distinct characteristic spectra. NMR has demonstrated the ability to discriminate IBD patients from controls through identification of dysregulated urine and plasma metabolites4; and distinguished IBD patients with active disease from those in remission5. Identification of genomic variation associated with disease severity markers, or biomarker profiles, can lead to targeted therapeutics and repurposing of known medications for new conditions6. Combining urine NMR spectra analysis with common variants identified through genome-wide association studies has previously been used to discover genetically determined metabolites in unselected samples7.
This study aimed to establish the discrete regions of the plasma metabolomic spectrum that specifically associate with inflammation measured using C-reactive protein (CRP) in paediatric CD patients. Following identification of a robust metabolomic signature of inflammation, we further aimed to compress the data underlying these discrete metabolomic peaks for correlation against exome sequencing data in order to identify genes and molecular pathways harbouring genetic variation that may explain altered the plasma metabolites. Ultimately, we wished to see how integrating metabolomic and genomic data could be used to stratify patients and inform therapeutic targeting.
Materials and methods
Patient samples
Patients aged < 18 years diagnosed with Crohn’s disease using the modified Porto criteria were recruited as part of the Genetics of IBD research study. Research blood samples were acquired during routine clinics.
Metabonomic and whole exome sequencing data were generated for a total of 228 patients diagnosed with CD. Where routine blood tests were clinically-requested on the same day as the plasma sample used for metabolomic analysis was acquired, these data were digitally retrieved from hospital records as previously described8. C-reactive protein (CRP) level was applied as our outcome measure to identify patients with actively inflamed disease. Patient medications at the time of plasma sampling were retrieved from electronic health records.
The study has ethics approval from Southampton & South West Hampshire Research Ethics Committee (09/H0504/125) and the study was conducted in accordance with relevant guidelines and regulations. All patients, or their parents/guardians, gave informed consent for participation in this study.
DNA and plasma extraction
Genomic DNA was extracted from peripheral venous blood samples collected in Ethylenediaminetetraacetic acid (EDTA) using the salting out method. Deoxyribonucleic acid (DNA) concentration was estimated using the Qubit® 2.0 Fluorometer and 260:280 ratio calculated using a nanodrop spectrophotometer. The average DNA yield obtained was 150 µg/ml and approximately 20ug of each patient DNA was extracted for next generation sequencing.
Plasma was isolated from peripheral venous blood by centrifuging samples for 10 min at 2000 RPM and 4 °C. After centrifugation, the plasma laying above the buffy coat was extracted and immediately frozen and stored at − 80 °C.
Genomic data processing
Whole exome sequencing data were generated using Agilent SureSelect exon capture kits and Illumina HiSeq sequencing platforms. Processing and targeted analyses of the whole exome sequencing data applied herein have been presented elsewhere9,10. Genomic data were transformed into per-patient gene pathogenicity scores using the GenePy algorithm11. GenePy integrates the effect of multiple variants in each gene incorporating information on variant zygosity, frequency and deleteriousness (inferred using CADD v1.5 scores12). GenePy scores were initially generated for all patients for all 19,229 RefSeq genes. Genes with a Gene Damage Index (GDI) above the recommended threshold (GDI_Phred > 13.84) were excluded as genes with values above this level are considered highly mutable but unlikely to be disease causing13. This resulted in a final matrix of 15,854 GenePy scores for all patients.
Metabolomics analysis of plasma
Plasma samples (200 µL) were mixed with deuterium water (D2O) (400 µL). The homogenized samples were centrifuged (10 min; 4 °C; 12,000 × g) and transferred to 5 mm NMR tubes for analysis by NMR spectroscopy. Plasma samples were processed into a single batch of 228 CD samples. NMR experiments applied a Bruker AV700 NMR instrument equipped with a 5 mm inverse CryoProbe™. A standard 1-dimensional NOESY-PR-1D experiment was performed on each sample, using a standard preset pulse sequence (noesypr1d90°). A Carr-Purcell-Meiboom-Gill (CPMG) experiment was applied (preset pulse sequence cpmgpr1d90°), where simple presaturation of the water signal was used. This experiment reduces the signal contribution from albumin and lipoproteins present in plasma and highlight signals from otherwise overshadowed smaller molecules. All samples were analysed at 297° K, 65 k data point spectrum (spectral width 19,607 Hz) was obtained by recording 256 scans (8 dummy scans). Phase and baseline of the spectra were corrected using MestreNova software v10.0m. NMR spectra were referenced to the glucose peak at δ 5.223 ppm.
Metabolomics statistical analysis
Full resolution spectra were processed using Matlab vR2017a. The residual water signal was removed. Relative spectra were mean-centred and scaled to unit variance. Principal component analysis (PCA) was used to compare samples and identify outliers. Orthogonal Projection to Latent Structure (OPLS) analysis was performed for the supervised stage of the analysis, where NMR spectra were used as a matrix of variables. Regression of continuous patient CRP measurements against their metabolome data matrix, assessed plasma metabolic profile alteration with active inflammation. Model prediction was evaluated using goodness-of-fit correlation coefficient R2, showing what percentage of variation is explained by the model, and goodness-of-prediction (Q2), constituting the percentage of that variance which can be predicted by sevenfold cross-validation (hence splitting the input data in 7 subsets and recursively fit the model on 6 subsets and test its performance on 1 the left-out subset until all subset are used as test-set). Loadings were presented as a pseudo-NMR spectrum, plotting the model back-scaled coefficients and the weight of the variables. Metabolites with an R2 weight > 0.4 were considered highly discriminatory14.
Machine learning classification
A random forest classifier (RF) of metabolic profiles was employed to predict patients with active inflammation as measured by CRP levels. While the metabolomic analysis utilised continuous CRP values to identify highly correlating peaks, the objective of the RF was to discriminate patients with negligible active inflammation from those with moderate/severe inflammation. Therefore, continuous CRP levels were binarised following the current WHO and FDA guidelines to classify patient bloods as either inflamed (CRP ≥ 5 mg/L) or non-inflamed (CRP < 5 mg/L)15,16.
The machine learning (ML) approach consisted of three phases (Fig. 1). The first phase involved the use of an RF classifier and a fivefold cross-validated recursive feature elimination approach (RFE-CV) to identify the regions of the NMR spectrum contributing to the non/inflamed patient classification (feature selection). This step recursively excludes 1% of the 38,470 datapoints comprising the metabolic profiles, until all the features are removed, and identifies datapoints consistently important for classification. The resultant selected regions were then employed to generate the final fivefold cross-validated RF model. Averaged metrics collected to assess performance include the F-1 statistic, precision, recall and balanced accuracy. From this final cross-validated model, features ranked within the top 5th percentile of importance were retained for further analysis.

NMR and genomic data integration. Phase (I) NMR spectra and patient CRP data were input to the RF model using (a) RFECV and (b) cross-validated methods to select spectral regions discriminating non/inflamed patients. Phase (II) Informative data-points were clustered and peaks reduced to a single eigenvector. Phase (III) Eigenvectors for each peak were individually correlated against all genes and tested for enrichment. Created with BioRender.com.
The selected points of the spectra were subsequently binned by their location on the NMR spectrum. Groups of ≥ 10 points observed in close proximity were defined as ‘peaks’ and their constituent variance summarised using PCA. The discriminatory power of each component in separating inflamed/non-inflamed patients was assessed using Wilcoxon rank sum test and p values adjusted using false discovery rate (FDR). Components with a corrected p value < 0.05 were combined by their sum, generating a single eigenvector for each peak. This process transformed the multiple points within each of the n peaks into a matrix of eigenvector scores for each patient (Fig. 1, phase II). Summing those components significantly discriminating the inflamed and non-inflamed classes after FDR correction allowed for integration into downstream analyses.
In phase III, the resulting eigenvector matrix was integrated with the GenePy-transformed genomic data. These steps resulted in two matrices for all patients summarising: (1) eigenvector scores representing the metabolomic data most discriminatory of inflammation status and; (2) genetic data summarising the pathogenic burden of mutation for each gene. Spearman’s rank was used to correlate each of the metabolomic eigenvectors against GenePy scores for 15,854 genes. Genes with a nominally significant correlation were tested for enrichment in human databases (Gene Ontology, KEGG pathways, REACTOME, Complexes (CORUM), Human Phenotypes (HPA), WikiPathway (WP) using gProfiler217. Enrichment scores (p values) were corrected using the SCS method embedded in the gProfiler2 model.
ML methods were applied using the Scikit-learn Python v3.7 library and R v4.0.3 packages.
Results
Patients were recruited during routine clinics and untargeted with respect to diseases state, duration or treatment. As expected for paediatric Crohn’s disease, our cohort is characterised by an excess of male patients. The cohort reflected heterogeneity expected within clinical service with respect to time since diagnosis, disease state and treatment. Retrospective interrogation of clinical records identified 30 children who had undergone (24-h liquid diet and 4-h) fasting in preparation for colonoscopy and 154 patients for whom same-day blood tests had been clinically requested (Table 1). Medication data were available for all patients, supplementary data 1. Twenty-seven of the 154 patients were on no medications, or only nutritional therapy, at the time of plasma sampling, Table 2.
Metabonomics
NMR-based blood-plasma metabolomic profiles were acquired for all 228 CD patients. Multivariate analysis of these samples identified a subset of patients whose metabolome was markedly characterised by elevated concentrations of ketone bodies (3-hydroxybutyrate, acetone and acetoacetate, Supplementary Fig. 1A). Cross referencing with clinical records revealed these patients had undergone bowel preparation for endoscopy procedure prior to the blood sampling used for NMR analyses. This metabolic perturbation was reflected in the OPLS model, which highlighted ketone bodies as strong discriminants, and overshadowed the importance of other discriminative peaks of the spectrum (Supplementary Fig. 1A, B and C). All 30 patients with documented evidence of bowel preparation for endoscopy prior to plasma collection were therefore excluded from subsequent analyses.
Metabolome was regressed against blood CRP readings for the 154 CD patients (R2Y = 0.63, Q2Y = 0.41; Fig. 2A). The corresponding loadings plot (Fig. 2B) highlights the peaks contributing most to that classification (weight > 0.4). Distinct signals (Table 3) associated with N-acetyl glycoprotein (δ 2.01–2.04 ppm), glycerol (δ 3.56, 3.64 ppm), phenylalanine (δ 7.33, 7.38, 7.43 ppm), and an unidentified lipid signal (δ 2.66 ppm) were identified at significantly higher concentration in plasma samples obtained from patients with higher systemic inflammation.

CRP prediction and spectra deconvolution. (A) OPLS scores plot. Each point represents one patient spectrum, colour-coded according to CRP levels. Strong correlation between T and Tcv indicates a robust model. (B) Loadings plot; colour-scale indicates the correlation magnitude of metabolites with the model scores (r2).
Machine learning classification
An RF model was employed to discriminate patient classes of actively inflamed (n = 58 patients with CRP ≥ 5 mg/L) versus uninflamed (n = 96 patients CRP < 5 mg/L) cases.
The first phase of modelling identified 23.1% percent (8934 datapoints) of the NMR spectrum as informative (Supplementary Fig. 2). On average, the model trained and tested on this fraction of the spectrum was effective in distinguishing the non/inflamed classes (mean F-1 statistic = 0.78 ± 0.05; balanced accuracy = 0.82 ± 0.04; precision = 0.84 ± 0.08 and; recall = 0.74 ± 0.05).
Figure 3A shows the regions of the spectrum identified by the RF model as most informative in discriminating patients with and without active inflammation.

Machine learning classification of patients using NMR data. (A) Most informative regions selected by RF model to discriminate patient inflammation status. (B) PCA of patient’s spectra using 258 most informative NMR datapoints. (C) Distribution of the selected most informative datapoints by their shift δ (ppm) and importance. Green and red dashed lines indicate the start and end of a peak.
PCA modelling of the subset of 258 points evaluated as having a relative importance measure within the top 5th percentile (Fig. 3A; Supplementary Fig. 3) shows reasonable separation between patients according to their inflammation status (Fig. 3B). The distribution of 199 of these points was concentrated within 6 discrete peaks containing ≥ 10 supporting datapoints (Fig. 3C). These six NMR spectrum peaks identified as highly informative in the classification of CD patients with/out active inflammation (Table 4) were compared with the peaks independently identified through OPLS modelling using continuous CRP levels. Reassuringly, peaks 2 and 3 defined by RF modelling recapitulate the OPLS findings of those peaks labelled as GlycA (δ 2.03–2.07 ppm); peak 4 defined by ML corresponds to one (δ 3.56–3.57 ppm) of the two spectral signatures that are noted in the OPLS modelling to depict glycerol; and peaks 5 and 6 in the RF model correspond to the two phenylalanine peaks (δ 7.20–7.26 ppm) as seen in Fig. 2B. Furthermore, the individual data points underlying ML derived peaks 3 and 4 are recognised as having the highest average discriminatory value for classification of inflamed status (average importance of 0.29 and 0.35 respectively) with peak 4 exhibiting the highest mean importance and also containing the single data point with the highest discriminative importance (Fig. 3C, Table 4).
Metabolomics-genomics integration
Single eigenvectors summarising the six RF peaks significantly discriminating the inflamed and non-inflamed classes after FDR correction (Table 4, Supplementary Fig. 4, Supplementary Fig. 5).
Correlation of each patient’s eigenvector scores for each of the six ML-defined metabolomic peaks against their GenePy gene scores was used to determine any relationship between metabolomic signatures of active inflammation and gene pathogenicity scores. This resulted in sets of nominally significant genes that were either positively or negatively correlated. In order to retain potentially informative biological insight, these genes were then grouped and assessed by direction of correlation (positive, negative, all). These gene sets were then interrogated for enrichment of specific functional pathways that might be useful in interpreting NMR peak signatures (Table 5).
Peak 1 is most significantly enriched for ‘g-protein coupled receptors (GPCRs)’ (WP:WP24; p = 0.004) when considering all correlated genes, although the signal remains significant when considering only genes that are negatively correlated with inflammation status. Specifically, patients designated as having non-inflamed status exhibit a higher burden of pathogenic variation in genes involved in GPCR signalling. Peak 2 is positively correlated with genes enriched to function within receptor complexes (p = 0.02) and regulate actin cytoskeleton (p = 0.03).
Metabolomic peak 4, identified by both OPLS and ML modelling to be most strongly associated with inflammation, contains the most significantly enriched functional gene-sets. One hundred and ten genes whose pathogenicity scores are significantly correlated with peak 4 are enriched to function within the ‘intrinsic component of membrane’ (p < 0.003; GO:0031224) and its subset-term ‘integral component of membrane’ (105 genes, p < 0.003; GO:0016021) (Supplementary Table 1). Interestingly, this is the same functional group identified as correlated with peak 3 suggesting a common biological mechanism might drive both metabolic signatures.
Of particular interest given our clinical cohort, is the set of seven genes correlated with peak 4 identified as enriching for molecular function in ‘inflammatory bowel disease’ (p < 0.003; KEGG:05321). This enrichment is specific to negatively correlating genes, indicating that CD patients with active inflammation are more likely to have a low burden of pathogenic variation within these genes. The seven nominally correlated genes that combine to define this IBD enrichment term are GATA3, IL12B, IL12RB2, IL6, MAF, NFKB1, RORC.
Peak 5 shows a distinct enrichment for the ESR (estrogen signalling receptor) signalling pathway (p < 0.005), an important molecular cascade involved in acute and chronic inflammation. Finally, peak 6 shows weaker enrichment for various enrichment terms many of which reflect of plasma membrane function echoed in peaks 3 and 4.
Discussion
This study combined untargeted metabolomics with whole gene pathogenicity burden scores derived from whole-exome sequencing data from paediatric patients diagnosed with Crohn’s disease. Assimilation of clinical and omic data for patient samples modelled NMR spectra into discrete peaks strongly associated with active inflammation detected in plasma. Individual patient differences in these metabolomic signatures of inflammation appear non-random with respect to functional capacity of genes that elicit the pro-inflammatory immune response for which targeted therapies exist18,19.
We used two approaches to determine the regions of patient metabolic spectra most associated with inflammation. Results of both OPLS modelling of continuous CRP and RF modelling of binarised CRP levels, culminated to identify the same regions of the spectrum typically assigned to N-acetyl glycoproteins (GlycA), glycerol and phenylalanine. The metabololomic signature of inflammation identified in this study of paediatric CD patients is consistent with that identified in other studies of adult inflammation20. GlycA is a composite signal reflecting glycoprotein acetylation of heterogeneous origin21,22. Our data independently corroborate this signal as an NMR-derived spectrometric biomarker of systemic inflammation. The same signal was recently highlighted in the context of acute febrile illnesses, chronic inflammatory and autoimmune diseases and found to strongly correlate with CRP, interleukin-6, fibrinogen, serum amyloid A, lipoprotein-associated phospholipase A2 and tumour necrosis factor23,24. While the data here presented reflects a single snapshot of patient’s inflammation course, previous studies indicated how the GlycA signature might evolve over time25—yet with unknown dynamic with respect to CRP—but confirming its role in systemic inflammation20.
CRP, produced by hepatocytes in response to IL-6, is a non-specific clinical marker of acute and chronic systemic inflammation. However, its efficacy as a single marker is limited by high inter- and intra-individual variability26. Although correlated, it has been suggested that the protein glycan biomarker GlycA and CRP may play distinct roles27. CRP levels increase in response to bacteria and intracellular antigens of damaged cells, as an early acute phase response, whereas haptoglobin, α1-acid glycoprotein, α1-antitrypsin and transferrin, that contribute the most to the GlycA signal, rise later stage of the inflammatory response28. GlycA measurement may represent an independent, more stable biomarker of acute response and systemic inflammation.
Regions of the metabolomic spectrum attributed to glycerol and phenylalanine were consistently associated with the inflammation in our CD patients. Phenylalanine is an aromatic amino acid previously linked to metabolic disturbance29 and a marker of systemic low grade inflammation possibly arising from liver disfunction, compromised uptake at the blood brain barrier or altered microbiota composition30. Glycerol has recently been described as a single molecule systemic biomarker of infection whereby increased glycerol in plasma reflects a metabolic adaptation to intestinal infection, as a provision of sufficient energy for survival31. This study provides evidence for a correlation between the NMR glycerol signal and genes known to be involved in the pathogenesis of inflammatory bowel disease. Our data demonstrate increased levels of glycerol in patient’s plasma negatively correlating with individual burden of pathogenic mutations in genes driving pro-inflammatory signalling i.e. patients with wild-type sequence across these genes exhibited a higher metabolic signature of inflammation suggesting a more intact and effective pro-inflammatory response. Despite our data modelling being blind to patient diagnosis, objective assessment of over fifteen thousand genes against the metabolomic signature of inflammation, ‘inflammatory bowel disease’ was amongst the most significantly enriched terms for correlated genes. The seven genes driving this result converge upon pro-inflammatory pathways and extensive data already support their role in IBD. The pathogenesis of Crohn’s disease is multi-factorial, but there appears to be a significant proportion of patients where the underlying genetic risk is related to a hypo-immune response (such as loss-of function variants in NOD2)30. This concept provides a framework for understanding why low burden of variation in pro-inflammatory ‘IBD’ genes correspond to high glycerol levels. We hypothesise that in these maintained pro-inflammatory pathways, chronic activation occurs due to alternative hypo-immune response to intestinal bacteria, resulting in chronic inflammation and the observed hyper-inflammatory response32,33.
Expression of GATA3, a mediator of Th2 cytokine response to inflammation is dependent on the p50 subunit of NFKB encoded by NFKB134. NFKB is activated by pattern-recognition receptors (PRRs) including the NOD-receptors and a master regulator of immune inflammation with an established role in perturbed mucosal inflammation in CD35,36. NFKB recruits several pro-inflammatory cytokines in response to microbial stimulation, including IL-12 and IL-23. IL6, in addition to promoting CRP, drives Th17 lineage development—plasticity of which is also influenced by RORC37,38. IL12B is an IBD-associated gene encoding the p40 subunit that is targeted by ustekinumab monoclonal antibody and common to both IL12 and IL2339,40. Functional studies in both mice39 and human patients41 proved how mutations in its coding sequence can alter the inflammatory response through the formation of the IL-12/IL-23 heterodimer. Although, IL12 and IL23 are both implicated in temporally distinct inflammatory responses to intestinal barrier impairment42, concurrent implication in our analysis of IL12RB that encodes the membrane receptor for the IL12 cytokine might suggest IL12 signalling is driving the inflammatory response in our paediatric cohort. Interestingly GlycA has been previously implicated as a tool for measuring inflammation, and specifically within IBD43,44. However this is a non-specific marker and the link to underlying genomic variation requires further investigation.
Future optimisation of the approach applied herein is possible. CRP levels are transient and fluctuate with disease state and treatment. Other than their attendance at routine tertiary clinics, our patients were unselected with respect to their disease state or clinical intervention. It is likely that standardising the patient cohort would further improve power to detect genetic signals. Superior power may be gained by focussing on treatment naïve individuals at point of diagnosis, although such samples can be difficult to attain and remain non-uniform with respect to underlying genetics, steroid and antibiotic use and duration of disease prior to first attendance.
In preparation for endoscopy, patients are restricted to a glucose-containing fluid-only diet from 24-h prior to the procedure and nil-by-mouth for the four hours immediately preceding endoscopy. Our data identified a metabolic signature highly inflated for ketone bodies in these patients that may warrant further clinical consideration.
Our data indicate patients with an altered burden of pathogenic mutation within genes critical to mounting the pro-inflammatory immune response following bacterial exposure, harbour a distinctive metabolomic signature (δ 3.56–3.57 ppm) reflecting inflammatorily active disease. This metabolomic signature and its correlated genes warrant further investigation as biomarkers to stratify CD patients into groups that may respond differently to targeted monoclonal antibodies. While our study focussed on children with a diagnosis of Crohn’s disease, we suggest mutations in these genes are unlikely to represent the primary CD disease trigger in many of these patients, but instead contribute to an individual genomic profile that substantially modulates the inflammatory response and disease progression.
Data availability
The datasets generated and/or analysed during the current study are available through direct collaborative agreements, in line with the informed consent gained from all participants.
References
Lee, J. C. et al. Genome-wide association study identifies distinct genetic contributions to prognosis and susceptibility in Crohn’s disease. Nat. Genet. 49, 262–268 (2017).
Khor, B. et al. Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Amiot, A. & Peyrin, B. L. Current, new and future biological agents on the horizon for the treatment of inflammatory bowel diseases. Ther. Adv. Gastroenterol. 8, 66–82 (2015).
Schicho, R. et al. Quantitative metabolomic profiling of serum, plasma, and urine by 1H NMR spectroscopy discriminates between patients with inflammatory bowel disease and healthy individuals. J. Proteome Res. 11, 3344–3357 (2012).
Dawiskiba, T. et al. Serum and urine metabolomic fngerprinting in diagnostics of inflammatory bowel diseases. World J. Gastroenterol. 20, 163–174 (2014).
Moschen, A. R., Tilg, H. & Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 16, 185–196 (2019).
Rueedi, R. et al. Metabomatching: Using genetic association to identify metabolites in proton NMR spectroscopy. PLOS Comput. Biol. 13, e1005839 (2017).
Ashton, J. J. et al. Analysis and hierarchical clustering of blood results before diagnosis in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 26, 469–475 (2020).
Ashton, J. J. et al. Genetic sequencing of pediatric patients identifies mutations in monogenic inflammatory bowel disease genes that translate to distinct clinical phenotypes. Clin. Transl. Gastroenterol. 11 (2020). Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7145023/. Accessed 9 May 2021.
Ashton, J. J. et al. TTC7A variants previously described to cause enteropathy are observed on a single haplotype and appear non-pathogenic in pediatric inflammatory bowel disease patients. J. Clin. Immunol. 40, 245–247 (2020).
Mossotto, E. et al. GenePy—A score for estimating gene pathogenicity in individuals using next-generation sequencing data. BMC Bioinform. 20, 254 (2019).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Itan, Y. et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. U. S. A. 112, 13615–13620 (2015).
Cloarec, O. et al. Statistical total correlation spectroscopy: An exploratory approach for latent biomarker identification from metabolic 1H NMR data sets. Anal. Chem. 77, 1282–1289 (2005).
Health C for D and R. Review criteria for assessment of C reactive protein (CRP), high sensitivity C-reactive protein (hsCRP) and cardiac C-reactive protein (cCRP) assays—Guidance for industry and FDA staff. U.S. Food and Drug Administration (2019). Available at https://www.fda.gov/regulatory-information/search-fda-guidance-documents/review-criteria-assessment-c-reactive-protein-crp-high-sensitivity-c-reactive-protein-hscrp-and. Accessed 20 April 2021.
World Health Organization. C-reactive protein concentrations as a marker of inflammation or infection for interpreting biomarkers of micronutrient status. Vitamin and Mineral Nutrition Information System. Available at WHO/NMH/NHD/EPG/14.7; http://apps.who.int/iris/bitstream/10665/133708/1/WHO_NMH_NHD_EPG_14.7_eng.pdf?ua=1 (2014).
Raudvere, U. et al. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019).
Toussirot, E. The IL23/Th17 pathway as a therapeutic target in chronic inflammatory diseases. Inflamm. Allergy Drug Targets Discontin. 11, 159–168 (2012).
Benson, J. M. et al. Discovery and mechanism of ustekinumab: A human monoclonal antibody targeting interleukin-12 and interleukin-23 for treatment of immune-mediated disorders. mAbs 3, 535–545 (2011).
Connelly, M. A. et al. Inflammatory glycoproteins in cardiometabolic disorders, autoimmune diseases and cancer. Clin. Chim. Acta 459, 177–186 (2016).
Bell, J. D. et al. Assignment of resonances for ‘acute-phase’ glycoproteins in high resolution proton NMR spectra of human blood plasma. FEBS Lett. 215, 311–315 (1987).
Otvos, J. D. et al. GlycA: A composite nuclear magnetic resonance biomarker of systemic inflammation. Clin. Chem. 61, 714–723 (2015).
Fuertes-Martín, R. et al. Characterization of 1H NMR plasma glycoproteins as a new strategy to identify inflammatory patterns in rheumatoid arthritis. J. Proteome Res. 17, 3730–3739 (2018).
Connelly, M. A. et al. Differences in GlycA and lipoprotein particle parameters may help distinguish acute kawasaki disease from other febrile illnesses in children. BMC Pediatr. 16, 151 (2016).
Ballout, R. A. & Remaley, A. T. GlycA: A new biomarker for systemic inflammation and cardiovascular disease (CVD) risk assessment. J. Lab. Precis. Med. 5 (2020). Available at https://jlpm.amegroups.com/article/view/5567. Accessed 6 July 2022.
Macy, E. M., Hayes, T. E. & Tracy, R. P. Variability in the measurement of C-reactive protein in healthy subjects: Implications for reference intervals and epidemiological applications. Clin. Chem. 43, 52–58 (1997).
Duprez, D. A. et al. Comparison of the predictive value of GlycA and other biomarkers of inflammation for total death, incident cardiovascular events, noncardiovascular and noncancer inflammatory-related events, and total cancer events. Clin. Chem. 62, 1020–1031 (2016).
Gabay, C. & Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454 (1999).
Batch, B. C. et al. Branched chain amino acids are novel biomarkers for discrimination of metabolic wellness. Metabolism 62, 961–969 (2013).
Libert, D. M., Nowacki, A. S. & Natowicz, M. R. Metabolomic analysis of obesity, metabolic syndrome, and type 2 diabetes: Amino acid and acylcarnitine levels change along a spectrum of metabolic wellness. PeerJ 6, e5410 (2018).
Le Roy, C. I. et al. Dissecting the role of the gut microbiota and diet on visceral fat mass accumulation. Sci. Rep. 9, 9758 (2019).
Ashton, J. J. et al. Deleterious genetic variation across the NOD signaling pathway is associated with reduced NFKB signaling transcription and upregulation of alternative inflammatory transcripts in pediatric inflammatory bowel disease. Inflamm. Bowel Dis. 28, 912–922 (2022).
Coelho, T. et al. Immunological profiling of paediatric inflammatory bowel disease using unsupervised machine learning. J. Pediatr. Gastroenterol. Nutr. 70, 833–840 (2020).
Das, J. et al. A critical role for NF-κB in Gata3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol. 2, 45–50 (2001).
Atreya, I., Atreya, R. & Neurath, M. F. NF-κB in inflammatory bowel disease. J. Intern. Med. 263, 591–596 (2008).
Liu, T. et al. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2, 1–9 (2017).
Khader, S. A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Ueno, A. et al. Th17 plasticity and its changes associated with inflammatory bowel disease. World J. Gastroenterol. 21, 12283–12295 (2015).
Glas, J. et al. Analysis of IL12B gene variants in inflammatory bowel disease. PLoS ONE 7, e34349 (2012).
Sandborn, W. J. et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N. Engl. J. Med. 367, 1519–1528 (2012).
Lee, H. W. et al. The correlation of serum IL-12B expression with disease activity in patients with inflammatory bowel disease. Medicine (Baltimore) 95, e3772 (2016).
Eftychi, C. et al. Temporally distinct functions of the cytokines IL-12 and IL-23 drive chronic colon inflammation in response to intestinal barrier impairment. Immunity 51, 367–380.e4 (2019).
Ritchie, S. C. et al. The biomarker GlycA is associated with chronic inflammation and predicts long-term risk of severe infection. Cell Syst. 1(4), 293–301. https://doi.org/10.1016/j.cels.2015.09.007 (2015).
Dierckx, T. et al. GlycA, a nuclear magnetic resonance spectroscopy measure for protein glycosylation, is a viable biomarker for disease activity in IBD. J. Crohns Colitis 13(3), 389–394. https://doi.org/10.1093/ecco-jcc/jjy162 (2019).
Acknowledgements
The authors would like to acknowledge: all patients and their families; Prof Niranjan for insightful comments; Nikki Graham for technical support; IRIDIS HPC Facility, University of Southampton; Chemical Analysis Facility, University of Reading.
Funding
This work was supported by European Society for Paediatric Research, Guts UK and by the National Institute for Health Research (NIHR) Southampton Biomedical Research Centre. JJA is funded by an ESPR post-doctoral grant and by an NIHR clinical lectureship.
Author information
Authors and Affiliations
Contributions
S.E. and S.P.C. conceived and designed the study. S.E. and R.M.B. led the recruitment to the study. E.M. and J.B. managed data, performed bioinformatics analyses and wrote the manuscript. J.B. processed metabolomics data. E.M. processed genomics data. J.J.A. and T.F.C. supported the recruitment. J.J.A., I.S.S., G.C., J.Baker, F.B. and H.T.T.P. helped collecting the data. All authors contributed to revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mossotto, E., Boberska, J., Ashton, J.J. et al. Evidence of a genetically driven metabolomic signature in actively inflamed Crohn’s disease. Sci Rep 12, 14101 (2022). https://doi.org/10.1038/s41598-022-18178-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-18178-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
