
Abstract
Hydrophilic interaction liquid chromatography (HILIC) has inherent merits over RP-HPLC in the analyzing of hydrophilic substances. Accordingly, an innovative HILIC-UV methodology is proposed for the simultaneous estimation of ethyl paraben (PRN), fluconazole (FLZ) and moxifloxacin hydrochloride (MOX) in raw materials and pharmaceutical eye gel. The separation process was conducted using Waters XBridge™ HILIC column (100 mm × 4.6 mm, 3.5 μm particle size) at room temperature. Isocratic mobile phase containing acetonitrile: 0.1% triethylamine buffer (90:10, v/v, pH 5.0), was pumped at flow rate 1.0 mL/min and detected at 260 nm. Under these optimized conditions, PRN, FLZ and MOX showed rectilinear relationships with the concentration ranges (0.5–6.0), (5.0–50.0) and (5.0–60.0) μg/mL, respectively. The developed method offered at least fivefold increase in sensitivity within shorter time than the reported methods. Three greenness assessment tools namely: Analytical eco-scale, GAPI and AGREE were exploited to investigate the method's impact on the environment and conduct a comparative study with the reported methods. International council of Harmonization (ICH) guidelines have been followed to calculate validation parameters. The statistical comparison between results of the suggested method and the comparison method showed no discrepancy confirming accuracy of the method.
Similar content being viewed by others


Introduction
Microbial keratitis is a common complication of ocular surface infections in which layers of the cornea are inflamed due to bacterial, viral or fungal invasion to the underlying layers through the spaces that occurred in the corneal surface. This leads to tissue necrosis, corneal inflammation and eventually destruction. It was reported that fungal keratitis can be more pernicious and destructive compared to that caused by bacteria. Fluoroquinolones are described as first-line therapy for keratitis; in addition to anti-fungal medications in suspicion of fungal invasion1,2. Topical eye gel combining fluconazole and moxifloxacin hydrochloride is preferred as a single dosage form in a such case3.
Fluconazole (FLZ) (Fig. 1a) chemically named as 2-(2,4-difluorophenyl)-1,3-bis(1H-1,2,4-triazol-1-yl) propan-2-ol4, is an antifungal with a triazole ring. It is described in treating fungal infections by stopping cell growth through inhibiting ergosterol synthesis in fungal cell membrane5.It is used to treat candidiasis, particularly vaginal, oropharyngeal and esophageal candidiasis. Also, it is used as a prophylactic therapy against candidiasis infection in bone marrow transplantation6.

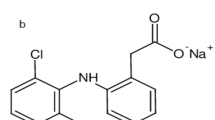
Structural formulae of: (a) FLZ. (b) MOX. (c) PRN.
Literature survey revealed different methods in FLZ determination including but not limited to: spectroflourimetry7, spectrophotometry8,9, HPLC10,11 and voltammetry12.
Moxifloxacin hydrochloride (MOX) (Fig. 1b) is chemically named as 1-cyclopropyl-6-fluoro-8-methoxy-7-[(4aS,7aS)-octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl]-4-oxo-1,4dihydroquinoline-3-carboxylic acid hydrochloride4. It is a broad spectrum fluoroquinolone with anti-bacterial activity. Its pharmacological action arises from inhibiting enzymes responsible for bacterial DNA synthesis and replication13. It has an excellent activity against gram negative bacteria in ocular infections14. It is described in treating acquired pneumonia, acute bacterial sinusitis and bacterial keratitis2,15
Variable methodologies have been described for determining MOX including: spectroflourimetry16,17, spectrophotometry18, HPLC19,20 and voltammetry21.
The assayed eye gel contains ethyl paraben (PRN) (Fig. 1c) as a preservative. It is chemically named as ethyl 4-hydroxybenzoate4.
To the best of our knowledge, two HPLC methods have been published for the simultaneous estimation of the eye gel components3,22. But, these methods have a low sensitivity with a long-time separation process. The aim of the developed study is to suggest a novel proposal for the implementation of hydrophilic interaction liquid chromatography (HILIC) technique for the simultaneous quantification of PRN, FLZ and MOX in their raw materials and laboratory-prepared pharmaceutical eye gel. Also, to compare the performance of the proposed method with the reported methods3,22.
Recently, HILIC has witnessed popularity and attention in separating and estimating food components and toxic contaminants, especially polar and ionized compounds. In contrast with RP-HPLC, both stationary phase (SP) and mobile phase (MP) in HILIC are polar. Also, the elution strength of the solvents is reversed, water is the strongest solvent in HILIC mode. SP in HILIC has been classified into different classes according to the charge of the functional groups of SP. This classification includes neutral, charged and zwitterion charged types. Ideal MP composition should contain at least 3% water and at least 60% organic solvent mostly acetonitrile (ACN). This high ratio of ACN lessened the overall viscosity of the used MP, which in turn decreases the operating pressure, allowing applying higher flow rate. In HILIC, elution order is related to the hydrophilicity of the analyte, the most hydrophilic component is the last eluted. Separation mechanisms suggested for HILIC are complex and contribute to various degrees. These include: partitioning, adsorption, ion exchange (IE) and hydrophobic retention. The separation mechanism can count on several factors concerned with physicochemical properties of SP, hydro-organic nature of MP and the structure of the assayed components23,24. Thus, a critical concern was introduced to optimize the separation conditions and to increase the selectivity of the SP toward the studied drugs.
Novelty of the suggested method originates from applying HILIC technique for the determination of the cited drugs which were only studied utilizing the traditional chromatographic techniques. It excelled over the reported methods in accomplishing the separation process in much less time and having a relatively high sensitivity with reasonable peak shapes. Applying the HILIC method allows overcoming the encountered limitations in the reported RP-HPLC methods.
Experimental
Apparatus
-
Shimadzu Prominence HPLC system (Shimadzu Corp., Kyoto, Japan) with an LC-20 AD pump and SPD-20A UV–Vis detector was used.
-
Waters XBridge™ HILIC column (100 mm × 4.6 mm, 3.5 μm particle size) (Ireland) was utilized to perform the proposed study. Mobile phase was degassed using Merck L-7612 solvent degasser.
-
Adjusting pH through the work was performed by Consort NV P-901 pH Meter (Belgium).
-
0.45 μm membrane filter (Millipore, Ireland) was utilized to filter MP.
Materials and reagents
-
FLZ was kindly obtained from Amoun Pharmaceutical Co. El-Obour City, Cairo, Egypt.
-
MOX was kindly provided by Al-Andalous Medical company, Second 6th of October, Giza, Egypt.
-
PRN and sodium chloride (analytical grade) were purchased from El-Nasr Pharmaceutical Chemicals Company, Egypt.
-
Poloxamer 407 (used as gelling agent), organic solvents (HPLC grade) and triethylamine (greater than or equal to 99.5%) were purchased from Sigma-Aldrich, Germany.
-
Orthophosphoric acid (85%, w/v) was acquired from RiedeldeHäen, Seelze, Germany.
Standard solutions
Stock standard solutions of FLZ and MOX were prepared separately as 200.0 μg/mL and as 100.0 μg/mL for PRN. The solutions were prepared in ACN for both FLZ and PRN and in methanol for MOX. Working solution of 50.0 μg/mL were prepared for PRN by appropriate dilution. Completing the stock solutions was aimed to be the same components of MP (ACN), but due to the lower solubility of MOX in ACN4, methanol was used instead. The solutions were all stable upon storing at 4 °C for 7 days.
Chromatographic conditions used during the separation procedure
Waters XBridge™ HILIC column (100 mm × 4.6 mm, 3.5 μm particle size) was used throughout the work. The wavelength was adjusted at 260 nm using a flow rate of 1.0 mL/min. MP composition was ACN: 0.1% triethylamine buffer (TEA) in ratio of (90:10, v/v), respectively. The pH was adjusted at 5.0 using 0.2 M orthophosphoric acid then filtered using a 0.45 μm membrane filter. The separation was performed at room temperature.
General procedure
Procedures for calibration curves
Different aliquots from stock standard solutions of FLZ and MOX and from working solution of PRN were separately transferred into 3 sets of 10 mL volumetric flasks covering concentration range of (5.0–50.0), (5.0–60.0) and (0.5–6.0) μg/mL for FLZ, MOX and PRN, respectively. The flasks were completed to the final mark with the MP. Under the optimum chromatographic conditions, 20 µl of each were injected as triplicates. The average peak area was plotted versus the corresponding concentration to get the concentration curves and to compute the corresponding regression equations.
Analysis of lab-synthetic mixtures
Into a series of 10 mL volumetric flasks, different aliquots of FLZ and MOX stock standard solutions and PRN working solution were transferred keeping the pharmaceutical ratio of (1:1:0.1), respectively3. The procedure for calibration curves was then adopted and the % recoveries were calculated utilizing the corresponding regression equations.
Analysis of laboratory-prepared co-formulated gel
The eye gel contains PRN, FLZ and MOX in a ratio of (0.1:1:1)3, so this ratio was followed to prepare the laboratory formulation by mixing 1.0, 10.0 and 10.0 mg for PRN, FLZ and MOX, respectively with the gel additives which were 2.4 gm sodium chloride and 15.5 gm poloxamer 407 all in 100 mL volumetric flask using ACN as a solvent. MOX was firstly dissolved in about 5.0 mL of methanol then mixed with other components. The flask was sonicated for 45 min and filtered using double Whatman® filter paper. Different concentrations were analyzed and the corresponding regression equations were utilized to compute percentage recoveries.
Results and discussion
HILIC is a novel variant for RP-HPLC technique in analyzing polar compounds23. Comprising high percent of the organic solvent (ACN) in MP decreases its viscosity and paves HILIC to be a technique of choice for separation of hydrophilic and polar components24. The chromatographic conditions of the developed HILIC method were investigated and optimized to obtain the best performance in a reasonable time. The order of separation of the studied components was found to be PRN, FLZ and MOX, respectively as shown in Fig. 2. Table 1 shows the optimum chromatographic parameters obtained for the separation of the studied drugs by the proposed method.

Typical chromatogram of laboratory prepared mixture of the studied components under the described chromatographic conditions. (a) PRN (0.5 μg/mL), (b) FLZ (5.0 μg/mL) and (c) MOX (5.0 μg/mL).
Optimization of critical parameters
Choice of appropriate wavelength
Upon recording the UV spectrum of PRN, FLZ and MOX, it was found that PRN, FLZ, and MOX have absorption maxima at 255, 260 and 295 nm, respectively. Due to the lower sensitivity of FLZ, 260 nm was selected as the most suitable wavelength for scanning the cited components (Fig. 3).

UV spectrum of: (a) PRN (5.0 μg/mL), (b) FLZ (20.0 μg/mL) and (c) MOX (5.0 μg/mL).
Mobile phase composition
An ideal MP composition in HILIC should composed of water-miscible polar organic solvents like ACN; Methanol is rarely used as water–methanol mixture is limited to induce retention to the stationary phase25,26,27. Choice of ACN is hypothesized to its miscibility with water and absence of hydrogen bond forming groups, diminishing its competition with water to solvate the surface of SP. Various modifications have been tried to achieve the best results for the three studied drugs. By trying ACN-water, MOX peak was eluted with high tailing. Replacing water by ammonium acetate buffer in different concentrations (5.0 and 10.0) mM, quietly decreased peak's tailing but still had a relatively high value. TEA was tried instead of ammonium acetate buffer and achieved the best result concerning MOX peak's tailing factor. Subsequently, the concentration of TEA was studied. Increasing the concentration higher than 0.1% didn't significantly improve the results, so 0.1% TEA was chosen as a suitable concentration.
pH of the mobile phase
Several pH values of the mobile phase (3.0–8.0) were studied, where pH 3.0 showed the least well-resolved peaks. Almost constant results were obtained in pH values between (4.0–6.0) with the best values according to tailing factor (Tf), resolution (Rs), and number of theoretical plates (NTP). pH values over than 6.0 showed high Tf for MOX peak and weren't preferred for column integrity, so pH 5.0 is chosen as a suitable pH. The results are summarized in Table S1.
Ratio of organic modifier
High percent of ACN in HILIC MP is required to induce a reasonable retention time23. Different ratios were tried starting from 80% ACN up to 97%. FLZ and PRN were eluted as one peak at 80% ACN. Weak resolution was observed at 85% ACN. Better resolution was achieved at 90%, further increasing showed no significant enhancement in chromatographic suitability parameters, so 90% ACN was selected as the most suitable ratio (Table S1).
Flow rate and column temperature
In this study, 0.8, 1.0 and 1.2 mL/min were investigated. Trying 0.8 mL/min increased the separation time, while 1.2 mL/min decreased Rs between PRN and FLZ. Accordingly, 1.0 mL/min was chosen as the best flow rate (Table S1). Different temperatures were tried including room temperature, 30 & 40 °C, increasing the temperature didn’t improve the results, so the separation was performed at room temperature.
Separation mechanism
Retention in HILIC is strongly related to the composition of MP. Incorporation of > 5% water in MP thickens the adsorbed aqueous layer on the SP and creates liquid-liquid partitioning system. Upon utilizing lower water percentage, the stagnant aqueous layer becomes thinner. This in turn downsides partitioning mechanism and allows direct interaction between the analyte and SP. In the suggested system, separation mechanism can be attributed to partitioning of the drugs between the stagnant aqueous layer on the ethylene bridged hybrid (BEH) SP and organic solvent-rich MP, and elution of the drugs according to their lipophilicity. Log P values for PRN, FLZ and MOX are 2.50, 1.0 and 0.01, respectively28,29. It explains the early elution of PRN and the late elution of MOX. Another explanation is polar surface area values which were 46.5, 81.6 and 82.1 Å2 for PRN, FLZ and MOX, respectively28. The highest polarity value for MOX explains its long retention on the polar SP and the lowest value for PRN explains its early elution.
Method validation
The suggested method was validated according to ICH guidelines30.
Under the prescribed chromatographic conditions, a good linearity was obtained by plotting average peak area against concentration for PRN, FLZ and MOX within concentration range (0.5–6.0), (5.0–50.0) and (5.0–60.0) μg/mL, respectively. The performance of the chromatographic method is listed in Table 2.
Intra- and inter-day precisions were investigated by analyzing three concentration levels of each component three times within the same day and within three consecutive days, respectively. Satisfactory S.D, %RSD and %Error values expressed method's precision as shown in Table S2.
Using of the intercept of the standard deviation and slope values, limit of detection (LOD) and limit of quantification (LOQ) were calculated following ICH mathematical equations30.
LOD was 0.14, 0.94, and 1.38 μg/mL for PRN, FLZ and MOX, respectively, while LOQ was 0.42, 2.84 and 4.19 μg/mL for PRN, FLZ and MOX, respectively.
Three concentration levels of each component were tested in their raw materials adopting the reported method22. The reported method includes HPLC determination of the drugs using ODS-C18 column and mobile phase containing TEA: methanol at (58:42, v/v, pH 3.2), respectively, where pH was adjusted with phosphoric acid. The wavelength was selected at 260 nm using 1.0 mL/min flow rate. The results of the suggested and the reported methods were statistically compared using Student's t-and variance ratio F-tests. The comparison showed that the calculated t and F values were lower than tabulated values assuring the method is accurate (Table 3). Specificity of the method was assessed through quantifying different concentrations of the studied components in the laboratory-prepared eye gel. The acceptable obtained %recoveries (Table 4) reflect absence of interference that may occur from the excipients in the formulation (poloxamer and NaCl). System suitability parameters were calculated to check the competence and feasibility of the developed methodology in terms of NTP, Tf and Rs. The results shown in Table 1 points out that, the values of the suggested method were in acceptance with USP criteria revealing efficiency of the evolved approach [The United States Pharmacopoeia 40 and National Formulary 35. US Pharmacopoeial Convention, Rockville. 2017].
Analysis of different matrices
The proposed method showed eligibility in the simultaneous determination of the studied components in different concentrations of their synthetic mixtures with accepted percentage recoveries, the results are shown in Table S3. Moreover, estimation of the studied components in the laboratory-prepared co-formulated gel by performing the developed methodology showed reasonable percentage recoveries. The reported method22 was also adopted to quantify the studied components in the laboratory prepared co-formulated gel. The results of the developed method were compared statistically with the reported one22 and showed no significant difference proving method's accuracy (Table 4)31.
Greenness evaluation and comparative study
Serious concern was raised toward method's impact on the environment, especially when such large volume of ACN was used. Studying the eco-friendly profile was performed using three assessment tools to study the all insights for the method’s greenness. These tools include: Analytical eco-scale, Green Analytical Procedure Index (GAPI) and Analytical GREEnness (AGREE). Analytical eco-scale is concerned with volume and hazard of both the used solvents and the resulted waste in addition to instrument's energy. Each parameter is given penalty points and the total score is subtracted from 100 (the ideal score)32. GABI is a more descriptive tool which encompasses almost all principles of green analytical chemistry (GAC). A pictogram with five pentagrams, colored with green, yellow or red reflecting good, intermediate or bad impact on the environment33. The recent AGREE tool is an easily-used software calculator for evaluating the greenness property. It is classified into 12 sections relying on GAC principles. It provides both qualitative and quantitative data. Each section colored with either green, yellow or red reflects the ecological impact, with a total score in the middle from 0 to 1 (1 is the greenest)34.
A comparison of the eco-friendly property between the suggested HILIC and the reported HPLC methods3,22, was performed using the aforementioned tools. The greenness profile comparison shown in Table S4 and Fig. 4 elucidate superiority of the proposed HILIC method over the reported methods.

Upon comparing the results of the suggested approach with the reported methods, it was found that the suggested method offers either a fivefold or a 20-fold increase in sensitivity more than the reported methods22 and 3, respectively. Moreover, it accomplishes the separation process within four minutes, much less time than 20 min in the reported methods. This decrease in the separation time offers consumption of fewer quantities of organic solvents, adding time and cost saving properties to the developed method, also producing less waste that helped in saving the environment. The developed method provides a greenness evaluation using three different metric tools which wasn't studied in the reported methods. As a consequence, applying the HILIC method allows overcoming the encountered limitations in the reported RP-HPLC methods.
Conclusion
Keeping up with the modern hyphenated analytical separation methods, evoked us to adopt the HILIC technique in our work. The developed methodology allowed the simulatenous estimation of PRN, FLZ and MOX in raw material and laboratory-prepared eye gel. Chromatographic conditions were optimized to get the best separation conditions. A comprehensive comparative study was performed between the suggested and the comparison methods' greenness profile using Analytical eco-scale, GAPI and AGREE assessment tools. Acceptable percentage recoveries were obtained from analyzing different concentrations of synthetic mixtures and laboratory-prepared eye gel proving method’s selectivity and specificity. In a nutshell, the proposed HILIC method's features make it a suitable candidate for quality control labs.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request (Eman Yosrey).
References
Ansari, Z., Miller, D. & Galor, A. Current thoughts in fungal keratitis: Diagnosis and treatment. Curr. Fungal Infect. Rep. 7(3), 209–218 (2013).
Ong, H. S. & Dart, J. K. Managing ocular surface disease: A common-sense approach. Community Eye Health 29(95), 44 (2016).
Wu, M. et al. Preparation and quality control of fluconazole/moxifloxacin in situ forming eye gel. China Pharm. 66, 17 (2012).
Pharmacopoeia B. London: Her Majesty's Stationary Office; 2009. Electronic version. 2003.
Manzouri, B., Wyse, R. K. & Vafidis, G. C. Pharmacotherapy of fungal eye infections. Expert Opin. Pharmacother. 2(11), 1849–1857 (2001).
Govindarajan, A., Bistas, K. G. & Aboeed, A. Fluconazole. In StatPearls [Internet] (StatPearls Publishing, Treasure Island (FL), 2020).
El-Bayoumi, A., El-Shanawany, A., El-Sadek, M. & El-Sattar, A. A. Synchronous spectrofluorimetric determination of famotidine, fluconazole and ketoconazole in bulk powder and in pharmaceutical dosage forms. Spectrosc. Lett. 30(1), 25–46 (1997).
Göğer, N. G. & Aboul-Enein, H. Y. Quantitative determination of fluconazole in capsules and IV solutions by UV spectrophotometric methods. Anal. Lett. 34(12), 2089–2098 (2001).
Pandey, S., Pandey, P., Dubey, S., Chaturvedi, U. & Rai, A. K. Facile derivative UV spectroscopy method: simultaneous estimation of tinidazole and fluconazole in combined tablet dosage form. Thai. J. Pharm. Sci. 36, 55–62 (2012).
Aloudah, N. M., Radwan, M. A., Al Omar, N. F. & Jacobs, S. HPLC assay of fluconazole and its application to patients with early septic shock. J. Liq. Chromatogr. Relat. Technol. 28(4), 571–580 (2005).
Roshdy, A., Elmansi, H., Shalan, S. & El-Brashy, A. Factorial design-assisted reversed phase-high performance liquid chromatography method for simultaneous determination of fluconazole, itraconazole and terbinafine. R. Soc. Open Sci. 8(2), 202130 (2021).
Gil, Éd. S., Cordeiro, D. D., Matias, A. E. & Serrano, S. H. Electrochemical behavior and determination of fluconazole. J. Braz. Chem. Soc. 22(4), 767–71 (2011).
Ezelarab, H. A., Abbas, S. H., Hassan, H. A. & Abuo-Rahma, G. E. D. A. Recent updates of fluoroquinolones as antibacterial agents. Archiv der Pharmazie 351(9), 1800141 (2018).
McDonald, M. & Blondeau, J. M. Emerging antibiotic resistance in ocular infections and the role of fluoroquinolones. J. Cataract Refract. Surg. 36(9), 1588–1598 (2010).
Keating, G. M. & Scott, L. J. Moxifloxacin. Drugs 64(20), 2347–2377 (2004).
Khan, M. N., Ali, W., Shah, Z., Idrees, M. & Gulab, H. A validated spectrofluorimetric method for the determination of moxifloxacin in its pure form, pharmaceutical preparations and biological samples. Anal. Sci. 19, 19P370 (2019).
Ocaña, J. A., Barragán, F. J. & Callejón, M. Spectrofluorimetric determination of moxifloxacin in tablets, human urine and serum. Analyst 125(12), 2322–2325 (2000).
Elbashir, A. A., Ebraheem, S. A., Elwagee, A. H. & Aboul-Enein, H. Y. New spectrophotometric methods for the determination of moxifloxacin. Acta Chim. Slov. 60(1), 159–165 (2013).
Czyrski, A., Sokół, A. & Szałek, E. HPLC method for determination of moxifloxacin in plasma and its application in pharmacokinetic analysis. J. Liq. Chromatogr. Relat. Technol. 40(1), 8–12 (2017).
Ibrahim, F. A., Elmansi, H. & Fathy, M. E. Green RP-HPLC method for simultaneous determination of moxifloxacin combinations: investigation of the greenness for the proposed method. Microchem. J. 148, 151–161 (2019).
Erk, N. Voltammetric behaviour and determination of moxifloxacin in pharmaceutical products and human plasma. Anal. Bioanal. Chem. 378(5), 1351–1356 (2004).
Wu, M. et al. Determination of fluconazole and moxifloxacin in situ forming of eye gel by HPLC. Chin. J. Mod. Appl. Pharm. 2, 266 (2012).
Buszewski, B. & Noga, S. Hydrophilic interaction liquid chromatography (HILIC)—A powerful separation technique. Anal. Bioanal. Chem. 402(1), 231–247 (2012).
McCalley, D. V. Is hydrophilic interaction chromatography with silica columns a viable alternative to reversed-phase liquid chromatography for the analysis of ionisable compounds?. J. Chromatogr. A 1171(1–2), 46–55 (2007).
Ali, M. S., Ghori, M., Rafiuddin, S. & Khatri, A. R. A new hydrophilic interaction liquid chromatographic (HILIC) procedure for the simultaneous determination of pseudoephedrine hydrochloride (PSH), diphenhydramine hydrochloride (DPH) and dextromethorphan hydrobromide (DXH) in cough-cold formulations. J. Pharm. Biomed. Anal. 43(1), 158–167 (2007).
Louw, S., Pereira, A. S., Lynen, F., Hanna-Brown, M. & Sandra, P. Serial coupling of reversed-phase and hydrophilic interaction liquid chromatography to broaden the elution window for the analysis of pharmaceutical compounds. J. Chromatogr. A 1208(1–2), 90–94 (2008).
Xu, Y., Xie, W., Miller-Stein, C. M. & Woolf, E. J. Hydrophilic interaction chromatography/tandem mass spectrometry for the simultaneous determination of three polar non-structurally related compounds, imipenem, cilastatin and an investigational β-lactamase inhibitor, MK-4698, in biological matrices. Rapid Commun. Mass Spectrom. 23(14), 2195–2205 (2009).
Wishart, D. S. et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 46(D1), D1074–D82 (2018).
Moffat, A. C., Osselton, M. D., Widdop, B. & Watts, J. Clarke’s Analysis of Drugs and Poisons (Pharmaceutical Press, 2020).
Guideline, I. H. T. Validation of analytical procedures: Text and methodology. Q2 (R1) 1(20), 05 (2005).
Miller, J. & Miller, J. C. Statistics and Chemometrics for Analytical Chemistry (Pearson Education, 2018).
Gałuszka, A., Migaszewski, Z. M., Konieczka, P. & Namieśnik, J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends Anal. Chem. 37, 61–72 (2012).
Płotka-Wasylka, J. A new tool for the evaluation of the analytical procedure: Green Analytical Procedure Index. Talanta 181, 204–209 (2018).
Pena-Pereira, F., Wojnowski, W. & Tobiszewski, M. AGREE—Analytical GREEnness metric approach and software. Anal. Chem. 92(14), 10076–10082 (2020).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
E.Y. carried out the lab work and statistical analysis. H.E., Z.A.S., and M.El-S. Metwally supervised the work and made the critical revision for the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yosrey, E., Elmansi, H., Sheribah, Z.A. et al. Implementation of HILIC-UV technique for the determination of moxifloxacin and fluconazole in raw materials and pharmaceutical eye gel. Sci Rep 12, 13388 (2022). https://doi.org/10.1038/s41598-022-17064-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17064-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
