
Abstract
Insectivorous bats consume a diverse array of arthropod prey, with diets varying by bat species, sampling location, and season. North American bat diets remain incompletely described, which is concerning at a time when many bat and insect populations appear to be declining. Understanding the variability in foraging is thus an essential component for effective bat conservation. To comprehensively evaluate local foraging, we assessed the spatial and temporal variability in prey consumed by the little brown bat, Myotis lucifugus, in New Hampshire, USA. We collected bat guano samples from 20 sites over 2 years and analyzed sequence data for 899 of these samples using a molecular metabarcoding approach targeting the cytochrome oxidase I subunit (COI) gene. Some prey items were broadly shared across locations and sampling dates, with the most frequently detected arthropod orders broadly similar to previous morphological and molecular analyses; at least one representative sequence variant was assigned to Coleoptera in 92% of samples, with other frequently detected orders including Diptera (73%), Lepidoptera (65%), Trichoptera (38%), and Ephemeroptera (32%). More specifically, two turf and forest pests were routinely detected: white grubs in the genus Phyllophaga (50%), and the Asiatic Garden beetle, Maladera castanea (36%). Despite the prevalence of a few taxa shared among many samples and distinct seasonal peaks in consumption of specific arthropods, diet composition varied both temporally and spatially. However, species richness did not strongly vary indicating consumption of a broad diversity of taxa throughout the summer. These data characterize little brown bats as flexible foragers adept at consuming a broad array of locally available prey resources.
Similar content being viewed by others

Introduction
North American insectivorous bats have highly flexible foraging strategies. Dietary analyses indicate consumption of a broad assortment of prey1, yet the composition of prey contents reported appears sensitive to temporal or spatial factors among little brown bats, Myotis lucifugus2–4, big brown bats, Eptesicus fuscus4–6, as well as Indiana bats, Myotis sodalis7. Similar patterns of temporal variability of arthropods in bat diets occur in their European relatives8–10. Thus, it is unsurprising that many of the factors associated with intraspecies variation in little brown bat diets—a species with extensive historical records—are connected with either location or season. These factors include variation in prey abundance4,11, bat age12, landscape features13, and ambient temperature14. Heterogeneity in sampling location or date can alter the community composition of prey available and therefore what is observed in bat diets2,4,6, suggesting that a more comprehensive understanding of the niche breadth of a species would greatly benefit from a sampling design surveying multiple populations simultaneously and repeatedly throughout the season.
The techniques used to describe diets also play a fundamental role in how we characterize the foraging habits of a species15,16. Historically, morphological analyses have described bat diets at the order-level due to taxonomic challenges with prey species identifications from guano1,17. Diet compositions of insectivorous bats in Eastern North America typically contain some ratio of Coleoptera, Diptera, and Lepidoptera, with smaller fractions of Trichoptera and Ephemeroptera and a few other taxa. For example, these visual identification methods indicate that North American bats like E. fuscus are beetle specialists5, while Myotis spp. more frequently consume flies and moths18–20. More recent studies using molecular methods support some of the historically observed order-level diet findings—E. fuscus still prefer beetles6,21 (but less so in Southwestern deserts22), and M. lucifugus continue to consume flies and moths2,3. Although not without their own biases (see Nielsen et al.16 for a review of various diet tracing techniques, and Alberdi et al.23 for molecular metabarcoding, specifically), molecular analyses generally have higher resolution of prey items relative to morphological techniques, and can reveal a much broader palate than previously described24. Rather than a small menu consisting of a few arthropod orders, molecular studies of insectivorous bats routinely describe hundreds of unique sequences detected with varying frequencies across numerous arthropod orders. In light of the superior taxonomic resolution of molecular metabarcoding, historical assessments may have significantly underestimated the niche and dietary breadth of the bat species described.
Detailed diet information provided by metabarcoding may be of particular importance for conserving North American bat populations that have exhibited drastic declines due to the fungal disease White-Nose Syndrome25,26. For bat populations that are recovering and for those that continue to decline, diet information can help characterize the local habitat resources required by these populations. Molecular diet information is scant for a few of the species impacted by the disease and entirely absent for others. At the same time, insect populations have recently exhibited dramatic declines worldwide27,28, with likely effects on aerial insectivores such as bats9. The first multi-year molecular analyses of the diets of North American bats observed local compositional variability within and between seasons at the same location in little brown bats2,3 and big brown bats6. Similarly, a recent evaluation of little brown bat diets across six sites in Wisconsin identified regional and local foraging preferences, with evidence of family-level diet turnover between weeks4. We were motivated to build on these foundational studies by significantly expanding on the number of sites sampled to better assess the potential diversity in little brown bat foraging patterns across sampling locations and years. We completed a two-year sampling regime that spanned 20 sites across southern and central New Hampshire, USA. Our study had three main objectives: first, to assess the dietary breadth of little brown bats throughout New Hampshire; second, to determine the extent with which these guano samples reveal potential forest or agricultural pests in an area; and third, to compare the extent with which bat diets vary along temporal and spatial gradients.
Results
Over 4200 guano samples were collected from June to August 2015, and April to October 2016, at 20 locations throughout central and southern New Hampshire, USA. These sites included a mixture of forest land cover classes, as well as water, wetland, agricultural, and urban landscape components (Fig. 1). We generated sequence data for 2521 samples, and ultimately retained 899 samples at 19 of 20 sites for diet analyses after filtering for arthropod-specific COI sequences due to strict data quality requirements (Tables S1 and S2). Because guano samples were collected passively, classification of COI sequences was also used to identify the likely bat species. A single species, the little brown bat (Myotis lucifugus), was identified in 579 guano samples and contained more than 99.99% of all bat-classified sequences. Thus, while it is possible that other bat species were present and contributing guano, we did not detect them.

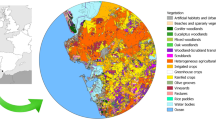
NH bat sample locations and landcover. (a) Guano samples collected throughout New Hampshire (dark state location of inset map) at particular locations (labels). Locations are abbreviated by 3-letter codes to reflect a particular New Hampshire town: ALS, Alsted; BRN, Brown Lane, Hollis; CHI, Chichester; CNA, Canterbury; CNB, Canterbury; COR, Cornish; EPS, Epsom; FOX, Fox State Forest, Hillsborough; GIL, Gilsum; GRN, Greenfield; HOL, Squam Science Center, Holderness; HOP, Hopkinton, MAP, Maple Hill, Hollis; MAS, Massabesic Audubon Center, Auburn; MTV, Mont Vernon; PEN, Penacook; SWZ, Swanzey; WLD, Willard Pond, Antrim; WLT, Wilton. (b) Fraction of land cover type within a 2500 m radius at collection site. Map created using a custom R script available at https://github.com/devonorourke/nhguano/blob/master/scripts/r_scripts/mapplots.R.
The largest fraction of arthropod sequence diversity was detected among the coleopteran order, with 766 sequence clusters (OTUs) identified among all samples, and at least one coleopteran OTU was identified among 92% of samples (Table 1). Note that ‘sequence clusters’, herein termed OTUs, are in fact denoised exact sequence variants (ASVs) clustered at 98.5% similarity (see Methods for details). Many samples also contained at least one OTU classified to Diptera (73% of samples), Lepidoptera (65%), Trichoptera (38%), and Ephemeroptera (32%). While we detected thousands of OTUs, just 51 were identified in at least 5% of samples (Table S3). Most of these frequently detected OTUs were prevalent in diets throughout most locations: 11 of the 19 sites contained at least half of these 51 OTUs. We grouped these frequent OTUs into shared genus labels rather than examining the taxonomic identity assigned to specific OTUs because several of these OTUs shared identical taxonomic labels, or alternatively, contained ambiguous species labels. Interestingly, the five most frequently detected genera are all coleopterans, and are present in at least 20% of all samples (Fig. 2). In fact, no other arthropod order had any genus represented in more than 20% of samples, although other orders contained multiple genera detected at lower frequencies. Overall, these data depict little brown bats consuming a diverse assortment of prey spanning many arthropod orders, although several beetle taxa are the most commonly consumed group in New Hampshire.

Proportion of samples with commonly detected genus labels organized by arthropod order. Genera shown represent labels present in at least 5% of samples across all New Hampshire sites. One particularly frequent taxon was missing a genus-specific label and is listed by its known family and generic OTU alias (f. Chironomidae OTU-19).
Two groups of beetles were frequently detected in guano samples that are listed as pests by US Forest Service (USFS) or US Department of Agriculture (USDA): white grubs of the genus Phyllophaga (detected in 52% of samples at 19 sites), and the Asiatic garden beetle, Maladera castanea (37% of samples at 18 sites). Only one other exact species match was identified as a pest in our dataset, the forest tent caterpillar moth, Malacosoma disstria (25 distinct samples detected at 13 sites). Because matching species identities in different databases can be complicated by ambiguous labels and incomplete records, we expanded our search to include any taxa we detected that shared the genus of a pest listed by USFS or USDA. Bats consumed a much wider variety of taxa when comparing at the genus level; 32 pest genera were identified, all of which were detected in at least 10 samples and at multiple sites (Table 2). While genus-level identification of pests is not definitive as to whether these bats are consuming particular insect species, these results suggest that bat guano sampling may provide a robust and rapid survey method for forest and agricultural pests, with refinements possible to focus on particular species.
Three separate analyses investigated how bat diets varied with temporal and spatial factors: (1) comparing effect of bat diet on sampling date only, by comparing samples collected at a single site across multiple sampling windows in a single year; (2) comparing effects between multiple sites and sampling windows in a single year; (3) comparing effects of multiple sites at a single sampling date between multiple years. Sampling windows are defined in the Methods section and were used break each season into periods of equal length.
First, we compared bat diets temporally, and examined samples collected at a single site (Fox State Forest in Hillsboro, NH, USA; the site with the most complete collection data) in 2016. Sample collection spanned 6.5 months at the site, beginning in sampling window 3 on April 7 and ending in sampling window 8 on October 21 (mean 13.5 ± 9.7 S.E. samples per window). We calculated the effective number of arthropod species in samples to determine whether the dietary richness varied by sample window using three metrics (Fig. S1): observed OTUs (SR), Shannon’s entropy (H), and Faith’s phylogenetic diversity (PD). We chose to apply multiple tests to contextualize our understanding of richness: while SR provides a measure of taxa richness, H and PD provide additional information about the evenness of a community (with PD further incorporating the phylogenetic signal driving that evenness), A Kruskal–Wallis test for group differences suggested modest group differences for sampling window for all three metrics: SR (H(5) = 13.35, p = 0.02); H (H(5) = 9.79, p = 0.082); PD (H(5) = 11.99, p = 0.035). While a post hoc Dunn’s test revealed a small number of significant pairwise differences in species richness between sampling windows for uncorrected data, no significant differences were detected after applying a Benjamini–Hochberg correction. This indicates there was little variation in the richness or evenness of species detected in the sampling windows throughout the entirety of the foraging season at this site.
However, we detected variability in diet composition, with significant main effect of sampling window for both non-phylogenetic (Dice-Sorensen; ADONIS: R2 = 0.22; P < 0.001; Table S4) and phylogenetic distance estimates (unweighted UniFrac; ADONIS: R2 = 0.19; p < 0.001) (Table S4). Evaluating diet composition using both phylogenetic along with non-phylogenetic distance measures helps contextualized what’s on the menu for these bats: with phylogenetic-weighted distances showing significant differences in diet, we understand that bats are not just eating different collections of any type of bugs, but they are consuming different collections of evolutionarily divergent types of bugs at different points of the season. Principal Coordinates Analyses (PCoA) using Dice-Sorensen (Fig. 3a) and unweighted UniFrac (Fig. 3b) distance measures clustered samples along expected temporal gradients, with early and late samples forming distinct groups between a third cluster of samples collected mid foraging season. The composition of frequently detected taxa at Fox State Forest was consistent with overall observations across New Hampshire sites, with coleopteran, dipteran, and lepidopteran orders representing the majority of OTUs detected (Fig. 3c). However, the dominant taxa shifted throughout the season, with beetle taxa being far more detected in early and mid-season samples, while flies were the most detected taxa in the later part of the foraging season. In particular, white grub beetles of the genus Phyllophaga were detected in the majority of samples at Fox State Forest in the early sampling windows, while non-biting midges in the genus Chironomus were common in later sampling windows (Fig. 3d).

Changes in bat diet arthropod composition at one site. Compositional change at Fox State Forest (Hillsboro, NH, USA) throughout an entire foraging season in 2016. Sampling windows define 37-day periods beginning in early April and ending in late October. PCoA ordinations shown for (a) Dice-Sorensen index and (b) unweighted UniFrac distance metrics are grouped into early, mid, and late foraging season clusters; ellipses depict 95% confidence intervals around window group median. (c) The proportion of detections per arthropod order in a sampling window shift from being most represented by coleopteran taxa in early season to dipteran taxa in late season. (d) The fraction of samples with particular genera detected at each sampling window suggest specific taxa are major diet targets at different points of the foraging season. For example, coleopteran (Phyllophaga) in early sampling windows and dipteran (Chironomus) in late sampling windows.
We next evaluated whether sample richness and species composition varied across both location and date within one season. We focused on samples collected from seven locations in 2016 during sampling windows 4–6 (15.8 ± 9.1 samples per site + window group) due to the completeness of this dataset. Species richness varied by site + window groups for all three measures: SR (H(20) = 49.59, p < 0.001); H (H(20) = 39.06, p < 0.005); and PD (H(20) = 91.12, p < 0.001). Species richness measures were broadly similar across various site + window groups for non-phylogenetic measures, but more variable among Faith’s PD diversity (Fig. S2). A post hoc Dunn’s test for pairwise differences illustrated that only a small fraction of possible group pairs were significantly different after multiple significance correction among non-phylogenetic measures and were largely attributed to a single site + window with elevated richness (of the 210 pairwise comparisons, 7 of 10 significant differences included the 5-EPS window + site group). Similarly, significant pairwise differences in phylogenetic diversity among site + window groups were largely attributed to just 3 of 21 potential site + window groups (4MAP, 6HOL, 6PEN). In particular, 4MAP was significantly lower in diversity compared to nine other site + window groups, while 6HOL and 6PEN groups were larger in phylogenetic diversity relative to 8 and 5 site + window pairs, respectively (Figure S3). Collectively, these comparisons suggest that limited variability in dietary richness over different sampling windows and across multiple New Hampshire sites, although a few particular sites and sampling periods differed.
Diet composition varied in both space and time, with significant main effects for sampling widow and site, as well as their interaction, for both Dice-Sorensen and unweighted UniFrac estimates (Table S5). However, a greater proportion of variation in each PERMANOVA calculation was attributed to sampling site than sampling window for both Dice-Sorensen (Adonis: site R2 = 0.13, p < 0.01; window R2 = 0.05, p < 0.01; Table S5) and unweighted UniFrac (Adonis: site R2 = 0.14, p < 0.01; window R2 = 0.05, p < 0.01; Table S5) distances. The first two principal component axes captured only a modest proportion of variation for Dice-Sorensen (23.5%) or unweighted-UniFrac (25%) distance measures (Fig. 4a), indicating that many diet components are shared throughout these New Hampshire sites and sampling windows. Nevertheless, particular site + window groups can be discriminated by particular taxa at both the order (Fig. 4b) and genus (Fig. 4c) levels. An indicator species analysis (of shared genus labels, thus really an indicator ‘genus’ analysis) was performed separately for sampling window, site, and site + window groups (Fig. 4c) and identified representative taxa from multiple orders significantly associated with groups in each test. The majority of significant associations occur in one sampling window (window 6, 60% of significant taxa), or were associated with a single sampling site (HOL, 50% of taxa), though every distinct site or sampling window contains at least one indicator taxa, as did 13 of 21 possible site + window groups (Table S6). Notably, taxa detected in these bat guano may be associated with particular combinations of sampling window, site, or site + window groups (Figure S3), often befitting the life histories of these arthropod prey items. For example, the commonly detected genus Phyllophaga, fittingly known as a May or June bug, was strongly associated with sampling windows 4 and 5 (spanning April through June) but not window 6 (early July to early August).

Changes in bat diet arthropod composition at multiple sites and sampling windows in 2016. (a) PCoA ordinations shown for Dice-Sorensen index and unweighted UniFrac distance metrics depict sampling site (point shape) or sampling window (color); ellipses depict 95% confidence intervals around window group median. (b) The proportion of detections per arthropod order in a sampling window for each site; arthropods with fewer than 2% of detections aggregated as “other” taxa. (c) Indicator species analysis performed at genus level to identify taxa associated with sampling window, or site, or site + window groups. Taxa with ambiguous genus labels identified by known arthropod family labels.
Samples collected from three sites in both 2015 and 2016 were compared within a single sampling window (window 6, July 5 to August 11) to assess the variability in bat diet richness and composition between years (13.5 ± 4.5 samples per site + year group). Species richness did not vary significantly among site + year groups for any metric: SR (H(5) = 6.54, p = 0.26); H (H(5) = 2.51, p = 0.77); and PD (H(5) = 3.56, p = 0.62). A PERMANOVA revealed that diet composition was significant for main effects of Site and Year, as well as their interaction for both metrics. However, little of the variation was captured in the model by either Dice-Sorensen (Adonis: residual R2 = 0.86) or Unweighted-UniFrac (Adonis: residual R2 = 0.87) distances. Likewise, ordinating the first two principal components of the PCoA explained a limited amount of variation for both Dice-Sorensen (17.7%) and Unweighted-UniFrac (20.6%) metrics, yet no clustering was apparent for either site, year, or site + year groups (Fig. 5a). Few taxa (at the genus level) were identified as significant indicators of a particular site + year group, with just 4 genera associated to a particular site + year, and 3 genera associated to some combination of site + year groups (Fig. 5b). Indeed, at the taxonomic order level, the proportion of taxa detected were largely consistent between years at a given site (Fig. 5c).

Changes in bat diet arthropod composition at multiple sites in a single sampling window between years 2015 and 2016. (a) PCoA ordinations shown for Dice-Sorensen index and unweighted UniFrac distance metrics depict sampling site (point shape) and sampling year (color); ellipses depict 95% confidence intervals around year group median. (b) Indicator species analysis performed at genus level to identify taxa associated with one or more site + year groups. (c) The proportion of taxa detected per arthropod order in each site + year group.
Discussion
The earliest work characterizing insectivorous North American bat diets was performed using morphological techniques—even within New Hampshire specifically19,29—yet these visual identifications were largely limited to classifying prey to the order or family level and may have underrepresented or misassigned taxa17. Our study reaffirms that bats in New Hampshire are indeed foraging on many of the same arthropods described in these earlier morphological analyses: principally beetles, flies, and moths. However, our molecular approach indicates a more expansive diversity of arthropod prey than the earlier visual analyses have described, with 12 orders of insects and spiders detected in at least 5% of our samples. This was not unsurprising, as molecular metabarcoding approaches are known to reveal taxa previously unrecognized by morphological techniques24,30. We also detected arthropod orders like Blattodea, Psocodea, and Megaloptera, including cockroaches such as Parcoblatta pennsylvanica, and fishflies such as Chauliodes pectinicornis—taxa endemic to New Hampshire, but absent from previous bat diet records employing visual techniques. Our molecular analyses provide strong evidence that little brown bats in New Hampshire bats are capable of consuming a more diverse assortment of prey than their historical depictions.
Our most frequently detected arthropod orders (beetles, flies, and moths) were similarly depicted in previous molecular diet analyses of little brown bats2,3 and big brown bats6 from eastern Canada, as well as little brown bats in Wisconsin4. However, we found that the particular proportions of these arthropod components are in contrast to the preferences ascribed to these earlier reports. Coleopteran, not lepidopteran taxa, were by far the most frequently detected arthropod order in our study, despite these previous investigations indicating that beetles are preferred by big brown bats, and moths preferred by little brown bats. While it is possible that particular compositional differences occur between studies because of different primers used to generate the COI amplicon data, previous comparisons of these primer sets show no evidence for bias towards amplifying coleopteran taxa31,32, and would likely only reduce the breadth of taxa in earlier studies instead of the selective amplification of beetle over lepidopteran sequences. Additionally, different methodologies between studies produced similar OTU richness estimates between our single guano pellet approach, other North American insectivore richness values sampling from individual bats22, or earlier studies of little brown bat3 and big brown bat6 diets that used a bulk sampling (i.e., pooled guano pellets) approach. It is also possible our passive sampling technique failed to account for big brown bat contributions mixed in with little brown bat guano, as our study did not physically capture the bat to confirm species identity. However, the preponderance of bat-classified COI sequences characterizes our samples as distinctly belonging to the little brown bat, with over 99.9% of all bat-classified COI sequences assigned to the little brown bat in hundreds of guano samples. It has also been suggested that the hardy carapace of a beetle increases the likelihood of detection over softer-bodied prey items, yet the same authors also demonstrate that molecular techniques can consistently detect other arthropod orders6. Indeed, while we detected five distinct genera of beetles in over 20% of all samples, we also frequently detected particular genera in other arthropod orders such as Diptera. For example, dipteran sequences classified to the genus Tipula were detected in 144 samples, while the genus Hexagenia (order Ephemeroptera) were detected in 105 samples. Additionally, beetles were not universally the most dominant taxa at all sites or sampling windows. For example, samples collected in Holderness, NH contained more ephemeropteran and dipteran detections than coleopteran (see HOL in Fig. 1A). Thus, it is likely that the inordinate fondness for beetles observed among these little brown bats in New Hampshire is driven by prey availability rather than particular methodological differences.
Our extensive sampling across multiple sites and time periods was motivated to understand how spatial or temporal factors vary with New Hampshire bat diets. First, we discovered that the extent of diet composition varying with time was related to the phenology of the available prey in question. Thus, the frequency of detections for particular taxa in particular sampling windows routinely matched the expected life history of the prey. For example, beetles classified to the Phyllophaga genus were the most frequently detected taxa in our entire study, yet the number of samples with Phyllophaga detected dropped from 90% of samples between late April to late May, to 77% of samples between late May to early July, to 29% of samples between early July and mid-August. The same pattern extended to other arthropods: the psocodean barklouse (genus Metylophorus) was detected only in late summer and mid-fall sampling periods, befitting their seasonally late adult aerial emergence; non-biting midges like the dipteran (genus Chironomus) were detected in 10% or fewer samples from April through early July, yet increased from 18% of samples by mid-August, to nearly 60% of samples between August and September, an expected period of peak mating swarm activity. Collectively, our evidence supports the notion that New Hampshire bat diet composition shifts throughout the foraging season because of the variation in available prey, which is itself a function of the particular life cycle of the taxa in question. It is therefore no surprise to see May and June bugs at the top of the menu in May and June months. Systematic sampling of prey availability was beyond the scope of our study but would improve our inferences about the strength of the connections between bat diets and arthropod phenology.
The phenology of prey also explains the relatively modest effect of the sampling period on diet composition. Our samples were organized in 37-day sampling windows, a period of time shorter than the adult lifespan of many insect taxa. Thus, taxa detected across multiple sampling windows reduce the overall effect of time in our model. We observed that shifts in diet composition were the least pronounced among proximal sampling windows and the greatest during the beginning and end of the foraging season among samples collected at a single site in 2016. Likewise, when comparing across multiple sites in 2016 between three sampling periods spanning late April through mid-August, sampling period explained no more than 5% of the observed variation in diet composition.
Diet also varied based on sampling site. However, the effect of sampling location on diet composition was complicated by the fact that most sites contained highly similar land cover attributes; most sites consisted of mixed, softwood, and hardwood forest land cover. Thus, the effect of site explained no more than 14% of the observed variation in diet composition among samples collected at the seven locations evaluated in 2016. However, the greatest number of compositional differences did occur at the site with the greatest land cover difference. Samples collected from Holderness, a location less than 500 m to Squam Lake (a lake over 27 km2), contained more than half of all taxa uniquely associated with a particular site in an indicator species analysis, the majority of which were aquatic invertebrates. In addition, after further restricting this indicator analysis to particular site + window groups, 13 of the 23 genera significantly associated to a single group were from Holderness.
With examples of regional persistence33,34 providing an opportunity at conserving bat populations decimated by White-Nose Syndrome (WNS)35,36, diet analyses can play a consequential role informing future management considerations. The earliest U.S. Fish and Wildlife Service national plans for managing WNS specifically outlined the need to identify research methods effective for monitoring and conserving affected populations37,38. These kinds of guano analyses offer a rapid and robust characterization of the particular habitat resources a population requires. Our work builds on earlier studies indicating that little brown bats are sufficiently flexible in foraging across a range of land cover types2–4, yet it remains unclear whether this foraging flexibility is equally robust among the many other bat species affected by WNS. We recommend that future management strategies include molecular diet analyses in regional and national plans.
Beyond providing information on habitat requirements, molecular diet analyses of bat guano can also identify economic benefits bats may be providing. Bats provide well-known ecosystem services39, and insectivorous bats in particular can provide significant economic benefits40 as consumers of pests in a variety of agricultural and forested environments31,41–43. We discovered that two of the most ubiquitous diet components were beetle pests: the Asiatic garden beetle in 36% of samples and white pine grubs in the genus Phyllophaga in 48% of samples. These are turf44 and forest/crop45,46 pests, respectively, though they are of limited ecological and economic concern, and both were known to exist in New Hampshire. Among taxa detected in New Hampshire bat guano that shared a common genus with pests of concern to the USDA or USFS, all were known to exist in the Northeast, although several had not been recorded in New Hampshire specifically according to records available through the Insect and Arachnid Collections at the University of New Hampshire47. In some cases, it may be evident that this ambiguous classification is not a concern. For example, while there are five known species in the Coleotechnites genus in New Hampshire, the significant forest pest from our study, Coleotechnites milleri, is not one of them, and the host tree does not exist on the east coast. However, in other cases such as with the genus Dendroctonus it is not clear whether this ambiguous sequence is derived from one of the three endemic bark beetles, or if it represents a more concerning species such as D. mexicanus. Because of the ease with which guano is collected and the relatively inexpensive manner that sequence data can be obtained, molecular diet analyses can serve a dual benefit: providing a robust assessment of the dietary breadth of a species, as well as leveraging the bats’ expansive foraging capacity to screen for potential pests of concern within agency frameworks such as the USDA Forest Service early warning system48. While detections of putative pests discovered using this molecular approach are not definitive, and unlikely to be conclusive for species-level identification without further targeted comparisons of vouchered specimens, our results demonstrate that the broad array of arthropod orders detected using this approach can reveal pests or other non-native insects that were previously undescribed in an area.
Methods
Sample collection
Individual guano pellets were passively collected each week at sites throughout New Hampshire (Fig. 1) beginning June 2015 and ending October 2016. We relied on citizen scientists to assist with collecting samples at 19 of the 20 sites. Locations consisted of a mix of forest and agricultural landscapes typical of the region. We obtained data from partners collecting guano at a nature center (HOL in Fig. 1), a forest research station (FOX), conservation lands (BRN, MAP, MAS, WLD), and privately owned homes (all other sites) from bat colonies occupying structures such as attics, barns, garages, and bat houses. Volunteers were provided with supplies (forceps, dust masks, nitrile gloves, ethanol wipes, plastic sheets), and were instructed to collect up to 12 fresh guano samples per week by transferring individual pellets into the pre-filled microcentrifuge tubes containing 1 mL storage buffer (3.5 M ammonium sulfate, 16.7 mM sodium citrate, 13.3 mM EDTA, pH 5.2). Plastic sheets were replaced weekly to avoid cross contamination between weeks, and samples were shipped in batches back to our lab approximately monthly. Samples were stored at −80 °C until DNA extraction.
All research and experimental protocols were approved and performed in accordance with relevant guidelines following the University of New Hampshire’s licensing committee (IACUC) protocols 160105 and 181209. Likewise, we obtained informed consent from our citizen scientist volunteers.
Metabarcoding
DNA was extracted from individual guano pellets using 96-well plate format of the Qiagen DNeasy PowerSoil Kit (Qiagen, Hilden, Germany) following manufacturer guidelines. Samples were eluted with 60 µL of elution buffer and up to eight extraction blanks were included per 96-well plate. Arthropod COI gene fragments were targeted for amplification using primers detailed in Jusino et al.31. We modified the original primer sequences to preserve the COI-specific regions, but integrated linker, pad, adapter, and barcode sequences into the oligo following Kozich et al.49. We used 25 µL reactions with 10 µL of extracted DNA, 1 µL each of 10 mM forward and reverse primer pairs, and 13 µL of AccuStart II PCR SuperMix (Quanta BioSciences, Gaithersburg, MD, USA). Reaction conditions consisted of an initial 2 min denaturation at 95 °C, followed by 30 cycles of 20 s at 95 °C, 15 s at 50 °C, and 60 s at 72 °C and finally a 10 min extension at 72 °C. PCR products were quantified using a PicoGreen assay (Invitrogen, Carlsbad, CA, USA) with a Tecan plate reader using excitation and emission wavelengths of 480 nm and 520 nm, respectively (Tecan Group, Männedorf, Switzerland). Samples were pooled in approximately equimolar ratios. The initial pool volume was reduced with a vacuum concentrator to approximately 2 mL and was cleaned with a QIAquick PCR purification kit (Qiagen, Hilden, Germany); libraries were eluted in 30 µL elution buffer. Libraries were quantified with a Qubit High Sensitivity assay (Thermo Fisher Scientific, Waltham, MA, USA) and fragment sizes were analyzed using TapeStation D1000 ScreenTape (Agilent Technologies, Santa Clara, CA, USA).
Libraries containing samples from 2015 were sequenced at the Hubbard Center for Genome Studies at the University of New Hampshire on an Illumina HiSeq (Illumina, San Diego, CA, USA) using v2 chemistry with 500 cycles of 2 × 250 bp paired-end reads. Samples collected in 2016 were sequenced on a MiSeq machine at TGen North using v3 chemistry with 600 cycles of 2 × 300 bp paired-end reads. Raw sequence reads are available at NCBI BioProject PRJNA560640.
Sequence processing
Raw demultiplexed sequences were trimmed using Cutadapt v-2.350 and imported into QIIME 251. Sequences were denoised using DADA2 v1.10.052 with the QIIME 2 q2-dada2 function ‘qiime dada2 denoise-paired’ with default settings except for the additional parameters ‘--p-trunc-len-f 181’ and ‘--p-trunc-len-r 181’, resulting in a set of representative amplicon sequence variants (ASVs) for each library. Library-specific sequences and ASV tables were merged into a single dataset using ‘qiime feature-table merge’ and ‘qiime feature-table merge-seqs’ commands, respectively. The resulting study-wide collection of ASVs were further clustered at 98.5% identity using ‘vsearch --cluster_size’ with default parameters. Note that these sequence clusters, herein referred to as operational taxonomic units (OTUs), represent the clustering of exact sequence variants, rather than a conventional notion of an OTU that is often derived from clustering raw sequence data directly.
Biological mock community samples were sequenced in eight of nine libraries shared with New Hampshire guano samples; these mock data were used in a separate experiment32. Both positive and negative control samples were removed from the dataset, as were the ASVs matching the expected biological mock sequences. The GitHub repository for this project provides additional details regarding data processing (‘sequence_processing.md’) and sequence quality control processes (‘contamination_evaluation.md’) https://github.com/devonorourke/nhguano/blob/master/docs/.
Representative sequences were classified using a custom database curated with reference sequences and taxonomic information obtained from the Barcode of Life Data System (BOLD) database53. The development and use of this curated reference set was described previously54. Briefly, we first obtained the BOLD COI sequences using a custom R script that queried the BOLD API using the 'bold' R package55. Reference sequences were filtered for nucleotide ambiguity and homopolymer runs (no more than 5 degenerate bases per sequence or 12 homopolymers) with 'qiime rescript cull-seqs', as well as for length (min 250 bp, max 1600 bp) with 'qiime rescript filter-seqs-length'. The resulting sequences and their associated taxonomic identities were dereplicated by applying a Least Common Ancestor (LCA) method that gave preference to sequences represented most frequently with 'qiime rescript dereplicate --p-mode 'super' --p-derep-prefix'. Remaining sequences were trimmed to boundaries defined by our COI primer sequences by performing multiple sequence alignment of reference and primer sequences with MAFFT56. The remaining primer-trimmed, nucleotide quality and length-filtered references were further filtered for length and subsequent gaps removed with 'qiime rescript degap-seqs', retaining only reference sequences with a minimum length of 170 bp. Finally, these reference sequences were dereplicated a second time with the same LCA method used earlier with 'qiime rescript dereplicate'. The database consists of 739,345 unique COI reference sequences and taxonomic labels.
We used a hybrid approach in classifying representative sequences, prioritizing exact matches from VSEARCH first, then retaining Naive Bayes classifications with sufficient information. The naive Bayes classifier was trained using the RESCRIPt command 'qiime rescript evaluate-fit-classifier', producing the QIIME object required for Naive Bayes classification. We then classified the representative sequences using VSEARCH and naive Bayes approaches separately. For VSEARCH, we required 100% identity across 94% query coverage, with 'qiime feature-classifier classify-consensus-vsearch', while default parameters were used in the naive Bayes classification process with 'qiime feature-classifier classify-sklearn'.
All bat-associated sequences classified were identified separately with naive Bayes and VSEARCH, with both read abundances and sample occurrences. The three OTUs classified as a bat by VSEARCH were identical to the taxonomic labels assigned by naive Bayes classifications, though the naive Bayes classifier had three additional distinct labels of similar bat species. While we lacked visual confirmation of species identity for all sites, these data suggest our diet analyses are restricted to the little brown bat, as it was the only bat species from this region identified in our dataset.
For both VSEARCH and naive Bayes-classified OTUs, we separately filtered the dataset to retain only those OTUs with taxonomic family information assigned to phylum "Arthropoda". Thus, an OTU may be included that lacked genus or species labels, provided it retained an unambiguous family label. To select a final list of OTUs, we first retained VSEARCH-classified OTUs that fit these filtering criteria. Among VSEARCH-classified OTUs that did not pass this filtering threshold, we then selected from the naive Bayes-classified OTUs that met the same standard, when possible. In all, 559 OTUs were selected using the VSEARCH method initially, and 2,627 subsequently from the naive Bayes method. We manually resolved a single label that was undefined by either classification method, OTU-1, which was classified as M. castanea after using NCBI BLAST57 and discovering an exact match. Among all OTUs remaining across all samples, we next normalized samples using a method of scaling with ranked subsampling using the SRS R package58. We retained only those samples (and their OTUs) with a per-sample minimum of 1000 arthropod-classified reads for subsequent diversity analyses. These sequences were further used to generate a rooted tree using the function 'qiime phylogeny align-to-tree-mafft-fasttree', applied for all phylogenetic-based estimates (Faith’s phylogenetic diversity and unweighted UniFrac).
Diversity analyses
Diversity analyses were conducted among all guano samples to identify the most frequent taxa consumed by little brown bats throughout New Hampshire, as well as among particular subsets of samples to evaluate whether dietary richness and community composition varied among with particular collection sites or dates. Samples were grouped into 37-day sampling windows to maximize the greatest number of samples in the smallest range of time shared across the most locations that generated sufficient sequence data (Table S2). For example, window 3 was a sampling period from March 16 to April 22, window 4 was April 23 to May 29, and window 8 was September 17 through October 24. Windows with a shorter time period, say monthly, did not contain enough samples to robustly compare across the season or spatially. We applied the same methods for each spatial and/or temporal investigation. Samples were assessed for species richness using three metrics: observed OTUs, Shannon’s entropy, and Faith’s phylogenetic diversity. Kruskal–Wallis tests were then applied to evaluate group differences, and a post hoc Dunn’s test (with Benjamini–Hochberg correction) assessed the statistical significance of particular pairwise comparisons between groups. Community composition was calculated following a binary presence-absence transformation of sequence counts with Dice-Sorensen and Unweighted UniFrac distance measures. We evaluated differences in group medians and group dispersions with ‘adonis2’ and ‘betadisper’ functions in Vegan59, respectively. Principal coordinates analyses (PCoA) were completed with the ‘ordinate’ function in Phyloseq60 for each distance matrix. Indicator species analyses were completed with the R function ‘multipatt’ from the R ‘indicspecies’ package61, and restricted our analyses to those arthropod orders detected in at least 2% of samples in at least one group. Further descriptions of the bioinformatic steps, associated scripts, and related files for database construction, classification, and diversity analyses are available in the ‘diversity_analyses’.md’ document in the project GitHub repository (see Data Accessibility below).
Pest analysis
To identify whether taxa classified in our dataset were considered forest or agricultural pests, we cross referenced lists maintained by the U.S. Forest Service (USFS) and the U.S. Department of Agriculture (USDA). We used a custom R script, pest_work.R, to perform the comparisons, which were restricted to first identifying how many sequence variants were exact species matches, then expanding the search to identify instances in which common genera were shared. This was done because there were instances in which the USDA did listed taxa as ambiguous species (e.g., Malacosoma sp.) thus even if we classified our taxa to a species level, an exact match was not possible.
Additional software
Note that additional software within the QIIME environment such as Pandas62 and BIOM63 were used, as was a suite of R libraries including ape64, btools65, cowplot66, decontam67, FSA68, qiime2R69, ggmap70, ggpubr71, ggrepel72, indicspecies61, landscapemetrics73, lubridate74, magicfor75, Matrix76, multcompView77, pairwiseAdonis78, phyloseq60, raster79, reshape280, scales81, sf82, SRS58, svglite83, tidyverse84, urbnmapr85, usedist86, and vegan59.
Data availability
Data are provided as private-for-peer review (shared publicly). All scripts, supplementary figures and tables, and metadata is currently stored at the following GitHub repository: https://github.com/devonorourke/nhguano. Large individual database files used for classification are available in the following directory hosted by Open Science Framework: https://osf.io/d4jra/. Raw sequence reads are published and currently hosted at NCBI BioProject PRJNA560640. The GitHub repository is the intended location where data will be permanently archived if the paper is accepted for publication. This submission uses novel code, which is provided in an external repository to be evaluated during the peer review process. Please see the scripts directory of the project’s GitHub repository: https://github.com/devonorourke/nhguano/tree/master/scripts. Supplementary tables and figures referred to within the manuscript are available at: https://github.com/devonorourke/nhguano/tree/master/supplementaryData.
References
Whitaker, J. O., McCracken, G. F. & Siemers, B. M. Food habits analysis of insectivorous bats. in Ecological and Behavioral Methods for the Study of Bats. 567–592. (2011).
Clare, E. L., Barber, B. R., Sweeney, B. W., Hebert, P. D. N. & Fenton, M. B. Eating local: Influences of habitat on the diet of little brown bats (Myotis lucifugus). Mol. Ecol. 20(8), 1772–1780. https://doi.org/10.1111/j.1365-294X.2011.05040.x (2011).
Clare, E. L. et al. The diet of Myotis lucifugus across Canada: Assessing foraging quality and diet variability. Mol. Ecol. 23(15), 3618–3632. https://doi.org/10.1111/mec.12542 (2014).
Wray, A. K. et al. Predator preferences shape the diets of arthropodivorous bats more than quantitative local prey abundance. Mol. Ecol. https://doi.org/10.1111/mec.15769 (2020).
Agosta, S. J., Morton, D. & Kuhn, K. M. Feeding ecology of the bat Eptesicus fuscus: ‘Preferred’ prey abundance as one factor influencing prey selection and diet breadth. J. Zool. 260(2), 169–177. https://doi.org/10.1017/S0952836903003601 (2003).
Clare, E. L., Symondson, W. O. C. & Fenton, M. B. An inordinate fondness for beetles? Variation in seasonal dietary preferences of night-roosting big brown bats (Eptesicus fuscus). Mol. Ecol. 23(15), 3633–3647. https://doi.org/10.1111/mec.12519 (2014).
O’Rourke, D. R. et al. Lord of the Diptera (and moths and a spider): Molecular diet analyses and foraging ecology of Indiana bats in Illinois. Front. Ecol. Evol. 9, 12 (2021).
Hope, P. R. et al. Second generation sequencing and morphological faecal analysis reveal unexpected foraging behaviour by Myotis nattereri (Chiroptera, Vespertilionidae) in winter. Front. Zool. 11(1), 39. https://doi.org/10.1186/1742-9994-11-39 (2014).
Vesterinen, E. J., Puisto, A. I. E., Blomberg, A. S. & Lilley, T. M. Table for five, please: Dietary partitioning in boreal bats. Ecol. Evol. 8, 10914–10937 (2018).
Vesterinen, E. J. et al. What you need is what you eat? Prey selection by the bat Myotis daubentonii. Mol. Ecol. 25, 1581–1594 (2016).
Barclay, R. M. R. Population structure of temperate zone insectivorous bats in relation to foraging behaviour and energy demand. J. Anim. Ecol. 60(1), 165. https://doi.org/10.2307/5452 (1991).
Fraser, E. E. & Fenton, M. B. Age and food hardness affect food handling by insectivorous bats. Can. J. Zool. 85, 985–993 (2007).
von Frenckell, B. & Barclay, R. M. R. Bat activity over calm and turbulent water. Can. J. Zool. 65, 219–222 (1987).
Kaupas, L. A. & Barclay, R. M. R. Temperature-dependent consumption of spiders by little brown bats (Myotis lucifugus), but not northern long-eared bats (M. septentrionalis), in northern Canada. Can. J. Zool. 96(3), 261 (2018).
Alberdi, A., Aizpurua, O., Gilbert, M. T. P. & Bohmann, K. Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol. Evol. 9, 134–147 (2018).
Nielsen, J. M., Clare, E. L., Hayden, B., Brett, M. T. & Kratina, P. Diet tracing in ecology: Method comparison and selection. Methods Ecol. Evol. 9, 278–291 (2018).
Kunz, T. H. & Whitaker, J. O. An evaluation of fecal analysis for determining food habits of insectivorous bats. Can. J. Zool. 61, 1317–1321 (1983).
Hamilton, I. M. & Barclay, R. M. R. Diets of juvenile, yearling, and adult big brown bats (Eptesicus fuscus) in Southeastern Alberta. J. Mammal. 79(3), 764. https://doi.org/10.2307/1383087 (1998).
Moosman, P. R., Thomas, H. H. & Veilleux, J. P. Food habits of eastern small-footed bats (Myotis leibii) in New Hampshire. Am. Midl. Nat. 158(2), 354–360 (2007).
Ober, H. K. & Hayes, J. P. Prey selection by bats in forests of Western Oregon. J. Mammal. 89(5), 1191–1200. https://doi.org/10.1644/08-MAMM-A-025.1 (2008).
Long, B. L., Kurta, A. & Clemans, D. L. Analysis of DNA from feces to identify prey of big brown bats (Eptesicus fuscus) caught in apple orchards. Am. Midl. Nat. 170(2), 287–297 (2013).
Gordon, R. et al. Molecular diet analysis finds an insectivorous desert bat community dominated by resource sharing despite diverse echolocation and foraging strategies. Ecol. Evol. 9, 3117–3129 (2019).
Alberdi, A. et al. Promises and pitfalls of using high-throughput sequencing for diet analysis. Mol. Ecol. Resour. 19, 327–348 (2019).
Clare, E. L. Molecular detection of trophic interactions: Emerging trends, distinct advantages, significant considerations and conservation applications. Evol. Appl. 7, 1144–1157 (2014).
Blehert, D. S. et al. Bat white-nose syndrome: An emerging fungal pathogen?. Science 323(5911), 227–227. https://doi.org/10.1126/science.1163874 (2009).
Frick, W. F. et al. Disease alters macroecological patterns of North American bats: Disease alters macroecology of bats. Glob. Ecol. Biogeogr. 24(7), 741–749. https://doi.org/10.1111/geb.12290 (2015).
Hallmann, C. A. et al. More than 75 percent decline over 27 years in total flying insect biomass in protected areas. PLoS ONE 12, e0185809 (2017).
Sánchez-Bayo, F. & Wyckhuys, K. A. G. Worldwide decline of the entomofauna: A review of its drivers. Biol. Conserv. 232, 8–27 (2019).
Anthony, E. L. P. & Kunz, T. H. Feeding strategies of the little brown bat, Myotis lucifugus, Southern New Hampshire. Ecology 58(4), 775–786. https://doi.org/10.2307/1936213 (1977).
Pompanon, F. et al. Who is eating what: diet assessment using next generation sequencing. Mol. Ecol. 21, 1931–1950 (2012).
Jusino, M. A. et al. An improved method for utilizing high-throughput amplicon sequencing to determine the diets of insectivorous animals. Mol. Ecol. Resour. 19, 176–190 (2019).
O’Rourke, D. R., Bokulich, N. A., Jusino, M. A., MacManes, M. D., & Foster, J. T. A total crapshoot? Evaluating bioinformatic decisions in animal diet metabarcoding analyses. Ecol. Evolut. https://doi.org/10.1002/ece3.6594 (2020).
Langwig, K. E. et al. Resistance in persisting bat populations after white-nose syndrome invasion. Philos. Trans. R. Soc. B Biol. Sci. 372, 2160044 (2017).
Maslo, B., Valent, M., Gumbs, J. F. & Frick, W. F. Conservation implications of ameliorating survival of little brown bats with white-nose syndrome. Ecol. Appl. 25, 1832–1840 (2015).
Frick, W. F. et al. An emerging disease causes regional population collapse of a common North American bat species. Science 329(5992), 679–682. https://doi.org/10.1126/science.1188594 (2010).
Turner, G. G., Reeder, D. M. & Coleman, J. T. H. A five-year assessment of mortality and geographic spread of white-nose syndrome in North American bats and a look to the future. Bat Res. News 52, 13–27 (2011).
Coleman, J. et al. A National Plan for Assisting States, Federal Agencies, and Tribes in Managing White-Nose Syndrome in Bats. https://s3.us-west-2.amazonaws.com/prod-is-cms-assets/wns/prod/b0634260-77d3-11e8-b37b-4f3513704a5e-white-nose_syndrome_national_plan_may_2011.pdf (2011).
Szymanski, J. A., Runge, M. C., Parkin, M. J. & Armstrong, M. White-Nose Syndrome Management: Report on Structured Decision Making Initiative. Vol. 51. http://pubs.er.usgs.gov/publication/70003465 (2009).
Kunz, T. H., Braun de Torrez, E., Bauer, D., Lobova, T. & Fleming, T. H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 1223, 1–38 (2011).
Boyles, J. G., Cryan, P. M., McCracken, G. F. & Kunz, T. H. Economic importance of bats in agriculture. Science 332(6025), 41–42. https://doi.org/10.1126/science.1201366 (2011).
Agosta, S. J. & Morton, D. Diet of the big brown bat, Eptesicus fuscus, from Pennsylvania and Western Maryland. Northeast. Nat. 10(1), 89–104 (2003).
Brown, V. A., Braun de Torrez, E. & McCracken, G. F. Crop pests eaten by bats in organic pecan orchards. Crop Prot. 67, 66–71 (2015).
Williams-Guillén, K., Perfecto, I. & Vandermeer, J. Bats limit insects in a Neotropical agroforestry system. Science 320(5872), 70–70. https://doi.org/10.1126/science.1152944 (2008).
Held, D. W. & Ray, C. H. Asiatic garden beetle Maladera castanea (Coleoptera: Scarabaeidae) grubs found in damaged turf in Alabama. Fla. Entomol. 92(4), 670–672 (2009).
Forschler, B. T. & Gardner, W. A. A review of the scientific literature on the biology and distribution of the genus Phyllophaga (Coleoptera: Scarabaeidae) in the Southeastern United States. J. Entomol. Sci. 25(4), 628–651. https://doi.org/10.18474/0749-8004-25.4.628 (1990).
United States Forest Service. White Grubs in Forest Tree Nurseries and Plantations. Vol. 4. https://www.fs.usda.gov/Internet/FSE_DOCUMENTS/fsbdev2_043588.pdf (1961).
Chandler, D. University of New Hampshire—Entomology Collection. UNH Insect and Arachnid Collections. https://duncan.unh.edu/ento/home.php (2020).
United States Forest Service. The Early Warning System for Forest Health Threads in the United States. https://www.fs.fed.us/foresthealth/publications/EWS_final_draft.pdf (2004).
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., & Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79(17), 5112–5120. https://doi.org/10.1128/AEM.01043-13 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Ratnasingham, S. & Hebert, P. D. N. bold: The barcode of life data system. http://www.barcodinglife.org. Mol. Ecol. Notes 7, 355–364 (2007).
Robeson, M. S. et al. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv. https://doi.org/10.1101/2020.10.05.326504 (2020).
Chamberlain, S. BOLD: Interface to BOLD Systems API. (2017).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30(4), 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Beule, L. & Karlovsky, P. Improved normalization of species count data in ecology by scaling with ranked subsampling (SRS): application to microbial communities. PeerJ 8, e9593 (2020).
Oksanen, J. et al. vegan: Community Ecology Package. (2018).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8(4), e61217. https://doi.org/10.1371/journal.pone.0061217 (2013).
Cáceres, M. D. & Legendre, P. Associations between species and groups of sites: indices and statistical inference. Ecology 90, 3566–3574 (2009).
McKinney, W. Data structures for statistical computing in Python. Proc. Python Sci. Conf. https://doi.org/10.25080/Majora-92bf1922-00a (2010).
McDonald, D. et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 1, 7 (2012).
Paradis, E., Claude, J. & Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 (2004).
Battaglia, T. btools: A Suite of R Function for All Types of Microbial Diversity Analyses. (2020).
Wilke, C. O. cowplot: Streamlined Plot Theme and Plot Annotations for ‘ggplot2’. (2017).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226 (2018).
Ogle, D. H. & Wheeler, P. FSA: Fisheries Stock Analysis. (2018).
Bisanz, J. E. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions. (2018).
Kahle, D. & Wickham, H. ggmap: Spatial visualization with ggplot2. R J. 5, 144–161 (2013).
Kassambara, A. ggpubr: ‘ggplot2’ Based Publication Ready Plots. (2018).
Slowikowski, K. ggrepel: Automatically Position Non-Overlapping Text Labels with ‘ggplot2’. (2018).
Hesselbarth, M. H. K., Sciaini, M., With, K. A., Wiegand, K. & Nowosad, J. landscapemetrics: an open-source R tool to calculate landscape metrics. Ecography 42, 1648–1657 (2019).
Grolemund, G., & Wickham, H. Dates and times made easy with lubridate. J. Stat. Softw. 40(3). https://www.jstatsoft.org/index.php/jss/article/view/v040i03/v40i03.pdf (2011).
Makiyama, K. magicfor: Magic Functions to Obtain Results from for Loops. (2016).
Bates, D. & Maechler, M. Matrix: Sparse and Dense Matrix Classes and Methods. (2018).
Graves, S., Piepho, H.-P. & Selzer, L. multcompView: Visualizations of Paired Comparisons. (2019).
Martinez Arbizu, P. pairwiseAdonis: Pairwise Multilevel Comparison using Adonis. (2017).
Hijmans, R. J. raster: Geographic Data Analysis and Modeling. (2020).
Wickham, H. Reshaping data with the reshape Package. J. Stat. Softw. 21(1), 1–20. https://doi.org/10.18637/jss.v021.i12 (2007).
Wickham, H. scales: Scale Functions for Visualization. (2018).
Pebesma, E. Simple features for R: Standardized support for spatial vector data. R J. 10, 439 (2018).
Wickham, H. et al. svglite: An ‘SVG’ Graphics Device. (2020).
Wickham, H. tidyverse: Easily Install and Load the ‘Tidyverse’. (2017).
Strochak, S., Ueyama, K. & Williams, A. urbnmapr: State and County Shapefiles in sf and Tibble Format. (2020).
Bittinger, K. usedist: Distance Matrix Utilities. (2020).
Acknowledgements
The project would without the diligent citizen scientists collecting guano throughout 2015 and 2016. Thank you to all the citizen scientists for their guano collection efforts, without which this project could not have been completed: Carol Drummond, Pete Smith and Celeste Barr, Hilary Nelson, Sarah Boyden, David and Shelley Brooks, Reegan Spinney, Anya Kattef, Charles and Jeanne Hawthorne, Katherine Heck, Barbara Kahn, Leslee Glidden, Catherine Skove, Inge Seaboyer, Katherine York, Kay Cushman, Hilary Nelson, William Schultz, Dave Erler, Amy O'Brien, and Elizabeth Smith. In addition, we thank Cynthia Nichols and Emily Preston connecting us with their volunteer network. This study was made possible thanks to funding by the New Hampshire Agricultural Experiment Station at the University of New Hampshire as part of USDA National Institute of Food and Agriculture McIntire-Stennis Project NH00080-M.
Author information
Authors and Affiliations
Contributions
D.R.O. and J.T.F. conceived the study. D.R.O., N.P.R., and K.L.P. acquired the data. D.R.O. wrote the manuscript and performed the analyses. All authors read, edited, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
O’Rourke, D., Rouillard, N.P., Parise, K.L. et al. Spatial and temporal variation in New Hampshire bat diets. Sci Rep 12, 14334 (2022). https://doi.org/10.1038/s41598-022-17631-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17631-z
This article is cited by
-
DNA metabarcoding reveals diet composition of invasive rats and mice in Hawaiian forests
Biological Invasions (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
