
Abstract
Carcinoma ex pleomorphic adenoma (CXPA) is a rare malignancy that transforms from PA. Early detection of the carcinoma by biopsy is difficult due to similar histopathology of the malignant and benign components. To address this, we investigated and compared the characteristic miRNA expression patterns across samples of the PA, carcinomatous portions (CA) of CXPA, as well as conventional PA. We selected 13 CXPA and 16 conventional PA FFPE samples, separated the PA and CA portions of CXPA samples and conducted miRNA profiling for each group. Among 13 transcripts that were differentially expressed between PA and CA of CXPA, eight miRNAs were up-regulated and five down-regulated in CA. Bioinformatic analysis revealed that the up-regulated miRNAs were related to cancer progression and down-regulated ones to tumor suppression. Additionally, seven miRNAs were significantly up-regulated in PA of CXPA compared to conventional PA, although they are histopathologically similar. Almost all of these transcripts interacted with TP53, a well-known tumor suppressor. In conclusion, we identified differentially expressed miRNAs in PA and CA of CXPA, which were closely associated with TP53 and various cancer-related pathways. We also identified differentially expressed miRNAs in the PA of CXPA and conventional PA which may serve as potential biomarkers.
Similar content being viewed by others

Introduction
Carcinoma ex pleomorphic adenoma (CXPA) is a rare malignant neoplasm of the major salivary glands, accounting for about 3–15% of all salivary gland malignancies1,2. In addition, CXPA has been shown to exhibit a rising incidence rate making it a growing concern for public health policy3. CXPA is the result of malignant transformation of the most common benign salivary gland tumor—pleomorphic adenoma (PA)—into various malignant histologies including salivary duct carcinoma (SDC), adenoid cystic carcinoma (AdCC), mucoepidermoid carcinoma (MEC), and myoepithelial carcinoma, amongst others4. This transformation generally occurs after the PA has been present for many years, usually decades. The prognosis of salivary malignancies evolving from PA tends to be worse than that of de novo salivary gland malignancies, with a reported 5-years overall survival-rate, as little as 25%5. Therefore, the diagnosis of the carcinomatous portion of CXPA is critical in determining the appropriate treatment modality and timing. However, accurate diagnosis of CXPA via biopsy is not easy because it is often difficult to accurately target the carcinomatous portion (CA) of these tumors and this diagnosis is usually possible only when the specimen contains clear adenoma and carcinomatous portions, which tends to be towards the later stages of progression.
Recent molecular analysis of CXPA has shed new light on the mechanisms of carcinogenesis identifying CXPA specific mutations in TP53, PLAG1, and HMGA25. However, for the most part, the landscape of mutational and copy number alterations in CXPA seems to be similar to non-malignant PAs, leaving the molecular processes involved in the transition from PA to CXPA largely unknown6.
MicroRNAs (miRNAs) are short non-coding RNAs, involved in the post-transcriptional regulation of gene expression7. Dysregulation of miRNA expression has been extensively reported in various types of tumors, including lung, breast, colon, stomach, and thyroid cancers8, where it is believed that they play a role in the oncogenic progression or suppression of these cancers. Several studies of salivary gland tumors (SGT) have tried, with varying degrees of success, to identify miRNA signatures for the differential diagnosis of benign and malignant SGTs in an effort to improve preoperative diagnosis using tissue or saliva samples9.
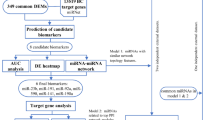
Here we hypothesized that the presence of the miRNAs involved in malignant transformation of CXPA might help predict the risk of carcinogenesis regardless of histology. Thus, we investigated the candidate miRNAs by comparing the PA and CA samples from CXPA biopsies in an effort to elucidate the pathophysiology of malignant transformation in these cancers. We then compared the miRNA profiles of the PA of CXPA and conventional PA and identified several candidate miRNAs which might be helpful for differential diagnosis between the two (Fig. 1).

Flowchart describing the selection of the samples and their evaluation in our study.
Results
Clinicopathological data
The mean age of the participants in CXPA group was 53.0 years (ranged 26–71 years) while it was only 44.7 years (ranged 28–75 years) in the conventional PA group. The mean tumor size was 2.6 cm (range 1.4–4.0 cm) in the conventional PA, and 3.5 cm (range 1.2–6.3 cm) in the CXPA groups. Our cases included four SDC, two epithelial-myoepithelial carcinoma, two MEC, three adenocarcinoma NOS, one clear cell carcinoma, and one oncocytic carcinoma presented with a clear CA portion. In addition, risk stratification revealed five patients with high-aggression, eight with low-aggression carcinomas. When we evaluated the extent of the invasion in each sample we were able to identify 5 patients with wide invasive, three with minimally invasive, and five with intracapsular tumors. All of these details are summarized in Table 1.
Identification of differently expressed miRNAs
Comparison between the PA and CA in CXPA samples
We identified 2588 miRNAs and 13 differentially expressed miRNAs in the CA and PA of CXPA groups when applying a two-fold change as the threshold (adjusted p-value < 0.05). We found five up-regulated (miR-455-3p, miR-140-5p miR-483-5p, miR-125b-5p, and miR-125a-5p) and eight down-regulated miRNAs (miR-21-3p, miR-183-5p, miR-182-5p, miR-425-5p, miR-96-5p, miR-200a-3p, miR-181a-3p, and miR-505-3p) in these CA relative to PA of CXPA samples (Table 2).
Comparision between conventional PA and PA/CA of CXPA samples
We identified a total of 17 differentially expressed miRNAs when comparing conventional PA and PA/CA of CXPA samples (fold change > 2, adjusted p-value < 0.05). We identified ten up-regulated miRNAs (let-7a-3p, miR-27a-3p, miR-9-5p, miR-135a-5p, miR-135b-5p, miR-455-5p, miR-218-5p, miR-181d-5p, miR-369-3p, and miR-132-5p) and seven down-regulated miRNAs (miR-196a-5p, miR-193a-3p, miR-193b-3p, miR-29c-3p, miR-331-3p, miR-361-3p, and miR-423-5p) in these evaluations (Table 3).
GO functional enrichment and KEGG pathway analyses
Evaluation of these miRNAs in the biological process category identified several target genes with differential expression in CA and PA of CXPA, which pulled in “apoptotic process”, “cell cycle arrest”, “negative regulation of transforming growth factor beta receptor signaling pathway”, “regulation of transcription from RNA polymerase II promoter”, and “positive regulation of transcription from RNA polymerase II promoter” as their top five GO terms (Table 4). Similarly, when comparing the differentially expressed miRNAs from PA of CXPA and conventional PA, the GO terms for the biological process category for the target genes included “peptidyl-serine phosphorylation”, “positive regulation of transcription, DNA-templated”, “protein phosphorylation”, and “negative regulation of transforming growth factor beta receptor signaling pathway” (Table 4).
Table 5 summarizes the most enriched KEGG pathways for each of the differently expressed miRNAs in both the CA vs PA of CXPA, and PA/CA of CXPA vs conventional PA evaluations. Many cancer-related pathways such as “viral carcinogenesis”, “Hippo signaling pathway”, “p53 signaling pathway”, and “proteoglycans in cancer” were amongst the most highly ranked processes in CA vs PA of CXPA. Interestingly, for the comparison between PA of CXPA and conventional PA, there was a similar pattern in the enriched pathways, including “proteoglycans in cancer”, “ECM–receptor interaction”, Hippo signaling pathway”, “AMPK signaling pathway”, “viral carcinogenesis”, and “TGF-beta signaling pathway”.
Target gene prediction, Protein–protein interaction (PPI) networks, and miRNA-Hub Gene network analyses
A total of 3082 genes were commonly identified by miRWalk, miRanda, RNA22, and Targetscan evaluations, which were then reduced to 383 genes with multiple identifications. We then used the STRING database to identify the PPIs among these 383 targets and identified those proteins that were most likely to interact with more than 10 other proteins and designated these as the hub nodes. Figure 2A shows the PPIs derived from the 15 hub nodes (CASP9, TP53, SMAD2, MAPK10, VEGFA, CXCL12, GNG12, PRKAR1A, IGF1R, SDC1, GNA01, ADCY1, ADCY6, ADCY9, and CAMK2A) for the targets of the differentially expressed miRNAs between PA of CXPA (conventional PA) and CA, with each network finally being constructed using Cytoscape. The most significant revelation from these networks was the identification of TP53 as a central node with more than 33 interactions in these networks.

Protein–protein interaction (PPI) network from the STRING database (https://string-db.org/) on Cytoscape software (version 3.8.2, https://cytoscape.org/). (A) The interaction network derived from the 15 hub nodes for the targets of the differentially expressed miRNAs between the pleomorphic adenoma portion (PA of CXPA) and carcinoma portion (CA) of carcinoma ex pleomorphic adenoma (CXPA). (B) The interaction network derived from 25 hub nodes identified for the targets of the miRNAs with altered expression between conventional pleomorphic adenoma versus PA/CA of CXPA.
Figure 2B shows the PPIs for the 25 hub nodes (PML, TP53, MAPK1, MAPK14, AKT2, AKT3, GSK3B, TNS1, PDPK1, BRAF, YWHAB, ADCY1, ADCY2, CAMK2A, CALM3, PRKCA, PRKCE, RPS6KB1, TNS1, IGF1, EGFR, PTPRJ, DNAJC5, KCNAB2, and SNAP25) identified for the targets of the differentially expressed miRNAs between conventional PA and PA of CXPA (CA). Interestingly, TP53 was once again identified as the central node with more interactions than any other target. Based on these findings, we created a final miRNAs-target gene network focused on their connections to the “pathways in cancer”, “p53 signaling pathway”, and “ErbB signaling pathway” terms obtained from the KEGG database (Fig. 3A,B).

Network of the miRNAs with the target genes related to “pathways in cancer”, “p53 signaling pathway”, and “ErbB signaling pathway” terms from the KEGG database using miRNet software (version 2.0, https://www.mirnet.ca). miRNA-target gene network which derived from the differentially expressed miRNAs from pleomorphic adenoma portion (PA of CXPA) and carcinoma portion of carcinoma ex pleomorphic adenoma (CA) (A), and conventional pleomorphic adenoma versus PA/CA of CXPA (B).
Discussion
CXPA is an uncommon SGT with relatively aggressive features that requires complete excision to prevent progression making its preoperative diagnosis a critical factor in improving clinical outcome. However, its mixed cellularity and the heterogeneity of benign and malignant components can make the detection of its CA component challenging, thus reducing the cytodiagnostic accuracy of CA in CXPA to roughly 50% and increasing false-negative diagnosis of PA to 38.5%10. Several studies have tried to identify novel biomarkers to facilitate better diagnostic accuracy in SGT. Many have evaluated the use of miRNAs to distinguish between patients with malignant and benign SGT using both tissue and circulating miRNA targets (saliva or blood). This is because miRNA profiling is expected to create significant diagnostic, prognostic, and/or predictive value, and to help uncover the etiological role of specific miRNAs in cancer initiation, progression, and/or metastasis in various cancers11,12,13,14,15,16. Numerous studies have identified a wide range of differentially expressed miRNAs in both benign and malignant SGTs, and a recent study attempted to create miRNA signatures for the diagnosis of 24 SGT patients (10 benign, and 14 malignancies) using a panel of 798 miRNAs and further suggested that 46 of them are likely to be differently expressed in benign and malignant tumors and may act as potential biomarkers for their differential diagnosis9. Another study evaluating 38 malignant and 29 benign parotid gland tumors identified four miRNA combinations (miR-132, miR-15b, miR-140, and miR-223) that could discriminate between malignant and benign parotid tumors using saliva samples17. On the other hand, a recent meta-analysis concluded that altered miRNA expression in SGTs may be useful for prognostication though their diagnostic value is still limited owing to the small sample sizes and pathological heterogeneity18.
Most of these studies include various kinds of malignant SGTs, while our study tried to identify differentially expressed miRNAs specifically associated with the PA and CA portions of CXPA tumors. Our study also compared the expression of these miRNAs between PA of CXPA and conventional PA in an attempt to understand the pathogenesis of malignant transformation in CXPA, and identify those precancerous miRNA changes that may facilitate the conversion from conventional PA to PA of CXPA.
Interestingly, both miR-21 and miR-96-5p are up-regulated in CA compared to PA of CXPA and both are known to act as onco-miRNAs in head and neck squamous cell carcinoma (HNSCC)19. This is further supported by the fact that miR-21 is commonly up-regulated in various malignant SGTs when compared to their benign tumor counterparts9,20. In addition, miR-96-5p forms part of the miR-96/182/183 cluster and its overexpression is also known to be found in patients with p53 mutations, where it acts to support cell migration and chemoradiation resistance via its targeting of PTEN and activation of the PI3K-AKT signaling pathway in HNSCC19. miR-425-5p is another well-known onco-miRNA which is upregulated in breast cancers where it targets PTEN21, and in MEC tissues, where the function remains unidentified22.
miR-125a-5p, miR-125b-5p, and miR-140-5p, which are down-regulated in the CA tissues when compared to PA of CXPA samples, are also known for their tumor suppressor activities in HNSCC19. Of these, both miR-125b-5p and miR-140-5p are also known to be significantly down-regulated in malignant SGT samples relative to their benign counterparts9. mir-455-3p has also been reported to act as a tumor suppressor gene downstream of TP53 inducing pro-apoptotic activity in pancreatic23, osteosarcoma24, and breast cancer25.
Unlike previous studies, we identified seven miRNAs (miR-196a-5p, miR-193a-3p, miR-193b-3p, miR-29c-3p, miR-331-3p, miR-361-3p, and miR-423-5p), which demonstrated specific differential expression in CA and PA of CXPA tissues when compared to conventional PA samples. Interestingly, some differentially expressed mRNAs (miR-196a-5p, miR-193b-3p, miR-423-5p, and miR-361-3p) were reported to be associated with oral cavity squamous cell carcinoma26,27,28. Moreover, miR-193a-3p was reported to be associated with MEC22, and miR-331-3p has been linked to AdCC29. These findings are particularly significant because, although PA of CXPA and conventional PA present with nearly indistinguishable histological profiles, their onco-miRNA profiles are significantly different. These unique results suggest that these miRNAs may be associated with some of the pre-malignant changes in these samples even when the pathology remains benign. Thus, we suggest that these pre-malignant miRNAs may facilitate better diagnostic accuracy for fine needle aspiration cytology or core needle biopsy in clinical settings. Based on these findings, we can infer that although the PA of CXPA was biopsied based on its heterogeneity of benign and malignant portions, the specific miRNAs can be expected to predict the possibility of malignancy.
Furthermore, the down-regulated miRNAs in PA/CA of CXPA relatively to the conventional PA samples were also shown to be well-known tumor suppressor miRNAs, including the let-7 family transcripts, miR-9-5p, miR-135a-5p and miR-135b-5p, amongst others. miR-9-5p, miR-135a-5p, and miR-135b-5p, were all shown to be down-regulated in malignant vs. benign SGT samples9, and the differential expression of both miR-132-5p and miR-140 was consistent with the results of other studies17.
GO enrichment and KEGG pathway analyses revealed that these differentially expressed transcripts were all linked to the regulation of several critical oncogenic signaling pathways, such as the p53 signaling pathway. Moreover, GO enrichment and KEGG pathway evaluations of the differentially expressed miRNAs from PA of CXPA vs. conventional PA also revealed their involvement in signal regulation with these transcripts being linked to various critical signaling pathways including the AMPK and TGF-beta signaling pathways. PLAG1 or HMGA2 fusions are also useful biomarkers for distinguishing between PA and various other pathologies suggesting that CXPA tissues are also likely to encode these PA-specific gene fusions30. In addition, the amplification of MDM2, mutations in TP53, gains and amplifications of MYC, Epidermal growth factor receptor (EGFR), HGF-A (scatter factor), c-Met (a proto-oncogene), Transforming growth factor alpha (TGF α), Fibroblast growth, factors (FGF)-2, and ErbB2 (HER2), amongst others have all been hypothesized to play a role in the progression and invasion of CXPA31,32,33,34. Thus, it was not surprising that our PPI evaluations included several of these genes, including ErbB2, TP53, HMGA2, PLAG1, HIF1A, EGFR, VEGFA, and MDM2, into the miRNA-Hub gene networks created for both conditions. The most significant hub gene in both the networks was found to be TP53, known to be implicated in the malignant transformation of CXPA. In addition, another recent study reported the molecular events underlying PA’s malignant transformation in one patient who experienced three bouts of recurrent disease before finally receiving a CXPA diagnosis, where they identified the sequential mutation of TP53 in these recurrent tumors, all of which are consistent with our results35. It must be noted that due to the rarity of this disease, we were forced to adopt a retrospective study design and were limited to a small cohort. This limited our ability to perform any validations in a second independent cohort. However, to the best of our knowledge, there have been no other studies evaluating the independent profiles of the PA and CA portions of CXPA nor any comparing the profiles of PA of CXPA and conventional PA. Given this we believe that our results offer a significant insight into the roles of miRNA during CXPA carcinogenesis. Further studies would be required for the validation of these target miRNAs in CXPA and to reveal their possible relationship with other well-known genes involved in CXPA such as PLAG-1 and HMGA2.
In conclusion, we identified several differentially expressed miRNAs in both the CA and PA of CXPA. Specifically, our analysis identified the well-known onco-miRNAs, miR-21 and miR-96-5p, and tumor suprressors miR-125a-5p, miR-125b-5p, and miR-140-5p to have altered expression in the malignant component of CXPA (CA) compared to the benign component (PA of CXPA). In addition, we found that they are related to TP53 and cancer-related pathways using target gene prediction and gene pathway analyses. Furthermore, our evaluations identified a handful of unique miRNAs that may help to differentiate between conventional PA and PA of CXPA, which may be valuable in the development of novel liquid biopsy tools. Taken together these results suggest that miRNAs affect tumor suppressor genes such as p53 during the malignant transformation of CXPA, and that there is a possibility that PA of CXPA may have the potential to progress to CXPA via the differential regulation of several key miRNAs.
Materials and methods
Patients and materials
This study was carried out in accordance with the tenets of the Declaration of Helsinki for research regarding human subjects. This present study was approved by the Seoul National University Institutional Review Board for application at both Bundang Seoul National University Hospital (No.B-1905-540-304) and Dongtan Sacred Heart Hospitals, Hallym University (No.2019-04-298). Bundang Seoul National University Hospital committee waived the need for written informed consent on 11 samples obtained before 2013, and another 18 subjects gave their written informed consent. All methods were carried out in accordance with relevant guidelines and regulations.
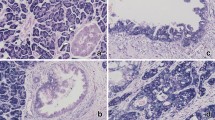
The sample collection was done as described in Fig. 1. A total of 13 CXPA and 16 conventional PA patients who underwent surgical resection at one of these two hospitals between 2010 and 2019 were enrolled in this study based on their pathological presentation. Their original hematoxylin–eosin (H and E)–stained slides were reviewed by the specialist head and neck pathologist (H.K), and the diagnoses in each case were categorized following the World Health Organization (WHO)’s Classification of Salivary Gland Tumors36. All CXPA were classified according to their invasiveness (intracapsular, minimally, and widely invasive) and histological subtype. As per the risk stratification of SGTs based on the WHO (2017) classification our cases include low aggression tumors namely epithelial-myoepithelial carcinoma, clear cell carcinoma, oncocytic carcinoma, adenocarcinoma NOS, and low grade MEC; the high aggression tumor, SDC, and high grade MEC. Macro-dissection of PA and CA portion of CXPA was performed under microscopic marking. Figure 4 shows all the pathologic findings of CA, PA portion of CXPA, and conventional PA.

Histopathologic findings of the carcinomatous portion (CA), pleomorphic adenoma portion (PA) of carcinoma ex pleomorphic adenoma (CXPA) (H & E, 100x) and conventional PA (H & E, 20x).
RNA isolation
Total RNA was extracted from paraffin-embedded tissues using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’ instructions. CXPA tissues were deparaffinization with xylene and the sections stained with H & E before the PA and CA portions of CXPA were selected and carefully dissected to minimize cross contamination. RNA quality was assessed using an Agilent 2100 bioanalyzer and an RNA 6000 Pico Chip (Agilent Technologies, Amstelveen, The Netherlands), and RNA quantification was performed using a NanoDrop 2000 Spectrophotometer system (Thermo Fisher Scientific, Waltham, MA, USA).
Library preparation and sequencing
Sequencing libraries were generated for both the control and test RNAs using the NEBNext Multiplex Small RNA Library Prep kit (New England BioLabs, Inc., USA) according to the manufacturer’ instructions. Briefly, 1 µg of total RNA from each sample was ligated to the adaptors before being transcribed to cDNA using reverse-transcriptase and adaptor-specific primers. Libraries were then amplified by PCR and prepared for sequencing using a QIAquick PCR Purification Kit (Qiagen, Inc, German) and AMPure XP beads (Beckman Coulter, Inc., USA). The yield and size distribution of the small RNA libraries were assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., USA) and high-throughput sequences were generated by single-end 75 sequencing using a NextSeq500 system (Illumina, San Diego, CA., USA).
Data analysis
Sequence reads were mapped using bowtie2 software (v2.4.2, http://bowtie-bio.sourceforge.net/bowtie2/) and the final bam file for evaluation (alignment file) was created. Mature miRNA sequences were then used as a reference for mapping. The read counts for each mature miRNA sequence were extracted from the alignment file using bedtools (v2.25.0) and Bioconductor programmed to use R (version 3.2.2; R development Core Team, 2011) as its base. Read counts were then used to determine the expression level of each miRNA and the quantile normalization method for between sample comparisons. We then identified the miRNA targets using the miRWalk 2.0 database (http://zmf.umm.uni-heiderlberg.de/apps/zmf/mirwalk2/)37,38. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were then applied using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) online tool (http://david.abcc.ncifcrf.gov/)39,40 and specific GO and KEGG pathway terms were identified using a p-value threshold of < 0.01. The protein–protein interaction data were obtained from the STRING database (http://string-db.org/) and visualized using Cytoscape software (version 3.8.2, https://cytoscape.org/). These analyses were then combined to create the miRNA-target gene networks for these samples using miRNet software (version 2.0, https://www.mirnet.ca).
Statistical analysis
All statistical analyses were completed in R (version 3.5.1) using R studio (version 1.1.383, https://www.rstudio.com). T-test was used to evaluate the differential expression between different sample types and a p-value of < 0.05 was considered statistically significant. The R “stats” package was used for all Benjamini–Hochberg corrections.
Ethics approval
The study was approved by the Committee on Seoul National University Bundang Hospital (No. B-1905-540-304) and Dongtan Sacred Heart Hospital (No. 2019-04-298), Republic of Korea.
Data availability
Data has been deposited to the Gene Expression Omnibus under the accession of GSE205670 (www.ncbi.nlm.nih.gov/geo).
References
Lüers, J. C., Wittekindt, C., Streppel, M. & Guntinas-Lichius, O. Carcinoma ex pleomorphic adenoma of the parotid gland. Study and implications for diagnostics and therapy. Acta Oncol. 48, 132–136 (2009).
Chen, M. M., Roman, S. A., Sosa, J. A. & Judson, B. L. Predictors of survival in carcinoma ex pleomorphic adenoma. Head Neck 36, 1324–1328 (2014).
Gupta, A., Koochakzadeh, S., Neskey, D. M., Nguyen, S. A. & Lentsch, E. J. Carcinoma ex pleomorphic adenoma: A review of incidence, demographics, risk factors, and survival. Am. J. Otolaryngol. 40, 102279 (2019).
Lewis, J. E., Olsen, K. D. & Sebo, T. J. Carcinoma ex pleomorphic adenoma: Pathologic analysis of 73 cases. Hum. Pathol. 32, 596–604 (2001).
Antony, J., Gopalan, V., Smith, R. A. & Lam, A. K. Carcinoma ex pleomorphic adenoma: A comprehensive review of clinical, pathological and molecular data. Head Neck Pathol. 6, 1–9 (2012).
El-Naggar, A. K. et al. Molecular genetic alterations in carcinoma ex-pleomorphic adenoma: A putative progression model? Genes Chromosom. Cancer 27, 162–168 (2000).
Bartel, D. P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004).
Hayes, J., Peruzzi, P. P. & Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 20, 460–469 (2014).
Denaro, M. et al. A microRNA signature for the differential diagnosis of salivary gland tumors. PLoS ONE 14, e0210968 (2019).
Klijanienko, J., El-Naggar, A. K. & Vielh, P. Fine-needle sampling findings in 26 carcinoma ex pleomorphic adenomas: Diagnostic pitfalls and clinical considerations. Diagn. Cytopathol. 21, 163–166 (1999).
Anfossi, S., Fu, X., Nagvekar, R. & Calin, G. A. MicroRNAs, regulatory messengers inside and outside cancer cells. Adv. Exp. Med. Biol. 1056, 87–108 (2018).
Graveel, C. R., Calderone, H. M., Westerhuis, J. J., Winn, M. E. & Sempere, L. F. Critical analysis of the potential for microRNA biomarkers in breast cancer management. Breast Cancer 7, 59–79 (2015).
Peng, Y. & Croce, C. M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 1, 15004 (2016).
Rupaimoole, R. & Slack, F. J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 16, 203–222 (2017).
Sempere, L. F. Fully automated fluorescence-based four-color multiplex assay for co-detection of microRNA and protein biomarkers in clinical tissue specimens. Methods Mol. Biol. 1211, 151–170 (2014).
Sempere, L. F., Azmi, A. S. & Moore, A. microRNA-based diagnostic and therapeutic applications in cancer medicine. Wiley Interdiscip. Rev. RNA 12, e1662 (2021).
Matse, J. H. et al. Discovery and prevalidation of salivary extracellular microRNA biomarkers panel for the noninvasive detection of benign and malignant parotid gland tumors. Clin. Cancer Res. 19, 3032–3038 (2013).
Dos Santos, E. S. et al. Diagnostic and prognostic value of miRNAs on salivary gland tumors: A systematic review and meta-analysis. Oral Maxillofac. Surg. 25, 445–456 (2021).
Vahabi, M., Blandino, G. & Di Agostino, S. MicroRNAs in head and neck squamous cell carcinoma: A possible challenge as biomarkers, determinants for the choice of therapy and targets for personalized molecular therapies. Transl. Cancer Res. 10, 3090–3110 (2021).
Cinpolat, O., Unal, Z. N., Ismi, O., Gorur, A. & Unal, M. Comparison of microRNA profiles between benign and malignant salivary gland tumors in tissue, blood and saliva samples: A prospective, case-control study. Braz. J. Otorhinolaryngol. 83, 276–284 (2017).
Xiao, S., Zhu, H., Luo, J., Wu, Z. & Xie, M. miR-425-5p is associated with poor prognosis in patients with breast cancer and promotes cancer cell progression by targeting PTEN. Oncol. Rep. 42, 2550–2560 (2019).
Binmadi, N. O. et al. miRNA expression profile of mucoepidermoid carcinoma. Oral Dis. 24, 537–543 (2018).
Zhan, T. et al. miR-455-3p functions as a tumor suppressor by restraining Wnt/β-catenin signaling via TAZ in pancreatic cancer. Cancer Manag. Res. 12, 1483–1492 (2020).
Yi, X., Wang, Y. & Xu, S. MiR-455-3p downregulation facilitates cell proliferation and invasion and predicts poor prognosis of osteosarcoma. J. Orthop. Surg. Res. 15, 454 (2020).
Li, Z. et al. MicroRNA-455-3p promotes invasion and migration in triple negative breast cancer by targeting tumor suppressor EI24. Oncotarget 8, 19455–19466 (2016).
Maruyama, T. et al. MicroRNA-196a-5p is a potential prognostic marker of delayed lymph node metastasis in early-stage tongue squamous cell carcinoma. Oncol. Lett. 15, 2349–2363 (2018).
Li, X., Rui, B., Cao, Y., Gong, X. & Li, H. Long non-coding RNA LINC00152 acts as a sponge of miRNA-193b-3p to promote tongue squamous cell carcinoma progression. Oncol. Lett. 19, 2035–2042 (2020).
Romani, C. et al. Genome-wide study of salivary miRNAs identifies miR-423-5p as promising diagnostic and prognostic biomarker in oral squamous cell carcinoma. Theranostics. 11, 2987–2999 (2021).
Zhu, Q. H. et al. Identification of pivotal microRNAs involved in the development and progression of salivary adenoid cystic carcinoma. J. Oral Pathol. Med. 51, 160–171 (2022).
Toper, M. H. & Sarioglu, S. Molecular pathology of salivary gland neoplasms: Diagnostic, prognostic, and predictive perspective. Adv. Anat. Pathol. 28, 81–93 (2021).
Röijer, E. et al. Translocation, deletion/amplification, and expression of HMGIC and MDM2 in a carcinoma ex pleomorphic adenoma. Am. J. Pathol. 160, 433–440 (2002).
Martins, C. et al. PLAG1 gene alterations in salivary gland pleomorphic adenoma and carcinoma ex-pleomorphic adenoma: A combined study using chromosome banding, in situ hybridization and immunocytochemistry. Mod. Pathol. 18, 1048–1055 (2005).
Furuse, C. et al. Study of growth factors and receptors in carcinoma ex pleomorphic adenoma. J. Oral Pathol. Med. 39, 540–547 (2010).
Hashimoto, K. et al. HER-2/neu gene amplification in carcinoma ex pleomorphic adenoma in relation to progression and prognosis: A chromogenic in-situ hybridization study. Histopathology 60, E131–E142 (2012).
Valstar, M. H. et al. Malignant transformation of salivary gland pleomorphic adenoma: Proof of principle. J. Pathol. Clin. Res. 7, 432–437 (2021).
Seethala, R. R. & Stenman, G. Update from the 4th edition of the World Health Organization classification of head and neck tumours: Tumors of the salivary gland. Head Neck Pathol. 11, 55–67 (2017).
Dweep, H., Sticht, C., Pandey, P. & Gretz, N. miRWalk–database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44, 839–847 (2011).
Dweep, H. & Gretz, N. miRWalk2.0: A comprehensive atlas of microRNA-target interactions. Nat. Methods. 12, 697 (2015).
Dennis, G. Jr. et al. DAVID: Database for annotation, visualization, and integrated. Discov. Genome Biol. 4, P3 (2003).
Huang, D., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2019R1G1A1009856).
Author information
Authors and Affiliations
Contributions
Conceptualization, H.K. and H.K.; methodology, H.K., S.E., and Y.J.B.; data analysis and interpretation, H.K., J.S.L., and H.K.; investigation, W.J., and S.A.; writing-original draft and review, H.K., J.S.L., and H.K.; supervision, S.E., W.J., and S.A.; funding acquisition, H.K. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, H., Eun, S., Jeong, WJ. et al. Identification of differentially expressed microRNAs as potential biomarkers for carcinoma ex pleomorphic adenoma. Sci Rep 12, 13383 (2022). https://doi.org/10.1038/s41598-022-17740-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17740-9
This article is cited by
-
Dynamic Role of miRNAs in Salivary Gland Carcinomas: From Biomarkers to Therapeutic Targets
Head and Neck Pathology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
