
Abstract
Goat warble fly infestation (GWFI) is a subcutaneous myiasis caused by larvae of Przhevalskiana silenus, an insect belonging to the order Diptera. The diagnosis of GWFI is challenging in the early larval instars (L1 and L2) as they are occult under the skin and hair coat causing prolonged economic loss in form of meat and hide damage. This necessitates early diagnosis for disease control at herd level and its prophylactic management to prevent economic losses. Hypodermins, a class of serine proteases from Hypoderminae subfamily have been used as serodiagnostic antigens for the past four decades for diagnosis of warble fly myiasis. In this study,the immunodominant antigen Hypodermin C (HyC) from P. silenus has been recombinantly expressed in E. coli and immunogenic characterisation of expressed protein was done. The protein shows hallmark residues in conserved cysteine and catalytic triad typical of serine proteases along with similar profile of immunoreactivity towards Hypoderminae infestation. The present study reports an optimised indirect-ELISA based on recombinant HyC derived from P. silenus for early diagnosis of GWFI. The optimised indirect ELISA provides a sensitive and specific immunodiagnostic for mass surveillance of the GWFI with diagnostic specificity and sensitivity of 96% and 100%, respectively and not showing any cross reactivity against other important parasitic and bacterial diseases of goats. This study presents the first report of indirect ELISA based on recombinant Hypodermin C antigen derived from P. silenus for the serosurveillance of goat warble fly disease.
Similar content being viewed by others

Introduction
Goat warble fly infestation (GWFI) is a subcutaneous myiasis caused by larvae of P. silenus, an oestrid fly belonging to the order Diptera, family Oestridae, subfamily Hypoderminae1. The disease is characterised by the presence of subcutaneous warbles on the dorsum and lumbar region of domestic and wild ruminants. GWFI occurs in many countries with considerable difference throughout the world due to geographical and meteorological variation and grazing pattern2,3,4. The disease is most prevalent in Mediterranean and Indian subcontinent in subtropical climatic zones ranging from northern India, Pakistan, Iran, Turkey and Southern Italy5,6,7,8,9. In India, the disease is prevalent in the northwestern Himalayan region, especially, in Bakerwali breed goats. The larvae infest the skin at lumbar and flank regions of goats causing sustained economic loss in form of reduced weight gain, hide value depreciation and degradation of meat quality by carcass trimming loss upon slaughter7,10,11. The GWFI is peculiar in the infestation as all the stages of larvae infest subcutaneous region of dorsum similar to H. acateon unlike other members of subfamily Hypoderminae (Hypoderma bovis and H. lineatum) which migrate to internal locations in the host12,13.
The life cycle of goat warble fly permits infestation of larvae for about 7–9 months within the host. The larval stages of P. silenus range in size from 2 to 7.9 mm at L1 stage, 8–9.9 mm at L2 stage and 10–18 mm L3 stage which all inhabit the subcutaneous region of dorsum until falling off the host for pupation in ground soil7,12.
The host infestation by first instar larvae remains occult from physical detection or visual observation by the farmer due to small size of L1 and subcutaneous presence without the formation of palpable warbles, externally on the dorsum. The infestation of advanced larval stages causes irreversible loss to hide and meat through L2 and L3 instars7,10,12.
The diagnosis of GWFI is mainly based on physicoclinical observation by palpation of warbles on the dorsum which are evident only after second and third larval instars have developed which occurs about 5–9 months after infestation. This has promoted the development of serological assays for the early diagnosis of hypoderminae infestation. Hypoderminae insects are known to possess three main serine proteases viz. Hypodermin A (HyA), Hypodermin B (HyB) and Hypodermin C (HyC). Hypoderminae origin HyC is a member of collagenolytic enzymes related to the trypsin family. The HyC is primarily secreted by L1 larvae to degrade the collagen at physiological conditions while entering the host tissue14,15,16, whereas HyA and HyB serve as immunomodulators to suppress host immune response and promote larval survival in the host17,18. The molecule of HyC has been characterised as a major immunodominant antigen and suitable candidate for detecting Hypoderma specific antibodies from cattle and other ruminants19,20,21.
The available serological tests have been based on Hypoderma spp antigens, primarily, from H. lineatum and H. bovis22,23,24,25,26,27,28.
The native HyC from H. lineatum and H. bovis has been widely used as antigen for serodiagnosis of hypodermosis in cattle population in several countries29,30,31. The recombinant hypodermin C (rHyC) from H. lineatum and H. bovis have been produced in heterologous expression system and used as a diagnostic antigen for detection of antibody from cattle sera26,27,28. The detailed molecular analyses of HyC has been performed to gain the information on sequence and biochemical features of HyC derived from H. lineatum15,32.
The comparative utility of rHyC antigen has been assessed against using native antigen preparation from H. lineatum and has been shown as effective alternative to the native antigen or crude preparations derived from the larval lysates in diagnostic application33. In a separate study, the comparative utility of purified HyC antigen has been shown over the use of crude lysate extract for detecting anti-Hypoderma antibodies in cervids34.
The cross reactivity of HyC from H. lineatum and H. bovis origin has been established and it is utilized for the diagnosis of other related H. sinense, H. actaeon, H. tarandi and P. silenus21,35,36,37,38. However, in India due to religious issues collection of Hypoderma species larvae from cattle is not possible and thus serodiagnosis of Hypoderminae cannot be achieved using native antigen and have to depend on costly commercial diagnostic kits. So, exploration of an antigen obtained from other Hypoderminae species different than H. lineatum and H. bovis is required. Moreover, at global level till date P. silenus antigen has not been explored for serodiagnosis of goat warble fly and other hypoderminae infestation in animals. Thus the present communication records the first attempt of recombinant HyC of P. silenus origin for serodiagnosis of GWFI.
Materials and methods
Parasite, cells and serum samples
First stage larvae were collected from the subcutaneous tissues of infected goats at the municipal abattoir of Jammu (India), washed with PBS, and identified as per keys of Zumpt1 and stored at − 80 °C for RNA isolation. E. coli BL21 (DE3)pLysS cells, pET32a(+) vector(Novagen) were used in expression study. Physicoclinical observation was kept as basis for determining reference serum. For sera, blood samples were collected from field and slaughterhouse and marked as positive and negative based on larval detection by palpation in live animals and by carcass examination.Serum samples from animals testing positive for coenurosis, paratuberculosis, enterotoxaemia, oestrosis, coccidiosis, cryptosporidiosis, haemonchosis, monieziosis, fasciolosis, amphistomosis and brucellosis were used in this study.
Construction of expression cassette in pET32a(+) vector
Total RNA was isolated from L1 stage larvae of P. silenus using RNeasy mini kit (Qiagen, Germany) as per manufacturer protocol. The cDNA was synthesized from total RNA using oligodT primer following the protocol described in cDNA synthesis kit (Revert Aid First strand cDNA synthesis kit, Thermo Scientific). The HyC gene of P. silenus was amplified using HyC gene of cattle warble fly (H. lineatum) specific expression primers based on the published sequences of HyC (EMBL Accession Number X74306) (AAGGATCCATAATCAATGGATACGAAG and ATCTCGAGTTAAAATATTATACCAGTATTTTG). The gene was amplified using a 25 µL reaction mixture containing 2 µL of cDNA, 2.5 µL of 10X Ex Taq buffer (TaKaRa,USA), 1 µL each of forward and reverse primers (10 pmol/µL), 1 µL of 10 mM dNTP mix, 0.5 µL of Ex Taq DNA Polymerase (TaKaRa, USA), and 17 µL nuclease free water. PCR reaction was carried out at initial denaturation of 94 °C for 5 min followed by 35 cycles of denaturation at 94 °C for 1 min, annealing at 56 °C for 1 min, extension at 72 °C for 1 min and a final extension at 72 °C for 10 min.
The amplified PCR product was analysed by 1% agarose gel electrophoresis. The amplified product was purified using Wizard™ SV Gel and PCR Clean-Up System (Promega, USA), directionally cloned into pET32a(+) expression vector at BamHI and XhoI restriction enzyme sites and transformed into TOP10 E. coli cells. The recombinant clone was selected on LB agar plate containing Ampicillin (100 μg/mL). The recombinant plasmid containing gene of interest was confirmed by colony PCR using gene-specific primers and restriction enzyme digestion. The recombinant plasmid was isolated using Plasmid extraction kit and sent for nucleotide sequencing to Agrigenome Pvt. Ltd (Kerala). The sequence received was primarily analysed using BioEdit software and submitted to Gen Bank. Further, the protein encoding nucleotide sequence was translated in silico using the Edit Sequence program of DNA Star (Laser gene Suite 6.0) and BLASTp (protein–protein BLAST) was performed. The sequence generated here was compared to the reference bovine sequences available in the public domain and aligned using CLC Genomics Workbench (Qiagen)39.
The deduced protein sequence of HyC originated from P. silenus was aligned with HyC of other Hypoderminae spp. viz H. lineatum, H. bovis and H. diana with accession numbers X74306, MK473847, EU999953, respectively. The protein sequences were aligned with CLC genomics platform version 20.01 (Qiagen).
Expression and purification of recombinant protein
The recombinant HyC plasmid was transformed into E. coli BL21(DE3)pLysS host cells and further confirmed by colony PCR. The positive clone was grown in LB broth containing ampicillin and chloramphenicol at 37 °C for overnight. The fresh LB broth was inoculated at 1:100 with overnight culture and incubated at 37 °C at 200 rpm till the culture reached an OD600 0.4–0.6. Henceforth, the expression was induced with 1 mM IPTG and harvested 1 mL culture at hourly interval up to 16 h. The collected fraction was pelleted by centrifugation and level of expression was analysed by SDS-PAGE. The rHyC protein was produced in bulk and the pellet was resuspended into denaturing buffer (8 M Urea) and sonicated at 25% amplitude with pulse of 15 s for 5 min. The lysate was clarified and the supernatant was purified by using Ni–NTA affinity chromatography under denaturing condition (8 M Urea). The Ni–NTA superflow gravity column was equilibriated with denaturing binding buffer containing 8 M urea in phosphate buffer (pH 7.8) followed by binding of clarified lysate with Ni–NTA Superflow® resin (Qiagen). The protein-bound resin was washed in subsequent steps with wash buffer at pH 6.0 and pH 5.3 upon which the bound target protein was eluted with elution buffer (100 mM PB/150 mM NaCl/8 M Urea) at pH 4.0. The eluates were analysed by SDS-PAGE, pooled and dialyzed under decreasing concentrations of urea 6 M, 4 M, 2 M and finally Tris buffer, pH 8.0. The purity of the recombinant protein was analysed by 12% SDS-PAGE gel after staining with Coomassie brilliant blue R-250 stain. The concentration of the recombinant protein was quantified by Bradford protein assay kit (VWR, USA) and stored at − 80 °C.
Western blotting
Purified recombinant protein was resolved under 12% SDS PAGE and blotted on nitrocellulose membrane (0.45 μm, Biorad) by Mini Trans-Blot® electrophoretic transfer cell (Bio-Rad). The membrane was blocked overnight with skimmed milk powder 5% (SMP) at 4 °C. Thereupon, the membrane was washed thrice with PBS-Tween-20 (0.05%). The individual lanes were cut and incubated with goat warble fly infected serum (1:50), negative control sera and Oestrus ovis positive sera (1:50), respectively at 37 °C for 1 h under gentle shaking. Upon triple wash with PBS-T, the membranes were incubated with anti-goat IgG HRP (1:5000 in PBS) for 1 h at 37 °C. The membranes were washed three times and developed with DAB substrate (VWR, USA).
Optimization of rHyC based iELISA
An indirect-ELISA using rHyC has been optimised and evaluated with known status of positive and negative GWFI sera to detect the anti-HyC antibodies. The optimum concentration of coating antigen, dilution of serum and conjugate, standard checkerboard titration was performed. A total of three different concentration of antigen i.e. 0.5 μg/mL, 0.25 μg/mL and 0.125 μg/mL (100 μL/well), three different dilutions of sera at 1:400, 1:800 and 1:1600 and two dilutions (1:5000 and 1:10,000) of anti-goat IgG HRP conjugate were used in checkerboard titration analysis using 5% SMP as blocking buffer. The maximum differentiation between positive and negative sera (P/N) measured at A490was selected for further analysis.
In brief, 96-well microtiter plate (Costar 3590, Corning-USA) were coated with 100 μL of rHyC (0.5 μg/mL) in 0.05 M carbonate bicarbonate buffer (pH 9.6). The plate was incubated at 4 °C overnight. The wells were washed twice with PBS-Tween 20 (0.075%), and then once with PBS, blocked with 5% skimmed milk prepared in PBS (pH 7.4) and incubated at 37 °C for 1 h. After washing, 100 μL of known positive and negative serum sample diluted in PBS (1:400) were added and incubated at 37 °C for 1 h. After washing the plate three times, 100 μL of anti-goat IgG HRP-conjugate (Sigma Aldrich, USA) at 1:10,000 dilution in PBS (pH 7.4) was added to each well and incubated at 37 °C for 1 h. Following washing, 100 μL of O-phenylenediamine dihydrochloride (OPD) substrate (Sigma Aldrich, USA) dissolved in citrate–phosphate buffer (pH 5.0) with 30% H2O2 was added to individual wells. The reaction was allowed to develop for 10 min in dark and reaction was stopped by adding 50 μL of 1 M H2SO4 to each well. Absorbance at 490 nm was measured by using microplate reader (iMarkmicroplate reader, Biorad).
Evaluation of rHyC based iELISA
After optimisation of the iELISA, serum samples with known status (15 positive and 25 negative) were screened in duplicate for determining the cut-off value. The physicoclinical evaluation was taken as a reference to distinguish the positive and negative samples. Negative reference serum samples were received from animals that had never grazed and belongs to Punjab state of North India, where GWFI has never been reported. The cut-off value was evaluated by the Receiver Operating Characteristic (ROC) curve, using MedCalc software, Mariakerke, Belgium40. Based on the checkerboard titration results, ROC curves and areas under the curve were plotted, and the sensitivity and specificity of iELISA were calculated using interactive dot diagrams. Cut-off value was calculated according to the Youden index (Youden = Se + Sp − 1) and used for immunodiagnosis of goat warble fly. In order to rule out the cross reactivity of optimised rHyC based iELISA with other economically important parasitic and bacterial diseases of goats, known sera positive for oestrosis, monieziosis, coccidiosis, fasciolosis, haemonchosis, amphistomiosis, coenurosis, cryptosporidiosis, Johne’s disease, brucellosis and enterotoxaemia were also screened.
Screening of random samples
A total of 421 serum samples collected from municipal slaughterhouses in Jammu and different parts of Union Territory of Jammu and Kashmir namely Jammu, Rajouri, Udhampur, Poonch, Samba,Kathua, Reasi, Kishtawar and Doda were tested by optimized rHyC based iELISA. All the samples were tested in duplicates. The sample showing a difference of more than 0.100 in raw OD values between duplicates were not considered in the present data.
Ethics approval
All the serum samples used in the study were collected by the research team under informed consent of goat farmers under the guidelines of Institutional Animal Ethics Committee [AU/FVSJ/AGB/17-18/415 Dated: 31-03-2018].
Results
Cloning and bioinformatics analysis of rHyc from P. silenus
PCR amplification of the HyC from P. silenus was obtained by using cDNA template produced from L1 larval instars with oligodT primers. The amplified product of expected size 706 bp was confirmed on 1% agarose gel electrophoresis (Fig. 1). The gel purified PCR product was cloned into pET32a( +) vector at BamHI and XhoI restriction enzyme sites and transformed into TOP10 E. coli cells. The transformants were screened on LB agar plates containing ampicillin. The recombinant plasmid was characterised by colony PCR and RE digestion that confirmed the presence of target gene and its orientation into the vector.

PCR amplification of CDS of hypodermin C gene of P. silenus. M1: 100 bp plus DNA ladder, 1: 706 bp PCR amplified product of hypodermin C CDS.
The recombinant plasmid was isolated, sequenced and the gene sequence was submitted to GenBank (Accession number MZ407908). Sequence similarity searches in Blastn revealed that the HyC from P. silenus (HyC_PS) was 86% identical to HyC of H. lineatum and H. bovis. The alignment of HyC_PS with HyC protein reported from H. lineatum (HyC_HL), H. bovis (HyC_HB) and H. diana (HyC_HD) revealed 18.695% variation in the polypeptides, with 43 substitutions out of total 230 amino acids (X74306.1) (Fig. 2). The HyC_PS with 232 residues with a predicted molecular weight of 25.150 kDa contains 22 strongly acidic, 12 strong basic, 79 hydrophobic residues and 78 polar amino acid residues with a predicted pI of 4.569. The molecule contains all the six cysteine residues as shown in sequence logo, at conserved position resulting in three disulphide bonds. The conserved residues of catalytic triad as His-45, Asp-88, Ser-180 are also present in the protein representing the functional feature of serine proteases (Fig. 2).

Protein sequence alignment of HyC derived from P. silenus (HYC_PS) with HyC from H. lineatum (HYC_HL), H. bovis (HYC_HB) and H. diana (HYC_HD).
Secondary structure prediction analysis indicated the trypsin domain structure for HyC with presence of three chymotrypsin peptidase regions in the HyC amino acid sequence. Secondary structure analysis also revealed that HyC_PS consists of 9% alpha-helix, 43% beta-strand and 16% unstructured coil regions, respectively41.
Expression of rHyC and its characterisation
The verified recombinant clone with HyC gene in BL21 cells was used for expression study. The expression of protein was induced through 1 mM IPTG at 37 °C and the samples collected at hourly interval were analysed by SDS-PAGE to check the maximum level of expression for the construct. The different fractions were compared for expression with the uninduced sample showed about 45 kDa of protein overexpressed upon 6 h post induction, maximally. The protein separated in SDS-PAGE was transferred on nitrocellulose membrane (NCM) for blotting and expressed recombinant HyC protein was confirmed by Ni–NTA HRP conjugate.
Purification and immunoreactivity of rHyC
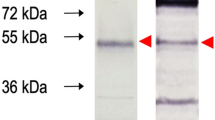
After optimisation, expression was scaled up by increasing the culture volume and purified under denaturing condition using Ni–NTA affinity chromatography. The eluted fractions were analysed by SDS-PAGE that indicated the presence of single band with about 45 kDa size with near homogeneity (Fig. 3). The yield of expressed fusion protein was at 2.8 mg/L of induced culture. The antigenicity of purified recombinant protein was confirmed by immunoblotting using GWFI positive, negative serum and Oestrosis positive sera. Recombinant HyC specificity was confirmed to GWFI serum. A distinct band of 45 kDa was observed with the expressed protein reacting strongly and specifically with the GWFI positive serum. No reactivity was observed with goat negative and oestrosis positive sera (Fig. 4).

Ni–NTA purification of recombinant hypodermin C fusion protein (45 kDa) M: Marker, Lane 1, 2: elutes of wash buffer Lane 3, 4, 5: elutes of purified protein.

Western blotting of rHyC protein M: Marker 1: immunoreactivity with natural warble fly positive goat serum, 2: commercial sterile goat serum, 3: Oestrus ovis positive serum.
Optimisation and evaluation of rHyC iELISA
Checkerboard titration revealed that at antigen concentration of 0.5 μg/mL (100 μL/well), serum dilution of 1:400, conjugate dilution of 1:10,000 with 5% SMP as blocking buffer were found to give maximum difference in the reactivity between positive and negative serum. ROC analyses using sera from 15 infested and 25 non- infested goats revealed an area under curve (AUC) of 0.992 (P < 0.001) for rHyC iELISA. Relative diagnostic sensitivity and specificity calculated by interactive dot analysis of MedCalc software were 100% and 96%, respectively (Fig. 5). A cut off value (OD) of 0.4835 for rHyC iELISA was fixed from the ROC curve and interactive dot diagram (Fig. 6) and used for serological screening of the random samples.

ROC curve analysis representing sensitivity and specificity of recombinant hypodermin C (rHyC) indirect ELISA.

Dot plot for different serum samples and cut-off for the recombinant hypodermin C (rHyC) indirect ELISA by ROC analysis.
The expressed protein showed strong reactivity with GWFI positive serum whereas it did not show any significant reactivity with other economically important parasitic and bacterial diseases of goat (Fig. 7). Further, screening of random serum samples collected from different parts of Union Territory of Jammu and Kashmir, North India were tested with optimised ELISA and the corrected OD values (Mean OD of duplicates − Mean OD of antigen blank) were calculated to differentiate between infested and healthy animals. Out of 421 serum samples screened through optimised iELISA for detecting anti-HyC antibodies, 59 were found to be positive and 362 tested negative for GWFI (Fig. 8).

Cross reactivity of optimized ELISA with most common parasitic and bacterial diseases of goat.

Distribution of ELISA results by sample classification as negative and positive.
Discussion
Goat warble fly disease is the cause of major economic losses to goat farmers with high prevalence in Mediterranean and Indian subcontinent. GWFI has been reported with varying prevalence rates in different countries. In North India, the disease has been reported to have a prevalence rate varying between 13 and 56.5% in Jammu region whereas the prevalence varies in Pakistan (5–75%), Turkey (53–94%), southern Italy (20–90%), Iran (7–19%) and Jordan4,6,7,11,12,43. The diagnosis of GWFI is based on physicoclinical examination of the suspected animal for presence of subcutaneous warble, caused by late stage of L2 and L3 larval instars of P. silenus, on dorsal palpation or carcass examination upon slaughter. In addition to physical observation, serodiagnosis has been used to detect antibodies against larval antigens. Physicoclinical detection of GWFI upon slaughter or field observation is a setback in early detection of infestation at L1 stage of larvae in the first 3 months of infestation by parasite. Goat warble fly, P. silenus causes subcutaneous myiasis with similar characteristics as exhibited by H. lineatum and H. bovis in cattle and other ruminants. P. silenus in family hypodermatinae also exhibit immunoreactivity with HyC antigen from H. lineatum and H. bovis19. Serological detection of infestation is facilitated by an immunoassay detecting the anti-P. silenus antibody in host (goat) at the first month of infestation37, similar to cattle antibody kinetics corresponding to H. lineatum and H. bovis for cattle hypodermosis30,37,43.
Hypodermin A, B and C are serine proteases from Hypoderminae insects which are the major immunogens in case of larval infestations. Hypodermins are reported to show overexpression in L1 larval instar as compared to L2 and L3 stages32. Hypodermin C has been established as the major antigen for serological detection of hypoderminae myiasis and shows cross-reactivity among Hypoderminae members (H. bovis, H. lineatum, H. tarandi, H.actaeon, H. sinense and P. silenus) due to shared epitopes as evident by several studies of cross reactivity and ELISA19,21,35,38,44,45. The HyC derived from P. silenus has shown several substitutions interspersed in the whole sequence. On comparison with other serine proteases reported from H. lineatum, H. bovis and H. diana, the HyC presents all the conserved features attributed to serine protease activity such as conserved cysteine residues and the catalytic triad at the designated sites (His-45, Asp-88, Ser-180)15,28,32. Although, the HyC protein derived from P. silenus show considerable variations in residues (18.695%, 43 substitutions/230 aa), the three dimensional structure shows strong homology in the predicted structure as compared to that of HyC from H. lineatum16.
ELISA has been promoted as surveillance tool in diagnosis of warble fly in various countries19,21,46,47,48,49. It is preferred over palpation or examination of carcass at slaughter49. Serology provides more sensitive detection of exposure to warble fly myiasis as a number of animals might resolve larvae over a period and can only present serological detection49.Thus, ELISA provides a tool of warble fly infestation study in occult stages as well as in case of early resolution of the larvae before warble development on the dorsum in case of GWFI. The antibody kinetics against H. lineatum, H. bovis, P. silenus, and H. tarandi in their hosts have revealed that it takes about one month to reach antibody peaks in the host sera, which implicates the utility of ELISA as diagnostic tool at L1 stage of infestation20,29,30,37.
Several in-house ELISA assays are used in different countries which are primarily based on native antigen derived from H. lineatum and H. bovis larvae. Native antigen and purified antigen preparations provide challenge in standardisation due to purification and requirement of larvae repeatedly. Recombinant protein may serve as antigen for better reproducibility and standardisation for a diagnostic test. Also, since the parasite availability for native antigen is decreasing and fluctuating upon progressive control measures in many countries, recombinant proteins serve as constant source of antigen for diagnostic evaluation of the population33. Recent study has provided rHyC based indirect ELISA derived from H. bovis for the diagnosis of hypodermosis in cattle28 whereas another study developed in-house ELISA protocol for the detection of GWFI9 which are based on the use of a larval crude lysate as antigen. A summary of selected ELISA for Hypoderminae myiasis detection is provided in Table 1. In natural infestations, larval development is not synchronous as individual hosts may have multiple oviposition events and this apparently causes episodic release of HyC limiting the utility of antigen-capture ELISA31,50,51. On the other hand, in case of competitive ELISA, sample dilution is permitting lower coverage of the herd surveillance in case of pooled sampling36.
The present study provides first molecular characterisation of HyC protein, the major antigen for serodiagnosis of warble fly myiasis, in case of goats, caused by P. silenus. The study characterised the HyC of P. silenus by cloning and heterologous expression in E. coli and assessed its immunoreactivity with GWFI positive sera along with other economically important goat diseases.
The HyC derived from P. silenus was amplified with gene specific primers and cloned into pET 32a(+) vector. The expected size of target protein is about 25 kDa whereas the complete construct used in this study presents around 45 kDa product as a fusion protein with thioredoxin tag and the same size was obtained as expected in SDS-PAGE analysis upon induction. The optimum level of expression was achieved at 6 h post-induction after which there was no improvement in the expression level. Purification was done under denaturing conditions with 8 M Urea in PBS with elution based on pH gradient in acidic condition. At a yield of 2.8 mg/L of induced culture, the recombinant fusion protein may be sufficient to screen approx. 2,800 serum samples in duplicate with the optimised protocol. The purified HyC protein reacted strongly with GWFI positive serum in ELISA format at antigen concentration of 0.5 μg/mL (100 μL/well) at 1:400 dilution of serum identified by checkerboard titration. The present ELISA showed no cross reaction with any other major parasitic and bacterial diseases of goats. The rHyC of P. silenus as an antigen in the ELISA provided a diagnostic sensitivity and specificity of 100% and 96% respectively, for serodiagnosis of goat warble fly disease The other iELISA established with rHyC derived from H. lineatum exhibited sensitivity of 85% and specificity of 98.2% whereas iELISA based on rHyC from H. bovis has shown sensitivity of 90% and specificity of 100%25,27. Also, the study comparing utility of native HyC (nHyC) and rHyC as antigen in ELISA for detection of anti-Hypoderma antibodies has shown a sensitivity of 95.8% and specificity of 95.7% for rHyC and 98.2% sensitivity as well as specificity for nHyC based iELISA32. On the other hand, competitive ELISA for detection of hypodermosis showed a sensitivity and specificity of 92.5% and 98.5%, respectively36.
The iELISA based on rHyC is optimised with a high sample dilution rate at 1:400 for GWFI serum samples. This provides an opportunity for a standardised test as a tool for mass surveillance of GWFI at national eradication program. Also, the high standard dilution rate permits scope of using pooled serum samples for the assessment of herd screening for GWFI52,53. It has also been used for the screening of random serum samples from different parts of Union Territory of Jammu and Kashmir, North India. The optimised assay is highly specific and sensitive for detection of antibodies against goat warble fly disease.
ELISA test has been adopted for mass epidemiology of hypodermosis as a tool of monitoring at national level eradication programs in countries like France, the UK, Belgium and Germany37. ELISA has proven an effective tool to establish antibody kinetics in both bovine hypodermosis and GWFI. This has further assisted the monitoring initiatives to facilitate appropriate timing of sample collection according to particular geographical and agroclimatic zones8,9,19,54. Durable control of GWFI requires repetitive monitoring for the detection of any reappearance of the disease in any geographical region. Serological monitoring is vital tool for aid in policy implementation of control and eradication programmes through the use of systemic insecticides52,53. As it is evident by immunological studies that Hypoderminae species do not provide complete protective immunity, it is plausible that constant monitoring and surveillance along with prophylactic treatment remains the practical solution for durable control of goat warble fly as has been observed in cattle hypodermosis49,51.
Data availability
All data generated or analysed during this study are included in this published article (and its supplementary information files). The sequences used for comparison are available under the accession numbers: X74306; MK473847; EU999953; MZ407908 from NCBI database at https://www.ncbi.nlm.nih.gov/.
References
Zumpt, F. Morphology, biology and pathogenesis of myiasis-producing flies in systematic order. In Myiasis in Man and Animals of the Old World: A Textbook for Physicians, Veterinarians and Zoologists (ed. Zumpt, F.) 205–214 (Butterworths, 1965).
Scholl, P. J. Biology and control of cattle grubs. Annu. Rev. Entomol. 38, 53–70 (1993).
Giangaspero, A. & Lia, R. Mapping goat warble fly distribution and related problems. Parassitologia 39, 423–426 (1997).
Oryan, A., Razavi, S. M. & Bahrami, S. Occurrence and biology of goat warble fly infestation by Przhevalskiana silenus (Diptera, Oestridae) in Iran. Vet. Parasitol. 166, 178–181 (2009).
Sayin, F. Incidence and seasonal activity of Przhevalskiana silenus (Brauer) in angora goats in Turkey. III Symposium on Medical and Veterinary Acaroentomology, Gdansk 25–28 September 1975. Wiadomości Parazytol. 23, 1–3 (1977).
Faliero, S. M. et al. Goat warble fly infestation by Przhevalskiana silenus (Diptera: Oestridae): immunoepidemiologic survey in the Basilicata region (southern Italy). Parassitologia 43, 131–134 (2001).
Yadav, A., Katoch, R., Khajuria, J. K., Katoch, M. & Agrawal, R. Prevalence and biology of goat warble fly infestation by Przhevalskiana silenus in Jammu province, India. Trop. Anim. Health Prod. 43, 1487–1492 (2011).
Liaquat, S. et al. Seroepizootiological investigation on goat warble fly infestation (Przhevalskiana silenus) in Pothwar Plateau, Pakistan. Trop. Biomed. 38, 1–8 (2021).
Bagheri, A., Madani, R., Navidpour, S. & Hoghoooghi-Rad, N. Prevalence and early detection of hypodermosis in goats using a competitive ELISA system in Lorestan Province. Arch. Razi Inst. 76, 69–77 (2021).
Yadav, A., Katoch, R., Khajuria, J. K., Katoch, M. & Rastogi, A. Economic impact of Przhevalskiana silenus infestation in native goats of Northern India. Trop. Anim. Health Prod. 44, 581–587 (2012).
Yadav, A., Panadero, R., Katoch, R., Godara, R. & Cabanelas, E. Myiasis of domestic and wild ruminants caused by Hypodermatinae in the Mediterranean and Indian subcontinent. Vet. Parasitol. 243, 208–218 (2017).
Otranto, D. & Puccini, V. Further evidence on the internal life cycle of Przhevalskiana silenus (Diptera, Oestridae). Vet. Parasitol. 88, 321–328 (2000).
Panadero, R. et al. Hypoderma actaeon: An emerging myiasis in roe deer (Capreolus capreolus): Hypoderma actaeon in roe deer. Med. Vet. Entomol. 31, 94–96 (2017).
Boulard, C. & Garrone, R. Characterization of a collagenolytic enzyme from larvae of Hypoderma lineatum (Insecta: Diptera, Oestridae). Comp. Biochem. Physiol. 59B, 251–255 (1978).
Lecroisey, A., Gilles, A. M., De Wolf, A. & Keil, B. Complete amino acid sequence of the collagenase from the insect Hypoderma lineatum. J. Biol. Chem. 262, 7546–7551 (1987).
Broutin, I. et al. 1.8 Å Structure of Hypoderma lineatum collagenase: A member of the serine proteinase family. Acta Crystallogr. D Biol. Crystallogr. 52, 380–392 (1996).
Chabaudie, N. & Boulard, C. Effect of hypodermin A, an enzyme secreted by Hypoderma lineatum (insect Oestridae), on the bovine immune system. Vet. Immunol. Immunopathol. 31, 167–177 (1992).
Panadero, R. et al. Immunomodulatory effect of Hypoderma lineatum antigens: In vitro effect on bovine lymphocyte proliferation and cytokine production. Parasite Immunol. 31, 72–77 (2009).
Otranto, D. et al. Serodiagnosis of goat warble fly infestation by Przhevalskiana silenus with a commercial ELISA kit. Vet. Rec. 144, 726–729 (1999).
Asbakk, K., Oksanen, A., Nieminen, M., Haugerud, R. E. & Nilssen, A. C. Dynamics of antibodies against hypodermin C in reindeer infested with the reindeer warble fly, Hypoderma tarandi. Vet. Parasitol. 129, 323–332 (2005).
Guan, G. et al. Sero-epidemiological surveillance of hypodermosis in yaks and cattle in north China by ELISA. Vet. Parasitol. 129, 133–137 (2005).
Boulard, C. & Petithory, J. Serological diagnosis of human hypodermosis: A preliminary report. Vet. Parasitol. 3, 259–263 (1977).
Sinclair, I. J. & Wassall, D. A. Enzyme-linked immunosorbent assay for the detection of antibodies to Hypoderma bovis in cattle. Res. Vet. Sci. 34, 251–252 (1983).
Colwell, D. D. & Baron, R. W. Early detection of cattle grub (Hypoderma lineatum and H. bovis) (Diptera, Oestridae) using ELISA. Med. Vet. Entomol. 4, 35–42 (1990).
Martínez-Moreno, F. J., Wassall, D. A., Becerra-Martell, C. & Hernández-Rodríguez, S. Comparison of the use of secretory and somatic antigens in an ELISA for the serodiagnosis of hypodermosis. Vet. Parasitol. 52, 321–329 (1994).
Casais, R., Martín Alonso, J. M., Boga, J. A. & Parra, F. Hypoderma lineatum: Expression of enzymatically active hypodermin C in Escherichia coli and its use for the immunodiagnosis of hypodermosis. Exp. Parasitol. 90, 14–19 (1998).
Boldbaatar, D. et al. Detection of antibodies to Hypoderma lineatum in cattle by Western blotting with recombinant hypodermin C antigen. Vet. Parasitol. 99, 147–154 (2001).
Atelge, M., Inci, A., Yildirim, A., Sozdutmaz, I. & Adler, P. H. First molecular characterization of hypodermin genes of Hypoderma bovis and serodiagnosis of bovine hypodermosis with recombinant hypodermin C antigen and a synthetic peptide containing its linear B-cell epitope. Vet. Parasitol. 292, 109394 (2021).
Panadero, R., López, C., Mezo, M., Morrondo, P. & Díez-Baños, P. Effect of early treatment with ivermectin and doramectin on the dynamics of antibody response in cattle naturally infested by Hypoderma lineatum and H. bovis. Vet. Parasitol. 73, 325–334 (1997).
Panadero-Fontán, R. et al. Detection of circulating hypodermin C: An antigen capture ELISA for diagnosis of cattle grub (Diptera: Oestridae) infestations. Vet. Parasitol. 108, 85–94 (2002).
Panadero, R. et al. Evaluation of an antigen capture ELISA for the early diagnosis of Hypoderma lineatum in cattle under field conditions. Vet. Parasitol. 147, 297–302 (2007).
Moiré, N., Bigot, Y., Periquet, G. & Boulard, C. Sequencing and gene expression of hypodermins A, B, C in larval stages of Hypoderma lineatum. Mol. Biochem. Parasitol. 66, 233–240 (1994).
Panadero, R. et al. Assessment of a recombinant antigen versus natural hypodermin C for the serodiagnosis of hypodermosis in cattle. Parasitol. Res. 86, 67–68 (2000).
Panadero, R. et al. Use of a crude extract or purified antigen from first-instar cattle grubs, Hypoderma lineatum, for the detection of anti-Hypoderma antibodies in free-ranging cervids from southern Spain. Med. Vet. Entomol. 24, 418–424 (2010).
Boulard, C., Villejoubert, C. & Moiré, N. Cross-reactive, stage-specific antigens in the Oestridae family. Vet. Res. 27, 535–544 (1996).
Webster, K. A., Giles, M. & Dawson, C. A competitive ELISA for the serodiagnosis of hypodermosis. Vet. Parasitol. 68, 155–164 (1997).
Otranto, D., Giangaspero, A., Caringella, M. P. & Puccini, V. Hypoderma lineatum antigen and anti-Przhevalskiana silenus antibodies: Cross-reactivity and antibody kinetics in naturally infested goats. Parassitologia 40, 325–331 (1998).
Domínguez, J., Panadero, R. & de la Fuente-López, C. Detection of Hypoderma actaeon infestation in Cervus elaphus with ELISA and Western Blotting. J. Wildl. Dis. 46, 934–938 (2010).
QIAGEN CLC Genomics Workbench 20.0 (https://digitalinsights.qiagen.com/).
Garber, C. MedCalc software for statistics in medicine. Clin. Chem. 44, 1370–1370 (1998).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Abo-Shehada, M. N., Batainah, T., Abuharfeil, N. & Torgerson, P. R. Przhevalskiana silenus myiasis among slaughter goats in northern Jordan. Vet. Parasitol. 137, 345–350 (2006).
Caringella, M. P., Giangaspero, A. & Puccini, V. Individual kinetic development of anti-Hypoderma spp. antibodies in naturally infested cattle. Parassitologia 36, 29 (1994).
Pruett, J. H., Scholl, P. J. & Temeyer, K. B. Shared epitopes between the soluble proteins of Hypoderma lineatum and Hypoderma bovis first instars. J. Parasitol. 76, 881–888 (1990).
Monfray, K. & Boulard, C. Preliminary evaluation of four immunological tests for the early diagnosis of Hypoderma tarandi causing hypodermosis in reindeer. Med. Vet. Entomol. 4, 297–302 (1990).
Frangipane di Regalbono, A., Capelli, G., Otranto, D. & Pietrobelli, M. Assessment of cattle grub (Hypoderma spp.) prevalence in northeastern Italy: An immunoepidemiological survey on bulk milk samples using ELISA. Vet. Parasitol. 111, 343–350 (2003).
Simsek, S., Utuk, A. E., Koroglu, E. & Dumanli, N. Seroprevalence of hypodermosis in cattle in some provinces of Turkey. Res. Vet. Sci. 84, 246–249 (2008).
Rehbein, S., Holste, J. E., Smith, L. L. & Lloyd, J. L. The efficacy of eprinomectin extended-release injection against Hypoderma spp. (Diptera: Oestridae) in cattle. Vet. Parasitol. 192, 353–358 (2013).
Colwell, D. D. Out of sight but not gone: Sero-surveillance for cattle grubs, Hypoderma spp., in western Canada between 2008 and 2010. Vet. Parasitol. 197, 297–303 (2013).
Colwell, D. D., López, C., Diez-Baños, P., Morrondo, P. & Panadero, R. Impact of previous infestation on dynamics of circulating hypodermin C in cattle artificially infested with Hypoderma lineatum (Diptera: Oestridae). Vet. Parasitol. 154, 114–121 (2008).
Colwell, D. D. Hidden antigens from third instar Hypoderma lineatum: Impact of immunization on larval survival in artificial infestations. Vet. Parasitol. 175, 313–319 (2011).
Boulard, C. Durably controlling bovine hypodermosis. Vet. Res. 33, 455–464 (2002).
Boulard, C. et al. A successful, sustainable and low cost control-programme for bovine hypodermosis in France. Vet. Parasitol. 158, 1–10 (2008).
Reina, D. et al. Experimental bovine hypodermosis in Spain. J. Med. Entomol. 37, 210–215 (2000).
Acknowledgements
The authors are grateful to the Indian Council of Agricultural Research (ICAR), New Delhi, Government of India, for providing ICAR-National Fellow Scheme to A.Y. The authors are thankful to Sher-e-Kashmir University of Agricultural Sciences and Technology of Jammu (SKUAST-J) for providing the facilities. The authors also acknowledge the ICAR-Central Institute for Research on Goats, Mathura (India) for providing selected serum samples of goats. The authors are grateful to the farmers and Veterinary Officers of Union Territory of Jammu and Kashmir, India.
Funding
The funding for the research was provided by ICAR (New Delhi), Government of India, under ICAR-National Fellow Scheme [No: Agri. Edn./27/06/NP/2017-HRD; Dated: 01/05/2017].
Author information
Authors and Affiliations
Contributions
A.Y. conceptualised the project and obtained funds from the funding agency. A.Y., S.I.R., V.Y., A.K. performed the experiments, collected samples, analysed the data and prepared the manuscript. R.G., S.S. collected samples. A.Y., M.A.B., R.K. designed the experiments R.P. provided critical analysis and edited the final manuscript. S.I.R. and V.Y. contributed equally. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
A.Y. has received ICAR-National Fellow grant from ICAR. A.Y., S.I.R., V.Y., R.G., S.S.,R.K. are included in patent filed for the developed ELISA under patent application [No: 202111024154; Dated: 31/05/2021] at Controller General of Patents, Design, The Patent Office, Government of India, New Delhi. A.K., M.A.B. and R.P. declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yadav, A., Rafiqi, S.I., Yadav, V. et al. First report of Przhevalskiana silenus derived recombinant hypodermin C based indirect ELISA for serodiagnosis of goat warble fly myiasis. Sci Rep 12, 13440 (2022). https://doi.org/10.1038/s41598-022-17760-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17760-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
