
Abstract
The Candida antarctica lipase B (Novozym 435) is found to catalyze a novel decarboxylative Michael addition in vinylogous carbamate systems for the synthesis of 1,4-benzoxazinone derivatives. The reaction goes through Michael addition, ester hydrolysis and decarboxylation. A possible mechanism is suggested, with simultaneous lipase-catalyzed Michael addition and ester hydrolysis. The present methodology offers formation of complex products through multi-step reactions in a one pot process under mild and facile reaction conditions with moderate to high yields (51–90%) and no side product formation. The reaction seems to be is a great example of enzymatic promiscuity.
Similar content being viewed by others

Introduction
Heterocyclic compounds containing oxygen and nitrogen are of considerable importance due to their occurrence in various natural products and their potential biological activities1. 1,4-benzoxazinone derivatives are examples of such heterocycles, exhibiting a wide range of biological activities such as antioxidant2, anti‐Alzheimer3, antidiabetic4, antimalarial5, antimicrobial6, antibacterial7 and anticancer8.
1,4-Benzoxazinones belong to a reactive class of compounds known as vinylogous carbamates9,10. In recent years, various efficient transformations have been developed for synthesizing these heterocyclic derivatives. Peddinti and co-workers have reported synthesis of various compounds such as pyrrolobenzoxazine11,12, conjugate addition reactions with N-substituted maleimide derivatives13, and regioselective 1,6-conjugate addition of 1,4-benzoxazinone to p-quinone methides14. In other works, a chiral phosphoric acid catalyzed the enantioselective addition of indole to a ketimine ester and produced new derivatives of 1,4-benzoxazinones15,16.
The concept of green chemistry is intrinsically linked to enzymatic catalysis, as enzymes can be obtained from renewable sources, and are capable of catalyzing various chemical reactions, being an alternative to the classical chemical catalysis17,18,19,20,21. The ability of enzymes to catalyze multiple distinctly different reactions is referred to as enzyme promiscuity22,23,24, Lipases (EC 3.1.1.3, carboxylesterase enzyme)25,26 have previously shown unexpected activities and have been used in organic reactions such as Aldol condensation27, Hantzsch reaction28, Cannizzaro reaction29, Mannich reaction30, Baylis–Hillman reaction31, Knoevenagel condensation32, Michael addition33 and Ugi reaction34.
As the field of biocatalysis continues to expand and play a greater role in synthetic chemistry, it is reasonable to expect that the development of innovative one-pot enzymatic processes will likewise see continued growth. The carbon–carbon (C–C) bond constructs the ‘backbone’ of organic molecules, and so carbon–carbon bond formation is a fundamental transformation in organic chemistry. Michael reaction in which 1,4- addition of a carbon nucleophile to an alpha/beta unsaturated carbonyl compound occurs, usually need strong acids and bases. In this study, CAL-B enzyme catalyzes the reaction under mild conditions (NO strong acids or bases) to obtain the final product. Interestingly, application of lipase catalysis in decarboxylative aldol reactions has been demonstrated to circumvent the traditional harsh conditions, such as those afforded by strong bases or metal catalysts35.
Decarboxylation reactions in organic chemistry are often carried out under harsh conditions such as transition metals and high temperatures. These metals are toxic and produce many by-products. It is environmentally friendly to use the Novozym 435 to synthesize 1,4-benzoxazinone derivatives because toxic metals are not used in this reaction.
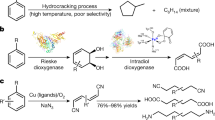
Feng et al. in 2009 reported a novel decarboxylative aldol and Knoevenagel reaction in the presence of Candida antarctica lipase B as a biocatalyst and acetonitrile/water as solvent (Fig. 1a)36. The authors investigated the catalytic effect of CAL-B by performing control experiments and found that the reaction of 4-nitrobenzaldehyde with ethyl acetoacetate in the absence of active CAL-B gave no product formation. Since experiments using acetone instead of ethyl acetoacetate also resulted in no product, it was concluded that decarboxylation was not occurring before addition. Evitt and Bornscheuer in 2011 objected to the report of Feng and coworkers (Fig. 1b), providing data suggesting the acetonitrile in Feng’s work was not adequately dry and may have contained sufficient water to promote ester hydrolysis, thereby allowing non-enzymatic aldol condensation with 4-nitrobenzaldehyde37. When the reaction was repeated with dry HPLC grade acetonitrile (MeCN) no aldol product and a very low formation of the Knoevenagel product were found.

CAL-B catalyzed decarboxylative aldol and Michael reaction.
However, in 2012 Kapoor et al. reinvestigated the reaction with different CAL-B formulations under presumably anhydrous conditions and found significant levels of aldol reaction38 (Fig. 1c), Without going in details with the mechanism, Feng et al. in 2014 reported a two-step sequential biocatalytic process for the synthesis of chiral hydroxyesters by a combination of lipase-catalyzed decarboxylative aldol reactions followed by lipase-catalyzed kinetic resolution of the secondary alcohols39 (Fig. 1d).
Consequently, inspired by previous works and our investigation, we presented new methods for the efficient synthesis of bioactive molecules. To the best of our knowledge, we have carried out for the first time a decarboxylation/Michael reaction between 1,4-benzoxazinone and chalcone derivatives in the presence of Novozym 435 as a biocatalyst (Fig. 1e).
Results and discussion
Initially a handful of commercially available enzymes and proteins were investigated as biocatalysts for the decarboxylative Michael reaction (Table 1). The reaction was performed with 1,4-benzoxazinon 1 and chalcone 2 in MeCN-H2O (100:1). The results showed that only Novozym 435 catalyzed the reaction (entry 5). Controls with urea-denatured Novozym 435 and bovine serum albumin (BSA) protein (entry 6, 7) showed no product formation.
Next, reaction conditions were optimized with Novozym 435 (Table 2). The reaction was monitored by thin-layer chromatography. Since the reaction medium affects the activity of enzyme has an important role in enzymatic reactions. For instance, some organic solvents decrease activity or inactive the enzyme. In this study different solvents dimethyl sulfoxide (DMSO), dimethylformamide (DMF), tetrahydrofuran (THF), dichloromethane (DCM), MeCN were investigated (entry 1–5). Accordingly, when the reaction was carried out in DMSO or DMF, no product could be detected, and in THF and DCM yields were 10, 15% respectively. MeCN was the superior solvent, producing the Michael product 3 in the highest yield after 24 h (entry 5). The temperature optimum seems to be 40–50 °C (entry 6–8). Increasing the amount of enzyme resulted in up to 70% yields (entry 9–11); when enzyme amount is increased, more active sites are involved in the reaction. And a further improvement was obtained by increasing water concentration in the system. As the amount of water increases, the rate of hydrolysis of the ester to acid increases. Also, water clustering on the surface of enzyme plays an important role in the catalytic activity of the biocatalyst and water molecules have tended binding site at the surface of the enzyme. As more water molecules bind to the enzyme, fewer substrate molecules approach the catalytic site of CALB, and thus the active site of the enzyme becomes closed. (entry 12–14). The optimized system (entry 13) resulted in with 75% yield.

Finally, the substrate scope of the reaction was explored by using various 1,4-benzoxazinons 1a–d and chalcones 2a–i (Fig. 2). A variety of 1,4-benzoxazinon and chalcone derivatives bearing electron-donating and electron-withdrawing groups at the benzene ring were amenable to the reaction and produced products 3a–n with 51–90% yields. The substitution on the aryl rings of 2a–i has a significant effect on the yield of the products. It is evident from Fig. 2 that when R3 is Cl, the yield increases compared to non-substituted chalcones. This increased yield may be attributed to the fact that chlorinated chalcones can be better Michael acceptors. When R1 on 1,4-benzoxazinone 1a–d is CH3, the yield was increased compared to non-substituted; however, with Cl and NO2 R1 substitutions yields decreased to 51% and traces, respectively. This shows electron-withdrawing groups on 1,4-benzoxazine decreased the nucleophilic capability of the Michael donor. By having withdrawing groups on the chalcon benzene ring (R3), it is suitable for nucleophilic attack due to its lower electron current. Furthermore, when the withdrawing groups are attached to the benzoxazinone ring (R1), the electron density and nucleophilicity of the compound decrease.

The reaction of 1,4-benoxazinone derivatives 1a–d (0.2 mmol) with chalcone derivatives 2a–i (0.2 mmol), MeCN (2 ml), H2O (40 µl), 40 °C, 24 h.
To investigate the mechanism of the reaction, several control experiments were performed.
At first the reaction was carried out with 1,4 benzoxazinone derivative (1a) in the absence of chalcone, in presence of MeCN and water as solvent (Fig. 3). Under these conditions, no ester hydrolysis or decarboxylation could be detected within 48 h reaction.

Control experiment for decarboxylative Michael reactions.
Secondly, the reaction was performed with dried acetonitrile and no added water. This reaction resulted in decarboxylative Michael addition product in 24 h, indicating hydrolysis prior to Michael addition is not critical for the reaction.
In a third control reaction, we performed the Michael addition of chalcone (2c) and 1,4-benzoxazinone derivative (1b) through chemical catalysis by BF3.OEt2 (Fig. 4). Subsequently the product (4) was incubated with Novozym 435, but after even 48 h no product was formed (according to TLC and HPLC comparison with our previous product).

Control experiment for BF3 catalyzed Michael addition.
In retrospect, it is not surprising that ester hydrolysis was not observed in control experiments 1 and 3, since both substrates contain bulky substitutions on the carboxylic acid side of the ester, something which most lipases, including CAL-B, struggle with40. In contrast to the broad spectrum of alcohol moieties accepted as substrates, only a limited spectrum of acids is accepted by CAL-B. For instance, acyl moieties with sterically demanding α- and β-substitutions yield significantly reduced specific activities41,42.
A proposal mechanism based on previous works30,43,44 and control experiments for the formation of the Michael adduct is presented in Fig. 5. The lipase catalyzed reaction involves the amino acids known as the catalytic triad composed of serine (Ser), histidine (His), and aspartate (Asp) in the enzyme active site. First, the carbonyl group in chalcone is activated by hydrogen bonding to serine, then 1,4-benzoxazinone is deprotonated by the His-Asp system, setting it up for nucleophilic attack of the vinylogous carbamate on the activated chalcone Michael acceptor (I). By considering the control experiments, it seems that the lipase-catalyzed ester hydrolysis occurs simultaneously in an intermediate (II) involving both chalcone and 1,4-benzoxazinone. In this way, the ester is first activated with the lipase, likely because of the charge delocalization facilitating nucleophilic attack of the serine residue. Subsequently, carbon dioxide is released from (VI) and Finally, the enamine (VII) is converted to the final product (VIII).

Suggested mechanism of decarboxylative Michael reaction.
Conclusion
In conclusion, we have successfully developed an efficient biocatalytic methodology for the synthesis of 1,4-benzoxazinone derivatives. The reaction seems to proceed via lipase catalyzed nucleophilic attack of 1,4-benzoxazinones to chalcone Michael acceptors and methyl ester hydrolysis followed by decarboxylation in the presence of Novozym 435. This reaction can be carried out under mild conditions with moderate to excellent yields of 1,4-benzoxazinone derivatives. This novel approach extends the already wide application of Novozym 435 in organic chemistry and provides an effective and environmentally friendly synthetic route for synthesis of 1,4-benzoxazinone derivatives.
Experimental
General information and methods
Commercial chemicals were used without further purification. Immobilized Candida antarctica lipase B (Novozym 435) was kindly donated by Novozymes Denmark. Porcine pancreas lipase (EC 3.1.1.3), Thermomyces lanuginosus lipase (EC 3.1.1.3), Amano lipase from Aspergillus niger (EC 3.1.1.3), and trypsin from porcine pancreas (EC 3.4.21.4) were purchased from Sigma-Aldrich. Analytical TLC (thin-layer chromatography) was performed on Merck pre-coated [silica gel 60 F254 20 × 20 cm)] plates. Melting points were determined with a melting point Thermo Scientific 9100 apparatus and are uncorrected. IR spectra were taken with a Bomem FT-IR MB spectrometer. NMR spectra were recorded in CDCl3 with 300 MHz Bruker DRX Avance spectrometers. Mass spectra were recorded with an Agilent Technologies (HP) 5975C mass spectrometer by electron ionization (EI) (20–70 eV).
Synthesis of 1,4-benzoxazinone derivatives 1a–d
1,4-Benzoxazinones were prepared according to Peddinti's work44, Hence aminophenols (2-Amino-4-methyl phenol, 2-amino-4-chlorophenol, 2-amino-4-nitrophenol) (5 mmol) and dimethyl acetylenedicarboxylate (5 mmol) were mixed in a glass beaker for 2–5 min with the help of a spatula to form a homogeneous paste. The reaction was completed within several minutes and afforded a solid product, which was washed with a few drops of methanol.
Synthesis of chalcone derivatives 2a–i
To a solution of ketone (acetophenone, 4-methylacetophenone, 2-acetylthiophene, 2-acetylfuran) (10 mmol) and aldehyde (benzaldehyde, 4-chlorobenzaldehyde, 2-chlorobenzaldehyde, and 4-methoxybenzaldehyde) (10 mmol) in 20 ml of methanol on an ice bath, 8 ml 10% NaOH was added dropwise over 10 min. After that, the reaction was stirred overnight at room temperature. The precipitate was separated by filtration and washed three times with a mixture of 1:1 ethanol:water. If needed, the crude product was recrystallized from hot ethanol.
General procedure for the synthesis of products 3a–n
To a mixture of 1,4-benzoxazinone 1a–d (0.2 mmol), chalcones 2a–i (0.2 mmol) in 2 ml of MeCN solvent, 40 µl H2O, Novozym 435 (30 mg) was added and the mixture was stirred at 40 °C (120 rpm) for 24 h. The reaction was monitored by TLC. The Novozym 435 was filtered and the solution concentrated under reduced pressure and the crude product was purified by thin layer chromatography on silica gel plates using n-hexane/ethylacetate (5:1) to yield pure compound 3a-n.
Characterization of products 3a–m
3-(4-oxo-2,4-diphenylbutyl)-2H-benzo[b][1,4]oxazin-2-one (3a)
Solid (light yellow), Isolated yield = 0.055 g [75%], Melting point: 124–126 °C; IR (νmax/cm−1): 3066, 1737, 1668; 1H NMR (300 MHz, Chloroform-d) δ 7.89–7.80 (m, 2H), 7.64–7.45 (m, 2H), 7.50–7.38 (m, 3H), 7.44–7.15 (m, 7H), 4.24 (m, 1H), 3.56 (dd, J = 17.1, 7.6 Hz, 1H), 3.49–3.34 (m, 2H), 3.23 (dd, J = 15.3, 8.2 Hz, 1H). 13C NMR (75 MHz, Chloroform-d) δ 198.6, 156.3, 153.0, 146.3, 144.0, 136.9, 133.0, 130.9, 133.5, 128.7, 128.6, 128.5, 127.9, 127.5, 126.7, 125.2, 116.3, 44.8, 40.7, 38.0; MS (EI, 70 eV): m/z = 369 [M+]. Anal. Calcd for C24H19NO3: C = 78.03, H = 5.18, N = 3.79; Found: C = 77.62, H = 5.21, N = 3.48.
3-(2-(2-Chlorophenyl)-4-oxo-4-(thiophen-2-yl)butyl)-2H-benzo[b][1,4]oxazin-2-one(3b)
Solid (light yellow), Isolated yield = 0.063 g [78%], Melting point: 114–116 °C; IR (νmax/cm−1): 3095, 1735, 1650; 1H NMR (300 MHz, Chloroform-d) δ 7.84–7.69 (m, 1H), 7.59 (t, J = 6.4 Hz, 2H), 7.41 (td, J = 17.2, 7.8 Hz, 3H), 7.26 (d, J = 10.6 Hz, 4H), 7.20–7.01 (m, 2H), 4.81–4.58 (m, 1H), 3.48 (dd, J = 16.6, 7.7 Hz, 1H), 3.39 (q, J = 9.4, 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 191.0, 155.5, 152.9, 146.3, 144.1, 140.7, 133.9, 133.8, 131.9, 130.9, 130.6, 129.9, 128.8, 128.0, 127.9, 127.1, 125.2, 116.3, 44.2, 38.6, 34.9; MS (EI, 70 eV): m/z = 409 [M+]. Anal. Calcd for C22H16ClNO3S: C = 64.47, H = 3.93, N = 3.42; Found: C = 64.15, H = 3.77, N = 2.99.
3-(2-(4-Chlorophenyl)-4-oxo-4-phenylbutyl)-2H benzo[b][1,4]oxazin-2-one (3c)
Solid (light brown), Isolated yield = 0.068 g [85%], Melting point: 138–140 °C; IR (νmax/cm−1): 3056, 1737, 1679; 1H NMR (300 MHz, Chloroform-d) δ 7.86 (d, J = 7.8 Hz, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.59–7.39 (m, 4H), 7.38 (dd, J = 21.1, 13.2 Hz, 6H), 4.23 (m, 1H), 3.53 (dd, J = 17.3, 7.2 Hz, 1H), 3.38 (dd, J = 16.2, 6.8 Hz, 2H), 3.22 (dd, J = 15.6, 7.9 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 198.2, 155.8, 153.0, 146.3, 142.4, 136.7, 133.2, 132.3, 130.9, 130.7, 129.0, 128.7, 128.5, 127.9, 125.3, 116.3, 44.7, 40.3, 37.3; MS (EI, 70 eV): m/z = 403 [M+]. Anal. Calcd for C24H18ClNO3: C = 71.38, H = 4.49, N = 3.47; Found: C = 71.53, H = 4.79, N = 3.51.
(4-(Furan-2-yl)-4-oxo-2-phenylbutyl)-2H-benzo[b][1,4]oxazin-2-one (3d)
Solid (light yellow), Isolated yield = 0.059 g [82%], Melting point: 127–129 °C. IR (νmax/cm−1): 3070, 1735, 1660; 1H NMR (300 MHz, Chloroform-d) δ 7.63 (d, J = 7.7 Hz, 1H), 7.54 (s, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.43 – 7.25 (m, 5H), 7.21 (q, J = 9.0, 7.5 Hz, 1H), 7.09 (t, J = 3.0 Hz, 1H), 6.79 (s, 1H), 6.49 (s, 1H), 4.19 (m, 1H), 3.44–3.16 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 187.6, 156.0, 152.7, 146.2, 143.6, 130.9, 130.6, 128.7, 128.6, 127.5, 126.7, 125.2, 117.0, 116.3, 112.2, 44.6, 40.4, 38.0; MS (EI, 70 eV): m/z = 359 [M+]. Anal. Calcd for C22H17NO4: C = 73.53, H = 4.77, N = 3.90; Found: C = 73.33, H = 4.85, N = 3.88.
3-(2-(4-Chlorophenyl)-4-oxo-4-(p-tolyl)butyl)-2H-benzo[b][1,4]oxazin-2-one (3e)
Solid (light yellow), Isolated yield = 0.067 g [80%], Melting point: 157–160 °C; IR (νmax/cm−1): 3075, 1735, 1675; 1H NMR (300 MHz, Chloroform-d) δ 7.76 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 1H), 7.47 (t, J = 7.9 Hz, 1H), 7.27 (td, J = 25.4, 7.9 Hz, 8H), 4.22 (m, 1H), 3.50 (dd, J = 17.1, 7.1 Hz, 1H), 3.36 (d, J = 17.5, 7.2 Hz, 2H), 3.21 (dd, J = 15.3, 7.8 Hz, 1H), 2.41 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 197.8, 155.8, 153.0, 146.3, 144.0, 142.4, 134.3, 132.3, 130.9, 130.6, 129.2, 129.0, 128.7, 128.7, 128.0, 125.3, 116.3, 44.5, 40.3, 37.4, 21.6; MS (EI, 70 eV): m/z = 417 [M+]. Anal. Calcd for C25H20ClNO3: C = 71.86, H = 4.82, N = 3.35; Found: C = 71.94, H = 4.73, N = 3.10.
3-(4-(Furan-2-yl)-2-(4-methoxyphenyl)-4-oxobutyl)-2H-benzo[b][1,4]oxazin-2-one (3f)
Solid (light yellow), Isolated yield = 0.060 g [78%], Melting point: 118–120 °C. IR (νmax/cm−1): 3019, 1732, 1604; 1H NMR (300 MHz, Chloroform-d) δ 7.63 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 9.4 Hz, 1H), 7.45 (t, J = 7.9 Hz, 1H), 7.29 (td, J = 12.1, 7.6 Hz, 4H), 7.10 (dd, J = 10.3, 3.7 Hz, 1H), 6.82 (d, J = 8.5 Hz, 2H), 6.58–6.35 (m, 1H), 4.15 (m, 1H), 3.78 (s, 3H), 3.41–3.15 (m,4H). 13C NMR (75 MHz, CDCl3) δ 187.7, 158.2, 156.1, 152.9, 152.7, 146.3, 135.5, 131.0, 130.6, 128.7, 128.5, 125.2, 117.0, 116.3, 113.9, 112.2, 55.1, 44.8, 40.6, 37.3; MS (EI, 70 eV): m/z = 389 [M+]. Anal. Calcd for C23H19NO5: C = 70.94, H = 4.92, N = 3.60; Found: C = 70.90, H = 4.79, N = 3.41.
3-(4-Oxo-2-phenyl-4-(p-tolyl) butyl)-2H-benzo[b][1,4]oxazin-2-one (3g)
Solid (light yellow), Isolated yield = 0.056 g [73%], Melting point: 142–146 °C; IR (νmax/cm−1): 3039, 1733, 1670; 1H NMR (300 MHz, Chloroform-d) δ 7.75 (d, J = 7.8 Hz, 2H), 7.60 (d, J = 7.8 Hz, 2H), 7.53–7.36 (m, 3H), 7.35–7.24 (m, 3H), 7.20 (d, J = 7.7 Hz, 3H), 4.23 (m, 1H), 3.53 (dd, J = 17.1, 7.7 Hz, 1H), 3.39 (dd, J = 14.3, 12.1, 8.5 Hz, 2H), 3.22 (dd, J = 15.3, 8.1 Hz, 1H), 2.40 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 198.2, 156.3, 153.0, 146.3, 144.0, 143.8, 134.4, 131.0, 130.5, 129.1, 128.7, 128.6, 128.1, 127.5, 126.7, 125.2, 116.3, 44.7, 40.7, 38.1, 21.6; MS (EI, 70 eV): m/z = 383 [M+]. Anal. Calcd for C25H21NO3: C = 78.31, H = 5.52, N = 3.65; Found: C = 78.27, H = 5.41, N = 3.42.
3-(4-Oxo-2-phenyl-4-(thiophen-2-yl)butyl)-2H-benzo[b][1,4]oxazin-2-one (3h)
Solid (light yellow), Isolated yield = 0.061 g [83%], Melting point: 157–160 °C; IR (νmax/cm−1): 3054, 1729, 1660; 1H NMR (300 MHz, Chloroform-d) δ 7.73–7.54 (m, 3H), 7.51–7.15 (m, 8H), 7.10 (t, J = 4.4 Hz, 1H), 4.23 (m, 1H), 3.43 (dd, J = 17.3, 16.9, 6.9 Hz, 2H), 3.25 (dd, J = 15.4, 7.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 191.4, 156.0, 146.3, 144.3, 143.6, 133.7, 131.8, 130.9, 130.5, 128.7, 128.6, 128.0, 127.5, 126.8, 125.2, 116.3, 45.5, 40.4, 38.3; MS (EI, 70 eV): m/z = 375 [M+]. Anal. Calcd for C22H17NO3S: C = 70.38, H = 4.56, N = 3.73; Found: C = 70.21, H = 4.51, N = 3.69.
3-(2-(4-Chlorophenyl)-4-oxo-4-(thiophen-2-yl)butyl)-2H-benzo[b][1,4]oxazin-2-one (3i)
Solid (light yellow), Isolated yield = 0.069 g [85%], Melting point: 139–142 °C; IR (νmax/cm−1): 3048, 1725, 1658; 1H NMR (300 MHz, Chloroform-d) δ 7.65 (s, 3H), 7.73–7.56 (m, 2H), 7.31 (q, J = 16.0, 12.0 Hz, 5H), 7.19–7.06 (m, 1H), 4.30–4.15 (m, 1H), 3.54 (m, 1H), 3.33 (m, 3H). 13C NMR (75 MHz, CDCl3) δ 190.9, 155.6, 152.9, 146.3, 144.1, 142.04, 133.9, 132.4, 131.9, 130.9, 130.7, 128.9, 128.7, 128.0, 125.3, 116.3, 45.3, 40.1, 37.6; MS (EI, 70 eV): m/z = 409 [M+]. Anal. Calcd for C22H16ClNO3S: C = 64.47, H = 3.93, N = 3.42; Found: C = 64.57, H = 3.68, N = 3.47.
6-Methyl-3-(4-oxo-2-phenyl-4-(thiophen-2-yl)butyl)-2H-benzo[b][1,4]oxazin-2-one (3j)
Solid (light brown), Isolated yield = 0.068 g [88%], Melting point: 131–135 °C; IR (νmax/cm−1): 3050, 1745, 1658; 1H NMR (300 MHz, Chloroform-d) δ 7.70 (d, J = 3.8 Hz, 1H), 7.59 (t, J = 4.1 Hz, 1H), 7.40 (d, J = 6.4 Hz, 3H), 7.37–7.18 (m, 4H), 7.12 (d, J = 12.8, 3.6 Hz, 2H), 4.23 (m, 1H), 3.36 (m, 4H), 2.40 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 191.3, 155.8, 153.2, 144.3, 144.2, 143.7, 135.1, 133.6, 131.8, 131.4, 130.7, 128.6, 127.9, 127.5, 126.7, 115.8, 45.5, 40.3, 38.3, 20.7; MS (EI, 70 eV): m/z = 389 [M+]. Anal. Calcd for C23H19NO3S: C = 70.93, H = 4.92, N = 3.60; Found: C = 70.89, H = 4.96, N = 3.14.
3-(2-(4-Chlorophenyl)-4-oxo-4-phenylbutyl)-6-methyl-2H-benzo[b][1,4]oxazin-2-one (3k)
Solid (light brown), Isolated yield = 0.075 g [90%], Melting point: 143–145 °C; IR (νmax/cm−1): 3058, 1724, 1679; 1H NMR (300 MHz, Chloroform-d) δ 7.90–7.81 (m, 2H), 7.55 (t, J = 7.0 Hz, 1H), 7.49–7.35 (m, 3H), 7.29 (q, J = 8.7 Hz, 5H), 7.15 (d, J = 8.3 Hz, 1H), 4.21 (m, J = 7.0 Hz, 1H), 3.52 (dd, J = 17.3, 7.0 Hz, 1H), 3.44–3.29 (m, 2H), 3.20 (dd, J = 15.6, 7.9 Hz, 1H), 2.41 (s, J = 3.8 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 198.1, 155.6, 144.2, 142.4, 136.7, 135.2, 133.1, 132.3, 131.6, 130.6, 128.9, 128.7, 128.5, 127.9, 115.9, 44.7, 40.3, 37.4, 20.7; MS (EI, 70 eV): m/z = 417 [M+]. Anal. Calcd for C25H20ClNO3: C = 71.86, H = 4.82, N = 3.35; Found: C = 71.94, H = 4.84, N = 3.30.
3-(4-(Furan-2-yl)-4-oxo-2-phenylbutyl)-6-methyl-2H-benzo[b][1,4]oxazin-2-one (3l)
Solid (light brown), Isolated yield = 0.065 g [87%], Melting point: 136–138 °C; IR (νmax/cm−1): 3102, 1741, 1670; 1H NMR (300 MHz, Chloroform-d) δ 7.53 (s, 1H), 7.47–7.34 (m, 3H), 7.28 (q, J = 8.5 Hz, 3H), 7.17–7.06 (m, 3H), 6.49 (d, J = 3.6 Hz, 1H), 4.19 (m,1H), 3.31 (m, 4H), 2.42 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 187.6, 155.8, 153.2, 152.7, 146.2, 144.2, 143.6, 135.1, 131.4, 130.7, 128.6, 128.5, 127.5, 126.7, 117.0, 115.8, 112.1, 44.6, 40.3, 38.0, 20.7; MS (EI, 70 eV): m/z = 373 [M+]. Anal. Calcd for C23H19NO4: C = 73.98, H = 5.13, N = 3.75; Found: C = 73.87, H = 5.24, N = 3.71.
6-Chloro-3-(4-oxo-2-phenyl-4-(thiophen-2-yl)butyl)-2H-benzo[b][1,4]oxazin-2-one (3m)
Solid (brown), Isolated yield = 0.041 g [51%], Melting point: 137–140; IR (νmax/cm−1): 3116, 1743, 1639; 1H NMR (300 MHz, Chloroform-d) δ 7.68 (d, J = 3.8 Hz, 1H), 7.56 (dd, J = 22.9, 3.7 Hz, 2H), 7.45–7.26 (m, 5H), 7.22 (t, J = 8.3 Hz, 2H), 7.11 (t, J = 4.4 Hz, 1H), 4.20 (m, 1H), 3.46 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 191.4, 157.5, 152.4, 144.9, 144.2, 143.5, 133.9, 131.9, 131.4, 130.4, 130.2, 128.7, 128.1, 128.0, 127.4, 126.9, 117.4, 45.5, 40.6, 38.2; MS (EI, 70 eV): m/z = 409 [M+]. Anal. Calcd for C22H16ClNO3S: C = 64.47, H = 3.93, N = 3.42; Found: C = 64.52, H = 3.91, N = 3.89.
Data availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Bhardwaj, V., Gumber, D., Abbot, V., Dhiman, S. & Sharma, P. Pyrrole: A resourceful small molecule in key medicinal hetero-aromatics. RSC Adv. 5, 15233–15266 (2015).
Sharma, V. et al. Microwave-assisted one-pot efficient synthesis of functionalized 2-oxo-2-phenylethylidenes-linked 2-oxo-benzo [1, 4] oxazines and 2-oxo-quino [1, 4] oxalines: Synthetic applications, antioxidant activity, SAR and cytotoxic studies. Acta Chim. Slov. 64, 988–1004 (2017).
Sharma, R. et al. Synthesis, antimicrobial activity, structure-activity relationship and cytotoxic studies of a new series of functionalized (Z)-3-(2-oxo-2-substituted ethylidene)-3, 4-dihydro-2H-benzo [b][1, 4] oxazin-2-ones. Bioorg. Med. Chem. Lett. 27, 4393–4398 (2017).
Bandyopadhyay, D., Mukherjee, S., Granados, J. C., Short, J. D. & Banik, B. K. Ultrasound-assisted bismuth nitrate-induced green synthesis of novel pyrrole derivatives and their biological evaluation as anticancer agents. Eur. J. Med. Chem. 50, 209–215 (2012).
Manohar, S., Khan, S. I. & Rawat, D. S. Synthesis of 4-aminoquinoline-1, 2, 3-triazole and 4-aminoquinoline-1, 2, 3-triazole-1, 3, 5-triazine Hybrids as Potential Antimalarial Agents. Chem. Biol. Drug Des. 78, 124–136 (2011).
Mashevskaya, I. et al. A comparative study of the antimicrobial activity of some quinoxalines, 1, 4-benzoxazines, and aza-analogs. Pharm. Chem. J 36, 32–34 (2002).
Bayaraa, T. et al. Discovery, synthesis and evaluation of a novel ketol-acid reductoisomerase inhibitor. Chem. Eur. J. 25, 8958–8968 (2020).
Pegklidou, K. et al. N-substituted pyrrole-based scaffolds as potential anticancer and antiviral lead structures. Med. Chem. 11, 602–608 (2015).
Sagar, A., Vidaycharan, S., Shinde, A. H. & Sharada, D. S. Hypervalent iodine (III)-promoted N-incorporation into N-aryl vinylogous carbamates to quinoxaline diesters: Access to 1, 4, 5, 8-tetraazaphenanthrene. Org. Biomol. Chem. 14, 4018–4022 (2016).
Naganaboina, R. T., Nayak, A. & Peddinti, R. K. Trifluoroacetic acid-promoted Michael addition–cyclization reactions of vinylogous carbamates. Org. Biomol. Chem. 12, 3366–3370 (2014).
Bisht, S. & Peddinti, R. K. FeCl3-mediated domino reaction of benzoxazinones with aroylmethylidene malonates: synthesis to functionalized pyrrolobenzoxazines. J. Org. Chem 82, 13617–13625 (2017).
Kundu, T. et al. Synthesis and biological assessment of pyrrolobenzoxazine scaffold as a potent antioxidant. J. Med. Chem. 62, 6315–6329 (2019).
Sharma, S., Kumar, P., Sharma, A. & Peddinti, R. K. BF3· OEt2-mediated synthesis of 2-arylthio-and (N-Aryl-2, 5-dioxopyrrolidin-3-yl)-substituted 1, 4-benzoxazine derivatives. Eur. J. Org. Chem. 2017, 3059–3071 (2017).
Dua, N., Ghosh, S. & Peddinti, R. K. Zn (OTf) 2-catalyzed 1, 6-conjugate addition of benzoxazinones to p-quinone methides: Access to 3, 3-diaryl-2-(2-oxo-2h-1, 4-benzoxazin-3-yl) propanoic acid esters. Synlett 32, 411–416 (2021).
Xun, W. et al. Regio and enantioselective organocatalytic Friedel-Crafts alkylation of 4-aminoindoles at the C7-position. Org. Lett 20, 590–593 (2018).
Kano, T., Takechi, R., Kobayashi, R. & Maruoka, K. Chiral Brønsted acid-catalyzed enantioselective addition of indoles to ketimines. Org. Biomol. Chem 12, 724–727 (2014).
Bell, E. L. et al. Biocatalysis. Nat. Rev. Methods Primers 1, 1–21 (2021).
Wu, S., Snajdrova, R., Moore, J. C., Baldenius, K. & Bornscheuer, U. T. Biocatalysis: Enzymatic synthesis for industrial applications. Angew. Chem. Int. Ed. 60, 88–119 (2021).
Yi, D. et al. Recent trends in biocatalysis. Chem. Soc. Rev. 50, 8003–8049 (2021).
Hauer, B. Embracing nature’s catalysts: A viewpoint on the future of biocatalysis. ACS Catal. 10, 8418–8427 (2020).
Braham, S. A. et al. Positive effect of glycerol on the stability of immobilized enzymes: Is it a universal fact?. Process Biochem. 102, 108–121 (2021).
Humble, M. S. & Berglund, P. Biocatalytic promiscuity. Eur. J. Org. Chem. 2011, 3391–3401 (2011).
Bornscheuer, U. T. & Kazlauskas, R. J. Catalytic promiscuity in biocatalysis: Using old enzymes to form new bonds and follow new pathways. Angew. Chem. Int. Ed. 43, 6032–6040 (2004).
Hult, K. & Berglund, P. Enzyme promiscuity: Mechanism and applications. Trends Biotechnol. 25, 231–238 (2007).
Ashjari, M., Garmroodi, M., Ahrari, F., Yousefi, M. & Mohammadi, M. Soluble enzyme cross-linking via multi-component reactions: A new generation of cross-linked enzymes. Chem. Commun. 56, 9683–9686 (2020).
Shahedi, M., Habibi, Z., Yousefi, M., Brask, J. & Mohammadi, M. Improvement of biodiesel production from palm oil by co-immobilization of Thermomyces lanuginosa lipase and Candida antarctica lipase B: Optimization using response surface methodology. Int. J. Biol. Macromol. 170, 490–502 (2021).
Guan, Z., Fu, J.-P. & He, Y.-H. Biocatalytic promiscuity: Lipase-catalyzed asymmetric aldol reaction of heterocyclic ketones with aldehydes. Tetrahedron Lett. 53, 4959–4961 (2012).
Wang, J.-L., Liu, B.-K., Yin, C., Wu, Q. & Lin, X.-F. Candida antarctica lipase B-catalyzed the unprecedented three-component Hantzsch-type reaction of aldehyde with acetamide and 1, 3-dicarbonyl compounds in non-aqueous solvent. Tetrahedron 67, 2689–2692 (2011).
Arora, B., Pandey, P. S. & Gupta, M. N. Lipase catalyzed Cannizzaro-type reaction with substituted benzaldehydes in water. Tetrahedron Lett. 55, 3920–3922 (2014).
Li, K. et al. Lipase-catalysed direct Mannich reaction in water: utilization of biocatalytic promiscuity for C–C bond formation in a “one-pot” synthesis. Green Chem. 11, 777–779 (2009).
Reetz, M. T., Mondiere, R. & Carballeira, J. D. Enzyme promiscuity: First protein-catalyzed Morita–Baylis–Hillman reaction. Tetrahedron Lett. 48, 1679–1681 (2007).
Bavandi, H., Habibi, Z. & Yousefi, M. Porcine pancreas lipase as a green catalyst for synthesis of bis-4-hydroxy coumarins. Bioorg. Chem 103, 104139 (2020).
Torre, O., Alfonso, I. & Gotor, V. Lipase catalysed Michael addition of secondary amines to acrylonitrile. Chem. Commun. 15, 1724–1725 (2004).
Kłossowski, S., Wiraszka, B., Berłożecki, S. & Ostaszewski, R. Model studies on the first enzyme-catalyzed Ugi reaction. Org. Lett 15, 566–569 (2013).
Magdziak, D. et al. Catalytic enantioselective thioester aldol reactions that are compatible with protic functional groups. J. Am. Chem. Soc. 127, 7284–7285 (2005).
Feng, X.-W. et al. Lipase-catalysed decarboxylative aldol reaction and decarboxylative Knoevenagel reaction. Green Chem. 11, 1933–1936 (2009).
Evitt, A. S. & Bornscheuer, U. T. Lipase CAL-B does not catalyze a promiscuous decarboxylative aldol addition or Knoevenagel reaction. Green Chem. 13, 1141–1142 (2011).
Kapoor, M., Majumder, A. B., Mukherjee, J. & Gupta, M. N. Decarboxylative aldol reaction catalysed by lipases and a protease in organic co-solvent mixtures and nearly anhydrous organic solvent media. Biocatal. Biotransform. 30, 399–408 (2012).
Zhang, W.-W., Wang, N., Feng, X.-W., Zhang, Y. & Yu, X.-Q. Biocatalytic synthesis of optically active hydroxyesters via lipase-catalyzed decarboxylative aldol reaction and kinetic resolution. Appl. Biochem. Biotechnol. 173, 535–543 (2014).
Naik, S. et al. Lipases for use in industrial biocatalysis: Specificity of selected structural groups of lipases. J. Mol. Catal. B Enzym. 65, 18–23 (2010).
Juhl, P. B., Doderer, K., Hollmann, F., Thum, O. & Pleiss, J. Engineering of Candida antarctica lipase B for hydrolysis of bulky carboxylic acid esters. J. Biotechnol. 150, 474–480 (2010).
Hollmann, F., Grzebyk, P., Heinrichs, V., Doderer, K. & Thum, O. On the inactivity of Candida antartica lipase B towards strong acids. J. Mol. Catal. B Enzym. 57, 257–261 (2009).
Svedendahl, M., Hult, K. & Berglund, P. Fast carbon−carbon bond formation by a promiscuous lipase. J. Am. Chem. Soc. 127, 17988–17989 (2005).
Branneby, C. et al. Carbon−carbon bonds by hydrolytic enzymes. J. Am. Chem. Soc. 125, 874–875 (2003).
Acknowledgements
The authors would like to acknowledge Iran National Science Foundation (INSF) Grant number 97008523 for financial support and Novozymes for kindly providing enzymes for this research.
Author information
Authors and Affiliations
Contributions
H.B. (conceptualization; investigation: software writing—original draft); M.S. (conceptualization; investigation, visualization); Z.H. (corresponding author; supervision; funding acquisition); M.Y. (corresponding author; supervision; writing, review & editing; project administration); M.M. (supervision: supporting; writing—original draft: supporting); J.B. (data curation; formal analysis; validation; writing—review & editing). All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bavandi, H., Shahedi, M., Habibi, Z. et al. Biocatalytic decarboxylative Michael addition for synthesis of 1,4-benzoxazinone derivatives. Sci Rep 12, 12713 (2022). https://doi.org/10.1038/s41598-022-16291-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-16291-3
This article is cited by
-
Immobilization of papain onto nitrogen graphene quantum dots and its use as an efficient catalyst for facile MW-assisted synthesis of polyhydroquinolines
Journal of the Iranian Chemical Society (2024)
-
Comparison of covalent and in situ immobilization of Candida antarctica lipase A on a flexible nanoporous material
3 Biotech (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
