
Abstract
Understanding the association of pyroptosis with tumor progression, prognosis and effect on immunotherapeutic response in breast cancer (BC) is limited. This study analysed forty pyroptosis-related genes to construct the pyroptosis score. Association of the pyroptosis score with the overall survival, clinical features, tumor mutation load, immune cell infiltration, and treatment sensitivity of patients with BC was analysed. Out of 983 BC samples, 304 (30.93%) had genetic alterations with the highest TP53 frequency. We identified three separate subtypes associated with pyroptosis action. These subtypes correlate with the clinicopathological characteristics, TME immune cell infiltration, and disease prognosis. Based on the expression levels of the pyroptosis genes, we divided the pyroptosis score into a high group and a low group. The immune-activated pyroptosis subtype had a higher score with a better prognosis. We also observed that the pyroptosis score correlates with the tumor mutation burden. The pyroptosis score and disease prognosis were directly proportional. A higher pyroptosis score indicated a better prognosis. Results suggest that the pyroptosis-related gene prognosis model is closely related to the immune cell infiltration of BC. The three pyroptosis subtypes associated with BC assist in accurately identifying the tumor subtype, the prognosis of immunotherapy drugs and the patient’s therapeutic response.
Similar content being viewed by others

Introduction
Breast cancer has become the most prevalent type of cancer in females globally by crossing the ratio of lung cancer, with approximately 2 million 300 thousand new cases. It has also become the fifth leading cause of mortalities due to malignancies, with 685,000 reported deaths1. Currently, there are four subtypes of BC based on the variable expression levels of PR, ER, Her-2, and Ki67. The most common subtype is the hormone receptor-positive breast cancer (HR+) which accounts for about 70% of the global breast cancer occurrence2. Presently, the major treatment options for breast cancer include surgery3, chemotherapy (neoadjuvant chemotherapy and postoperative adjuvant chemotherapy)4, radiotherapy5, endocrine therapy6, anti-HER-2 therapy7, and immunotherapy8. Breast cancer accounts for around 30% of all cancer deaths in women, the patients with breast cancer do not primarily die from cancer, rather, resistance to chemotherapeutic drugs, metastasis, and recurrence are the leading causes of the increased mortality related to it. Once cancer spreads as a result of metastasis, the 5-years survival rate is only 25%9.
Pyroptosis is a type of programmed cell death that includes the formation of cell pores mediated by the Gasdermin protein family, causing cell swelling and eventual rupture due to which inflammatory mediators such as interleukin (IL)-1β, IL-18, HMGB1 are released thereby promoting an inflammatory response10. The pyroptosis activation pathway is sorted into the classical cell pyroptosis pathway in which caspase-1 is activated by the inflammatory bodies and the non-classical cell pyroptosis pathway in which cytoplasmic lipopolysaccharide (LPS) activates caspase-4/5/1111. Pyroptosis is not only known for its significant role in inflammatory diseases such as acute severe pancreatitis12, lupus nephritis13, gastrointestinal inflammation14, sepsis15, but it also plays a substantial role in the inhibition of tumors or drug targeting in malignant tumors like lung cancer16, malignant melanoma17, liver cancer18, and esophageal squamous cell carcinoma19. The correlation between pyroptosis and the immune system depends on complex cellular interactions, including pyroptosis and immune cells20. The pyroptosis of normal cells has the ability to alter the tumor microenvironment (TME) and accelerate immune escape21. Meanwhile, there are therapeutic techniques that engage the immune system and cause the pyroptosis of cancer cells22. The pyroptosis in TME produces inflammatory bodies and cytokines that play a potential tumor-promoting role23. The altered and abnormal expression of the regulatory genes involved in pyroptosis is closely correlated with immune regulation and the progression of malignant tumors.
Previous studies indicate that breast cancer has poor immunogenicity. However, with the development in research, the immunological features of breast cancer have caught further attention24. Initial studies have shown that breast cancer has a high tumor mutation load (TMB)25. Later, it was reported that some breast cancer patients had higher expression of programmed cell death ligand-1 (PD-L1). In Triple negative breast cancer (TNBC), the positive rate of PD-L1 was about 20%26, and the expression of PD-L1 was positively correlated with the density of tumor-infiltrating lymphocytes (TILs)27. Tumor immunotherapy works by killing the tumor cells through the reactivation of the body's anti-tumor immune response. The research in the area of breast cancer immunotherapy mainly focuses on chimeric Antigen Receptor T-Cell Immunotherapy (CAR-T), vaccines, and immunosuppressive point inhibitors (ICIs). In breast cancer, the Gasdermin E (GSDME) expression level has a close correlation to the ER status. Oftentimes, hypermethylation of the GSDME gene is noticed in the ER-positive breast cancer cells which are positively correlated with lymph node metastasis of the BC28. The objective of this study is to explore the subtypes of BC based on the regulatory genes related to pyroptosis, construct a pyroptosis scoring system and prognosis model, evaluate the differences of immune cell infiltration, and deeply understand the characteristics of TME cell infiltration controlled by various pyroptosis genes. This could aid in better learning of the mechanism of BC carcinogenesis as well as the prediction of immunotherapy response.
Materials and methods
Data sources
The RNA sequencing (RNA-Seq) and tumor mutation burden (TMB) data of 1098 breast cancer patients were collected along with 113 normal breast samples from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov). The IPS of CTLA-4 and PD-1 blockers in BC were downloaded from the TCIA database. The website of the University of California, Santa Cruz (UCSC) was used to obtain copy number variation (CNV) data. The GSE20685 (contains 327 breast cancer cases)29, GSE88770(contains 117 breast cancer cases)30, GSE41119(contains 287 breast cancer cases)31 and GSE42568(contains 104 breast cancer cases and 17 normal breast cases)32 breast cancer dataset was taken from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). The ComBat function normalized all the RNA-Seq data. Forty pyroptosis genes related to BC were provided by GSEA. The CNV data, differential expression, mutation, gene type, and correlation of pyroptosis-related genes (PRGs) in breast cancer were analyzed. All analyses were performed according to the publication guidelines provided by TCGA and GEO databases. From this dataset, we obtained a total of 40 pyroptosis-related genes (Table 1) using a molecular signature database (http://www.gsea-msigdb.org/gsea/), and involved the Pyroptosis (M41805) and prior review33.
Survival analysis
Univariate Cox regression analysis was used to analyze the prognostic method and P < 0.05 was regarded as statistically significant. Correlation of pyroptosis genes to BC (Table 2). The survival curve was plotted by the Kaplan–Meier.
Consistency cluster analysis and differential gene analysis
Consensus clustering software package (https://bioconductor.org/) was used for clustering and typing on the basis of the co-expression of 40 pyroptosis regulatory genes. We used the “sva” package to merge GSE88770, GSE41119 and GSE42568 GEO datasets as a validation dataset, then analyzed and validated our pyroptosis subtypes. A single sample gene set enrichment analysis (ssGSEA) algorithm helped in measuring the immune cells (28 kinds of immune cells) content following a comparison of the infiltration of immune cells among different types of pyroptosis34. Differentially expressed genes (DEGs) were obtained from the TCGA dataset of the GEO dataset. Subsequently, univariate Cox analysis was performed, P < 0.05 was filtered to obtain DEGs related to patients’ prognosis, and consistent cluster classification was carried out according to the prognostic DEGs to the pyroptosis subtype based on DEGs. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes35 (KEGG) were used for the analysis of DEGs.
Scoring construction and analysis of immune-related indexes of pyroptosis
Principal component analysis36 (PCA) was used for the evaluation of the prognostic DEGs to attain a P-score (Pyroptosis-score = PCA1 + PCA2). The relationship between P-score and immune cells, TMB, different clinical features (Gender, Age, Stage, T, N, M), and PD-L1 was analyzed.
Construction of pyroptosis related prognosis model
We performed a single factor Cox regression analysis on DEG. All BC patients were randomly classified in a ratio of 1:1 into two separate groups called the training group (n = 708) and a test group (n = 708). Furthermore, we used the Lasso Cox regression algorithm, the “glmnet” R package helped in minimizing the risk of overfitting, and the risk prediction model was established by 10× cross-validation37,38. Multivariate Cox analysis helped in the selection of candidate genes (Supplemental Table S1) and also in the establishment of a risk score (risk score) = Σ (EXPI × coefi)), coefi while the EXPI represents the risk coefficient and expression of each respective gene. The 708 patients in the training group were split into two groups based on their median risk score: a low-risk group and a high-risk group. We then ran the Kaplan–Meier survival analysis and derived a receiver operating characteristic curve.
Comparison of pyroptosis-related gene Signature with other breast cancer pyroptosis models
To determine whether our 16 genes associated with pyroptosis genes are superior to other breast cancer pyroptosis models, we used the subject work curve (ROC) to compare 17-gene Signature39, 15-gene Signature40, and 3 gene Signature41. The 1, 3, and 5-year ROC curves constructed for the all TCGA cohort were compared with 16 genes Signature associated with scorch death in this study to assess the advantages and disadvantages of each model. Compare C-index and RMS at the same time.
Evaluate the immune status between high and low-risk groups
CIBERSORT42 was used to measure the quantity of 22 invasive immune cells in heterogeneous samples of the low and high-risk groups in order to estimate the ratio of TICs in TME. The correlation between the P-score and 22 types of infiltrating immune cells, as well as the association between TMB and P-score was investigated in this study.
Analysis of gene mutation and drug sensitivity
The “maftools” R package was used to build the mutation annotation format (MAF) in the TCGA database for observing somatic mutations in BC patients between the high and low-risk categories43. In both groups, the TMB score was also measured for each BC patient. To observe the difference in the efficacy of chemotherapeutic drugs between the two groups, the “pRRophetic” package was used to calculate the half maximal inhibitory concentration (IC50) of these drugs that are widely used for treating BC.
Establishment and verification of nomograph scoring system
Using the “rms” software tool, the clinical traits and risk scores were combined to build a prediction nomogram44. A score was assigned to each variable in the nomograph scoring system, and then added the scores of all variables for each sample to get the total score. For 1, 3, and 5-year survival, time-dependent ROC curves were used to evaluate Nomograms. The values between the anticipated 1, 3, and 5-year survival events and the observed results were described using the nomogram calibration plot.
Ethical approval
As this work is a bioinformatics analysis, ethical approval is not required. All methods were performed in accordance with the relevant guidelines and regulations.
Statistical analysis
We normalized all RNA-Seq data by the ComBat function in the sva software package. Wilcoxon rank sum test was performed to check the difference of gene expression between normal tissues and tumor tissues. The survival curve was drawn with the help of the Kaplan–Meier method, clustering classification was carried out by consensus clustering software package, and the ssGSEA algorithm helped in the evaluation of tumor-infiltrating immune cells. All statistics were completed using the R language software package (https://www.r-project). We considered P-value < 0.05 as significant.
Consent for publication
All author knows the situation and agrees to publish.
Results
Variation and prognosis of pyroptosis regulatory genes in BC
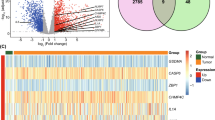
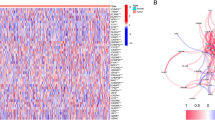
Initially, we studied the mutation frequency of CNV, insertion, and deletion of copy number of 40 pyroptosis regulatory genes found in BC (Fig. 1A,B). At the same time, researchers looked at the expression of 40 pyroptosis genes in malignancies and normal tissues. CASP5, CHMP4A, CHMP7, GSDMA, HMGB1, IL1A, NAIP, NLRC4 and TP53 expressions were determined to be the same (Fig. 1C). Changes in pyroptosis regulating genes were detected in 304 of 983 BC patients, with a frequency of 30.93%. Missense mutations, splice-site mutations, and nonsense mutations are the most common types of mutations. The most frequently mutated gene was TP53, which was followed by CASP8 and DHX9 (Fig. 1D). To learn more about how the pyroptosis regulatory genes interact, we developed a network diagram of survival and interaction between pyroptosis regulatory genes (Fig. 1E). Survival analysis revealed that 29 PRGs were closely associated with prognosis. Patients with high expression of AIM2, CASP1, CASP4, CASP8, CHMP2A, CHMP4B, CHMP6, CHMP7, ELANE, GSDMD, GZMA, GZMB, IL1A, IL1B, IL18, IRF1, IRF2, NAIP, NLRC1, TP63 and ZBP1 had a better prognosis, while patients with low expression of these genes had a better prognosis. Patients with low expression of BAK1, CHMP2B, CHMP4C, CYCS, DHX9, GSDMB, GSDMC and NLRC4 had a better prognosis (Supplementary Fig. S1). These findings reveal that the expression of pyroptosis regulating genes differ significantly between normal and malignant tissues. Simultaneously, it has been established that pyroptosis regulating genes influence the prognosis of BC patients.

Transcriptional and genetic alterations of PRGs in BC. (A) Frequency of CNV gain, loss and non-CNV with PRGs. (B) Location of CNV alterations in PRGs on 23 chromosomes. (C) Distribution of 40 PRGs expression in normal and BC tissues. (D) Frequency of mutations in 40 PRGs in 1098 BC samples from the TCGA cohort. (E) Interaction between PRGs in BC. The lines connecting the PRGs represent their interactions and the thickness of the lines indicates the strength of the association between the PRGs. Green and pink represent negative and pink-positive associations, respectively. PRGs pyroptosis-associated genes, BC breast cancer, TCGA cancer genome atlas, CNV copy number variant.
Pyroptosis subtypes and gene subtypes based on regulatory genes
The TCGA-BC data set and GSE20685 data set were clustered on the basis of 40 pyroptosis regulatory genes. It is categorized into three pyroptosis subtypes based on the cumulative distribution function (CDF) curve and area under the curve (AUC) of consensus score (Fig. 2A). The three pyroptosis regulating gene cluster groupings demonstrated statistical differences in survival (Fig. 2B). The major components of pyroptosis regulating genes were evaluated using the PCA. Three pyroptosis subtypes were discovered to be easily distinguishable (Fig. 2C). Similar results can be obtained for our validation data (Supplementary Fig. S2A–D). We next used ssGSEA to look at the number of immune cells infiltration in BC tumor samples and examined the difference between the three pyroptosis subtypes. It was observed that all three pyroptosis subtypes had a considerable number of immune cells and a high level of infiltration (Fig. 2D). The link between the three pyroptosis subtypes, pyroptosis regulatory genes, and clinicopathological parameters was then investigated. The survival rate of stage I patients increased dramatically in the pyroptosis cluster C age ≤ 50 years, and the expression of the pyroptosis gene increased substantially (Fig. 2E). Pyroptosis cluster C demonstrated enrichment pathways related to immune activation, including T cell receptor signal pathway, B cell receptor signal pathway, nod like receptor signal pathway, toll-like receptor signal pathway, chemokine signal pathway, cytokine receptor interaction, and JAK/STAT signal pathway according to GSVA enrichment analysis based on the three subtypes of pyroptosis regulatory genes (Fig. 2G,H). Pyroptosis cluster B was greatly linked with immunosuppression (Fig. 2F). We identified pyroptosis cluster C as an immunoinflammatory phenotype defined by adaptive immune cell infiltration and immunological activation based on the results of the aforementioned analysis. Pyroptosis cluster B is classified as an immunosuppressive phenotype known for its immunosuppression. Pyroptosis cluster A serves as a link between Pyroptosis cluster C and B. After that, gene analysis was carried out within the pyroptosis cluster groups, 2256 DEGs obtained by univariate Cox analysis (Fig. 3A). The consistency clustering was repeated, and three gene clustering types were identified as gene cluster A, B, and C (Fig. 3B). The results of survival analysis revealed a significant variation in prognosis (P = 0.002, Fig. 3C). At the same time, researchers analyzed the expression of 40 pyroptosis genes in tumors and normal tissues. The expression of the remaining 35 genes was different, except for APIP, CHMP6, CYCS, ELANE and TP53, which had no difference in expression (Fig. 3D). DEG-based analysis revealed that these subgroups had distinct clinicopathological features. Gene cluster A has a favorable prognosis for T1, N0, and stage I cancers (Fig. 3E).

PRGs subtypes and clinicopathological features. (A) Consensus matrix heat map defining the three clusters (k = 3) and their associated regions. (B) Survival analysis of the three subtypes versus OS. (C) PCA analysis showing significant differences in transcriptomes between the three subtypes. (D) Association of the three subtypes with immune cells infiltration. (E) Differences in the three subtypes in relation to clinicopathological features and expression levels of PRG. (F–H) GSVA of biological pathways between the three different isoforms, where red and blue represent activating pathways and blue represent inhibiting pathways, respectively. PCA principal component analysis, OS overall survival.

Gene subtypes based on PRGs. (A) Venn diagram showing DEGs for the three pyroptosis subtypes. (B) Consensus matrix heat map defining the three genetic subtypes subtypes (k = 3). (C) Survival analysis of the three genetic subtypes versus overall survival. (D) Expression distribution of the 40 PRGs in the three genetic subtypes. (E) Differences in expression levels of the three genetic subtypes in relation to clinicopathological features and PRGs. (F,G) GO and KEGG enrichment analysis of DEGs in the three genetic subtypes.
Enrichment function of DEGs and clinical correlation analysis
Through GO enrichment analysis of DEGs, the top 5 biological processes containing T cell activation, regulation of cell–cell adhesion, leucocyte cell–cell adhesion, regulation of T cell activation, regulation of leucocyte cell–cell adhesion. The top five cell components containing external side of the plasma membrane, secret granular membrane, membrane microdomain, membrane raft, and specific granular. And the molecular functions which include cytokine receptor binding, immune receptor activity, cytokine activity, cytokine receptor activity, and cytokine binding (Fig. 3F). The cytokine–cytokine receptor interaction pathway, chemokine signaling pathway, Epstein–Barr virus infection pathway were considerably enriched in KEGG pathway analysis (Fig. 3G).
Construction of pyroptosis score and its clinical significance
The pyroptosis regulatory gene has been discovered to have a regulatory effect on the breast cancer prognosis, cytokines, and immune infiltration of breast cancer. These conclusions, however, are predicted by the BC results. Presently, they cannot predict the pattern of pyroptosis regulatory genes accurately in a single BC patient. As a result, pyroptosis-score (P-score = PCA1 + PCA2) was used to quantify the pattern of pyroptosis regulatory genes in individual BC patients, and predict the patient’s treatment response and prognosis using PCA based on DEGs. The pyroptosis subtype was linked to the gene subtype’s pyroptosis score, with individuals having a higher score having a better prognosis (P = 0.024, Fig. 4A). A positive prognosis was indicated by Cluster A and Cluster C as well as higher pyroptosis scores (Fig. 4B–D). Simultaneously, the chosen DEGs regulate T cell activation and regulation, as well as cytokines and chemokines, and are closely linked with the clinicopathological characteristics. Activated CD4+ T cells, CD8+ T cells, B cells, dendritic cells, natural killer T cells, regulatory T cells, T follicular Helper cell, and type 1 are the immune cells among which the pyroptosis score was favorably associated with T helper cells (Fig. 4E). TMB is closely linked to a patient’s prognosis. The prognosis of low and medium TMB in BC patients is better according to this study (Fig. 4F). Patients with a high pyroptosis score, even if they have a high TMB, have a better overall prognosis (Fig. 4G). TMB, genotyping, and pyroptosis scores are all positively linked (R = 0.19, Fig. 4H). 84 of 119 BC patients with high pyroptosis scores had gene mutations with a frequency of 70.59% (Fig. 4I). 625 of 853 BC patients with low pyroptosis scores had gene mutations with a frequency of 73.27% (Fig. 4J). A high pyroptosis score is a consistent predictor of outcome in BC patients under the age of 50 who are female and have no lymph node metastasis (Supplementary Fig. S3A). On the other side, we discovered a link between pyroptosis score and age, gender, lymph node metastasis, stage, and size of the tumor (Supplementary Fig. S3B).

Construction of the pyroptosis score. (A) Prognostic analysis of the pyroptosis score. (B) Relationship between pyroptosis score and pyroptosis subtypes. (C) Relationship between pyroptosis score and genotyping. (D) Sankey plots of different pyroptosis subtypes (A–C), pyroptosis genetic clustering subtypes and pyroptosis score in relation to overall survival status. (E) Correlation analysis of pyroptosis score with 28 immune cell types. (F) Prognostic analysis of TMB. (G,H) Prognosis and correlation between pyroptosis score and tumor mutational load. (I,J) Waterfall plot of somatic mutation characteristics in high and low pyroptosis scores. Each column represents an individual patient. The upper bar graph shows TMB and the numbers on the right indicate the frequency of mutations in each gene. The bar on the right shows the proportion of each mutation type.
Comparison of genetic features associated with other models of prognosis of pyroptosis in breast cancer
To determine whether our 16 pyroptosis related genes model were superior to other breast cancer pyroptosis models, by comparing them with the 17-gene model, the 15-gene model, and the 3 gene model, our pyroptosis model AUC values at 1, 3, and 5 years that were statistically significant (Supplemental Fig. S4A–D), The OS of the pyroptosis model was s statistically significant (SupplementalFig. S4E–H). Our C-index and RMS index (Supplemental Fig. S4G–I) were higher than those of the other three models. Indicate that our model predicts well.
Immunotherapy
Breast cancer immunotherapy now focuses primarily on PD-L1 and CTLA4. The expression of PD-L1, CTLA4, PDCD1, PDCD1LG2, and HAVCR2 was found to be significantly higher in the high pyroptosis score group (Fig. 5A–E). We discovered that when CTLA4 was positive or PD-L1 was positive, and both CTLA4 and PD-L1 were positive, the immunotherapy scores were higher than in patients with negative CTLA4 and PD-L1 (Fig. 5F–I).

Correlation between pyroptosis score and immune blocking sites. (A–E) Correlation analysis between PD-L1, CTLA4, PDCD1, PDCD1LG2, HAVCR2 expression and pyroptosis score. (F–I) Correlation between IPS immunotherapy and pyroptosis score.
Construction of prognosis prediction model of pyroptosis gene
A single variable lasso technique and Cox regression analysis were used to examine survival-related pyroptosis genes. To create a BC predictive risk model, Lasso regression analysis was used to create 16 pyroptosis genes risk model (Fig. 6A,B). The survival duration of the low-risk group was much longer than that of the high-risk group, according to a Kaplan Meier analysis (Fig. 6C–E). With a median risk score of 1.96 (Fig. 6I–K), the number of deaths in the high-risk group increased dramatically (Fig. 6L–N). In order to assess the risk model’s predictive value in the BC cohort. The risk score ROC curve for 1, 3, and 5-year survival time (Fig. 6F–H) was further examined, indicating that it has high sensitivity and specificity for survival prediction. Meanwhile, the risk model’s expression of 16 genes was assessed (Fig. 6O–Q).

Predictive model for pyroptosis-associated genes. (A,B) LASSO Cox regression analysis of 16 pyroptosis-associated genes. (C–E) Overall survival analysis of high/low risk patients. (F–H) Time-dependent ROC analysis of risk scores in predicting survival. (I–K) Risk scores for each patient in the total cohort, training group, and validation group. (L–N) Number of deaths in the high- and low-risk groups in the cohort. (O–Q) Expression patterns of 16 pyroptosis-related genes in the high- and low-risk groups.
Construct a nomogram of pyroptosis risk score
A predictive nomogram incorporating risk score and clinicopathological aspects was developed to predict the prognosis of BC patients based on the difference in risk score between distinct clinicopathological variables (Fig. 7A). The calibration curve approximates the diagonal, indicating that in our nomogram, 1, 3, and 5-year OS have a good predictive capacity in our nomogram (Fig. 7B). The risk score and nomogram are effective predictors (Fig. 7C). The low-risk group was shown to be more susceptible to cisplatin and docetaxel. The low-risk group was more sensitive to both Cisplatin and Docetaxel (Fig. 7D,E). We determined through univariate and multivariate Cox regression analysis that the risk score was an independent prognostic factor influencing breast cancer patients (Supplement Fig. S5A,B).

Construction and validation of a nomogram. (A) Nomogram for predicting the 1-, 3-, and 5-year OS of BC patients. (B) Calibration curves of the nomogram for predicting of 1-, 3-, and 5-year OS in all BC patients. (C) ROC curves of risk scores, nomogram and clinicopathological features for all BC patients. (D,E) Sensitivity of high and low risk to two chemotherapeutic agents, cisplatin and docetaxel.
Correlation between risk score and characteristics of tumor immune microenvironment
It demonstrates that the prediction model’s risk score is closely linked to immunity. Further analysis revealed that the risk score was positively correlated with M0 macrophages, M2 macrophages, mast cell activation, and NK cell resting content, and negatively correlated with B cells naive, dendritic cells resting, CD4 memory T cell activation, T cells CD8, T cells regulatory (Tregs) and M1 macrophages, implying that immune cell infiltration in the high-risk group was reduced, resulting in a decline of immune function (Fig. 8A). The relationship between 16 genes in the model and the number of immune cells was evaluated. It was discovered that these 16 genes were highly linked to the majority of immune cells (Fig. 8B). The analysis indicated that a low TME score is strongly associated with a high immune score, while a high TME score is closely related to a high matrix score in order to further investigate if the risk score may be utilized as an immune index (Fig. 8C). And in the high-risk group TMB was higher (Fig. 8D), TMB also positively connected with the high-risk score (Fig. 8E), implying that the high-risk group is more likely the failure immunotherapy.

Correlation between risk scores and characteristics of the tumor immune microenvironment. (A) Correlation between risk score and immune cell type. (B) Correlation between the abundance of immune cells and the 16 genes in the risk model. (C) Correlation between risk scores and immune cell and stromal cell scores. (D) Correlation between risk score and TMB and genotyping. (E) The role of TMB in different risk score groups.
Discussion
Breast cancer is the major cause of cancer-related fatalities in women (approximately 15% of all cancer-related deaths in women)1. In addition to surgery, targeted therapy and chemotherapy are frequently used to control/shrink bigger tumors and lower the risk of recurrence and metastasis45. After treatment, most tumors trigger programmed cell death46, and associated cell death killing breast cancer cells. Caspase-3 activation by chemotherapeutic medicines causes secondary necrosis/pyroptosis of cancer and normal cells and plays a significant role in cancer chemotherapy47. Pyroptosis is the formation of pores on the plasma membrane, leading to the destruction of the cell's permeability barrier and subsequent cell enlargement48. Active caspase-3 cleaves GSDME to create the N-terminal fragment of GSDME (GSDME-NT) when apoptosis commences. GSDME-NT will translocate and perforate, causing pyroptosis49. CNV is a structural variation that accounts for 4.8% to 9.5% of human genome diversity50. Some CNVs in TNBC can indicate poor prognosis and can act as prognostic markers, and they may be connected to lymph node metastasis51. Based on the TCGA cohort and GEO dataset, we initially looked at pyroptosis regulating gene mutations in BC. BC demonstrated a unique somatic mutation due to its heterogeneity, according to the findings. The pyroptosis regulatory genes of CNV had varying degrees of deletion and amplification and the mutation rate of somatic cells was as high as 30.93%, showing that pyroptosis regulatory gene mutations play an essential role in breast cancer.
To validate the accuracy of our findings using a single dataset, we carried out further association analysis using multiple other GEO datasets by cluster analyses. These datasets with different molecular features are combined to achieve improved normalization52. Association outcomes reflect the differences between single and grouped/cluster dataset analysis. Clustering analysis of each subtype offers concentrated groups and amplifies the molecular typing differences. Clustering analysis also improved the accuracy of tumor subtype classification53.
Increasing data suggest that tumor pyroptosis is related to tumor formation and progression. Pyroptosis has been to slow the growth of lung cancer tumors54, gastric cancer55, and colorectal cancer56. Pyroptosis can activate the innate immune system, inhibit the development of tumor cells by changing TME, and even directly kill tumor cells39. DEGs screened according to three pyroptosis subtypes were shown to be involved in T cell activation and cytokine interaction in this validation, indicating that breast cancer is intimately linked to inflammation and immunological modulation. Inflammatory bodies and IL-1 have been linked in recent studies showing that they play a vital function in promoting breast cancer growth and metastasis. Tumor growth is linked to an elevated level of IL-1 in the tumor microenvironment in mice mammary tumor models and human breast cancer tissues, which increases the infiltration of myeloid cells like tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells57.
This study discovered that there was a clear enrichment of immune cells in Cluster C, as well as enrichment associated with immune activation, implying that localized death may play a role in breast cancer immune regulation. We also developed a pyroptosis score quantitative approach to identify different pyroptosis regulatory gene subtypes and serve as a guide for individual evaluation and treatment choices in this investigation. The immune activation pyroptosis pattern had a higher score and a better prognosis, according to the findings. TMB is one of the newest biomarkers in the field of cancer immunotherapy58, TMB is considerably greater in ER-negative BC individuals, particularly in TNBC patients59. The pyroptosis score is linked to the number of TMB in this study. The better the prognosis of patients with high TMB, the higher the pyroptosis score, showing that the pyroptosis score can be utilized as an independent prognostic marker.
According to the results of the analysis, the survival prognosis of the high pyroptosis score in our model was better than the low score, it was found that in the pyroptosis model of gastric cancer60, melanoma61, head and neck squamous cell carcinoma62, patients with high scorch death scores also had a good prognosis, and the immunotherapy effect was better than that of the low pyroptosis score group, which was consistent with our research results. However, the prognosis of low scores in low-grade gliomas63 indicates that the scorch death score may play an important role in different tumors, which can better predict the TME status of tumors.
It has been found that PD-L1 is significantly more expressed in breast cancer tissues, especially in triple-negative breast cancer than in normal breast tissue, and the safety and efficacy of PD-L1 inhibitor pembrolizumab in triple-negative breast cancer, hormone receptor positive, HER2-negative, local recurrence or metastatic breast cancer are significantly enhanced64,65.
In this study, the expression of PD-L1, CTLA4, PDCD1, PDCD1LG2 and HAVCR2 in high focus death score was found to increase, and combined with the results of the literature, it can be shown that immunotherapy in patients with PD-1/PD-L1-positive breast cancer has significant therapeutic advantages and clinical efficacy. Similar therapeutic effects were also found in lung cancer66 and melanoma67, consistent with our findings. Clinical trials have shown that tumor cells with higher levels of TMB are more easily recognized by the immune system and therefore have a stronger immune response to immune checkpoint inhibitors. If the tumor mutation burden is greater, there may be a good response to immunotherapy drugs (PD-1/PD-L1 inhibitors) Nivolumab, Pembrolizumab and Atezolizumab68,69.
Pyroptosis enhances immune activation and function, resulting in tumor clearance. Furthermore, tumor cells can activate pyroptosis in a variety of ways, and some immune cells can directly generate it, implying that pyroptosis is implicated in the positive feedback control of anti-tumor immunity70. In BALB/c mice treated with NP-Gsdma3 and Phe-BF3, the number of CD4+, CD8+, natural killer (NK), and M1 macrophages increased. Monocytes, neutrophils, myeloid-derived suppressor cells, and M2 macrophages all decreased, implying that pyroptosis may play a role in tumor immune control71. GSDMB-mediated pyroptosis can function downstream of GZMA, and cytotoxic lymphocytes can transmit GZMA to GSDMB-expressing cancer cells, enhancing antitumor immunity72.
This study was a retrospective analysis using information from the database. Selective bias could skew the results; therefore, more data from BC patients undergoing immunotherapy is needed to confirm the study's conclusions. Clinical data such as surgery, neoadjuvant chemotherapy, radiation, and chemotherapy are not studied, which could alter the immune response and pyroptosis prognosis.
Conclusion
In this investigation, we genotyped BC samples using 40 pyroptosis genes to assess the effect on tumour immune matrix milieu, clinicopathological characteristics, and prognosis in BC patients. The therapeutic benefits of different subtypes and immunotherapy were investigated using a pyroptosis prognostic model. This study adds to our knowledge of the regulatory role of pyroptosis genes in BC. Findings also provide a valuable reference for guiding personalised immunotherapy and BC prognosis.
Data availability
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
Abbreviations
- BC:
-
Breast cancer
- PRGs:
-
Pyroptosis-related genes
- TMB:
-
Tumor mutation burden
- UCSC:
-
The University of California, Santa Cruz
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene Expression Omnibus
- CNV:
-
Copy number variation
- TME:
-
Tumor microenvironment
- GSEA:
-
Gene Set Enrichment Analysis
- HMGB1:
-
High mobility group box 1
- LPS:
-
Lipopolysaccharide
- TNBC:
-
Triple negative breast cancer
- TILs:
-
Tumor-infiltrating lymphocytes
- CAR-T:
-
Chimeric Antigen Receptor T-Cell Immunotherapy
- ICIs:
-
Immunosuppressive point inhibitors
- GSDME:
-
Gasdermin E
- DEGs:
-
Differentially expressed genes
- PCA:
-
Principal component analysis
- MAF:
-
Mutation annotation format
- CDF:
-
Cumulative distribution function
- AUC:
-
Area under the curve
- ssGSEA:
-
Single sample gene set enrichment analysis
- TICs:
-
Tumor-infiltrating immune cells
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PD-L1:
-
Programmed cell death-Ligand 1
- CTLA4:
-
Cytotoxic T-lymphocyte-associated protein 4
- OS:
-
Overall survival
- Treg cells:
-
Regulatory T cells
- ROC:
-
Receiver Operating Characteristic Curve
- IC50:
-
Half maximal inhibitory concentration
- TAMs:
-
Tumor-associated macrophages
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. https://doi.org/10.3322/caac.21492 (2018).
Gianni, L. et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): Follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol. 15, 640–647. https://doi.org/10.1016/s1470-2045(14)70080-4 (2014).
Jatoi, I. & Kemp, Z. Surgery for breast cancer prevention. JAMA 325, 1804. https://doi.org/10.1001/jama.2021.1647 (2021).
Park, Y. et al. Chemotherapy induces dynamic immune responses in breast cancers that impact treatment outcome. Nat. Commun. 11, 6175. https://doi.org/10.1038/s41467-020-19933-0 (2020).
Vaidya, J. et al. Intraoperative radiotherapy for breast cancer: Powerful evidence to change practice. Nat. Rev. Clin. Oncol. 18, 187–188. https://doi.org/10.1038/s41571-021-00471-7 (2021).
Yung, R. & Davidson, N. Optimal adjuvant endocrine therapy for breast cancer. Lancet Oncol. 22, 1357–1358. https://doi.org/10.1016/s1470-2045(21)00420-4 (2021).
Hatschek, T. et al. Neoadjuvant trastuzumab, pertuzumab, and docetaxel vs trastuzumab emtansine in patients with ERBB2-positive breast cancer: A phase 2 randomized clinical trial. JAMA Oncol. 7, 1360–1367. https://doi.org/10.1001/jamaoncol.2021.1932 (2021).
Savas, P. & Loi, S. Expanding the role for immunotherapy in triple-negative breast cancer. Cancer Cell 37, 623–624. https://doi.org/10.1016/j.ccell.2020.04.007 (2020).
Miller, K. et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 69, 363–385. https://doi.org/10.3322/caac.21565 (2019).
Jorgensen, I. & Miao, E. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142. https://doi.org/10.1111/imr.12287 (2015).
Rühl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science (New York) 362, 956–960. https://doi.org/10.1126/science.aar7607 (2018).
Wu, J. et al. Treatment of severe acute pancreatitis and related lung injury by targeting gasdermin D-mediated pyroptosis. Front. Cell Dev. Biol. 9, 780142. https://doi.org/10.3389/fcell.2021.780142 (2021).
Cao, H. et al. Novel effects of combination therapy through inhibition of caspase-1/gasdermin D induced-pyroptosis in lupus nephritis. Front. Immunol. 12, 720877. https://doi.org/10.3389/fimmu.2021.720877 (2021).
Watanabe, D., Guo, Y. & Kamada, N. Interaction between the inflammasome and commensal microorganisms in gastrointestinal health and disease. EMBO Mol. Med. 13, e13452. https://doi.org/10.15252/emmm.202013452 (2021).
Jiao, Y. et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit. Care (London) 25, 356. https://doi.org/10.1186/s13054-021-03775-3 (2021).
Yuan, R. et al. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacol. Res. 170, 105748. https://doi.org/10.1016/j.phrs.2021.105748 (2021).
Ahmed, F. et al. Repurposing melanoma chemotherapy to activate inflammasomes in the treatment of BRAF/MAPK inhibitor resistant melanoma. J. Investig. Dermatol. 142, 1444–1455. https://doi.org/10.1016/j.jid.2021.09.030 (2022).
Shangguan, F. et al. A novel mechanism of cannabidiol in suppressing hepatocellular carcinoma by inducing GSDME dependent pyroptosis. Front. Cell Dev. Biol. 9, 697832. https://doi.org/10.3389/fcell.2021.697832 (2021).
Zheng, Z. et al. STAT3β disrupted mitochondrial electron transport chain enhances chemosensitivity by inducing pyroptosis in esophageal squamous cell carcinoma. Cancer Lett. 522, 171–183. https://doi.org/10.1016/j.canlet.2021.09.035 (2021).
Legrand, A., Konstantinou, M., Goode, E. & Meier, P. The diversification of cell death and immunity: Memento mori. Mol. Cell 76, 232–242. https://doi.org/10.1016/j.molcel.2019.09.006 (2019).
Chauhan, D., Vande Walle, L. & Lamkanfi, M. Therapeutic modulation of inflammasome pathways. Immunol. Rev. 297, 123–138. https://doi.org/10.1111/imr.12908 (2020).
Okondo, M. et al. DPP8 and DPP9 inhibition induces pro-caspase-1-dependent monocyte and macrophage pyroptosis. Nat. Chem. Biol. 13, 46–53. https://doi.org/10.1038/nchembio.2229 (2017).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24, 541–550. https://doi.org/10.1038/s41591-018-0014-x (2018).
Moaaz, M., Lotfy, H., Motawea, M. & Fadali, G. The interplay of interleukin-17A and breast cancer tumor microenvironment as a novel immunotherapeutic approach to increase tumor immunogenicity. Immunobiology 226, 152068. https://doi.org/10.1016/j.imbio.2021.152068 (2021).
Gao, C. et al. Tumor mutation burden and immune invasion characteristics in triple negative breast cancer: Genome high-throughput data analysis. Front. Immunol. 12, 650491. https://doi.org/10.3389/fimmu.2021.650491 (2021).
Corti, C., Nicolò, E. & Curigliano, G. Novel immune targets for the treatment of triple-negative breast cancer. Expert Opin. Ther. Targets 25, 815–834. https://doi.org/10.1080/14728222.2021.2006187 (2021).
Rizzo, A. & Ricci, A. Biomarkers for breast cancer immunotherapy: PD-L1, TILs, and beyond. Expert Opin. Investig. Drugs 31, 549–555. https://doi.org/10.1080/13543784.2022.2008354 (2022).
Xia, X. et al. The role of pyroptosis in cancer: Pro-cancer or pro-"host"? Cell Death Dis. 10, 650. https://doi.org/10.1038/s41419-019-1883-8 (2019).
Kao, K., Chang, K., Hsu, H. & Huang, A. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 11, 143. https://doi.org/10.1186/1471-2407-11-143 (2011).
Metzger-Filho, O. et al. Genomic grade adds prognostic value in invasive lobular carcinoma. Ann. Oncol. 24, 377–384. https://doi.org/10.1093/annonc/mds280 (2013).
Prat, A. et al. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist 18, 123–133. https://doi.org/10.1634/theoncologist.2012-0397 (2013).
Clarke, C. et al. Correlating transcriptional networks to breast cancer survival: A large-scale coexpression analysis. Carcinogenesis 34, 2300–2308. https://doi.org/10.1093/carcin/bgt208 (2013).
Chen, J. et al. Signature construction and molecular subtype identification based on pyroptosis-related genes for better prediction of prognosis in hepatocellular carcinoma. Oxid. Med. Cell. Longev. 2022, 4494713. https://doi.org/10.1155/2022/4494713 (2022).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Kanehisa, M., Furumichi, M., Sato, Y., Ishiguro-Watanabe, M. & Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551. https://doi.org/10.1093/nar/gkaa970 (2021).
Abbas, L. et al. Revealing novel, latent subsets of patients with morphoea through principal component analysis. Br. J. Dermatol. 186, 193–195. https://doi.org/10.1111/bjd.20748 (2022).
Liu, C., Wang, X., Genchev, G. & Lu, H. Multi-omics facilitated variable selection in Cox-regression model for cancer prognosis prediction. Methods (San Diego) 124, 100–107. https://doi.org/10.1016/j.ymeth.2017.06.010 (2017).
Li, C., Pak, D. & Todem, D. Adaptive lasso for the Cox regression with interval censored and possibly left truncated data. Stat. Methods Med. Res. 29, 1243–1255. https://doi.org/10.1177/0962280219856238 (2020).
Wu, J., Zhu, Y., Luo, M. & Li, L. Comprehensive analysis of pyroptosis-related genes and tumor microenvironment infiltration characterization in breast cancer. Front. Immunol. 12, 748221. https://doi.org/10.3389/fimmu.2021.748221 (2021).
Yu, H. et al. A novel pyroptosis-related signature predicts prognosis and response to treatment in breast carcinoma. Aging 14, 989–1013. https://doi.org/10.18632/aging.203855 (2022).
Xu, L., Hu, Y. & Liu, W. Pyroptosis-mediated molecular subtypes are characterized by distinct tumor microenvironment infiltration characteristics in breast cancer. J. Inflamm. Res. 15, 345–362. https://doi.org/10.2147/jir.S349186 (2022).
Newman, A. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. https://doi.org/10.1038/nmeth.3337 (2015).
Bi, F., Chen, Y. & Yang, Q. Significance of tumor mutation burden combined with immune infiltrates in the progression and prognosis of ovarian cancer. Cancer Cell Int. 20, 373. https://doi.org/10.1186/s12935-020-01472-9 (2020).
Balachandran, V., Gonen, M., Smith, J. & DeMatteo, R. Nomograms in oncology: More than meets the eye. Lancet Oncol. 16, e173–e180. https://doi.org/10.1016/s1470-2045(14)71116-7 (2015).
Kaboli, P., Imani, S., Jomhori, M. & Ling, K. Chemoresistance in breast cancer: PI3K/Akt pathway inhibitors vs the current chemotherapy. Am. J. Cancer Res. 11, 5155–5183 (2021).
Mohammad, R. et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. https://doi.org/10.1016/j.semcancer.2015.03.001 (2015).
Zhang, Z. et al. Caspase-3-mediated GSDME induced pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J. Cell Mol. Med. 25, 8159–8168. https://doi.org/10.1111/jcmm.16574 (2021).
Robinson, N. et al. Programmed necrotic cell death of macrophages: Focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 26, 101239. https://doi.org/10.1016/j.redox.2019.101239 (2019).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128. https://doi.org/10.1038/ncomms14128 (2017).
Zarrei, M., MacDonald, J., Merico, D. & Scherer, S. A copy number variation map of the human genome. Nat. Rev. Genet. 16, 172–183. https://doi.org/10.1038/nrg3871 (2015).
Pariyar, M., Johns, A., Thorne, R., Scott, R. & Avery-Kiejda, K. Copy number variation in triple negative breast cancer samples associated with lymph node metastasis. Neoplasia (New York) 23, 743–753. https://doi.org/10.1016/j.neo.2021.05.016 (2021).
Prabakaran, I. et al. Gaussian mixture models for probabilistic classification of breast cancer. Can. Res. 79, 3492–3502. https://doi.org/10.1158/0008-5472.Can-19-0573 (2019).
Mei, J., Zhao, J. & Fu, Y. Molecular classification of breast cancer using the mRNA expression profiles of immune-related genes. Sci. Rep. 10, 4800. https://doi.org/10.1038/s41598-020-61710-y (2020).
Xie, B. et al. Combination of DNA demethylation and chemotherapy to trigger cell pyroptosis for inhalation treatment of lung cancer. Nanoscale 13, 18608–18615. https://doi.org/10.1039/d1nr05001j (2021).
Wang, Y. et al. GSDME mediates caspase-3-dependent pyroptosis in gastric cancer. Biochem. Biophys. Res. Commun. 495, 1418–1425. https://doi.org/10.1016/j.bbrc.2017.11.156 (2018).
Wang, J. et al. Gasdermin D in different subcellular locations predicts diverse progression, immune microenvironment and prognosis in colorectal cancer. J. Inflamm. Res. 14, 6223–6235. https://doi.org/10.2147/jir.S338584 (2021).
Guo, B., Fu, S., Zhang, J., Liu, B. & Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 6, 36107. https://doi.org/10.1038/srep36107 (2016).
Chan, T. et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 30, 44–56. https://doi.org/10.1093/annonc/mdy495 (2019).
Luen, S., Virassamy, B., Savas, P., Salgado, R. & Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast (Edinburgh) 29, 241–250. https://doi.org/10.1016/j.breast.2016.07.015 (2016).
Xiang, R. et al. Pyroptosis patterns characterized by distinct tumor microenvironment infiltration landscapes in gastric cancer. Genes 12, 1535. https://doi.org/10.3390/genes12101535 (2021).
Meng, J. et al. Pyroptosis-related gene mediated modification patterns and immune cell infiltration landscapes in cutaneous melanoma to aid immunotherapy. Aging 13, 24379–24401. https://doi.org/10.18632/aging.203687 (2021).
Deng, H. et al. Pyroptosis patterns and immune infiltrates characterization in head and neck squamous cell carcinoma. J. Clin. Lab. Anal. 36, e24292. https://doi.org/10.1002/jcla.24292 (2022).
Zhou, Z. et al. Comprehensive characterization of pyroptosis patterns with implications in prognosis and immunotherapy in low-grade gliomas. Front. Genet. 12, 763807. https://doi.org/10.3389/fgene.2021.763807 (2021).
Winer, E. et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 22, 499–511. https://doi.org/10.1016/s1470-2045(20)30754-3 (2021).
Pérez-García, J. et al. Pembrolizumab plus eribulin in hormone-receptor-positive, HER2-negative, locally recurrent or metastatic breast cancer (KELLY): An open-label, multicentre, single-arm, phase II trial. Eur. J. Cancer (Oxford) 148, 382–394. https://doi.org/10.1016/j.ejca.2021.02.028 (2021).
Pérol, M. et al. Effectiveness of PD-(L)1 inhibitors alone or in combination with platinum-doublet chemotherapy in first-line (1L) non-squamous non-small-cell lung cancer (Nsq-NSCLC) with PD-L1-high expression using real-world data. Ann. Oncol. 33, 511–521. https://doi.org/10.1016/j.annonc.2022.02.008 (2022).
Ribas, A. et al. PD-L1 blockade in combination with inhibition of MAPK oncogenic signaling in patients with advanced melanoma. Nat. Commun. 11, 6262. https://doi.org/10.1038/s41467-020-19810-w (2020).
Marabelle, A. et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365. https://doi.org/10.1016/s1470-2045(20)30445-9 (2020).
Nie, W. et al. STK11Atezolizumab prolongs overall survival over docetaxel in advanced non-small-cell lung cancer patients harboring or mutation. Oncoimmunology 10, 1865670. https://doi.org/10.1080/2162402x.2020.1865670 (2021).
Loveless, R., Bloomquist, R. & Teng, Y. Pyroptosis at the forefront of anticancer immunity. J. Exp. Clin. Cancer Res. 40, 264. https://doi.org/10.1186/s13046-021-02065-8 (2021).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426. https://doi.org/10.1038/s41586-020-2079-1 (2020).
Zhou, Z. et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science (New York). https://doi.org/10.1126/science.aaz7548 (2020).
Acknowledgements
The author would like to thank all the authors for their contributions to this article. We also acknowledge the TCGA data and GEO data for providing data.
Author information
Authors and Affiliations
Contributions
G.L. and J.C. designed the project. G.H. and J.Z. wrote the paper. G.H., J.C. and J.Z. download data and perform bioinformatics analysis. G.L. has rigorously revised the final manuscript. All authors also read and agree to release versions of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, G., Zhou, J., Chen, J. et al. Identification of pyroptosis related subtypes and tumor microenvironment infiltration characteristics in breast cancer. Sci Rep 12, 10640 (2022). https://doi.org/10.1038/s41598-022-14897-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14897-1
This article is cited by
-
Dissection of pyroptosis-related prognostic signature and CASP6-mediated regulation in pancreatic adenocarcinoma: new sights to clinical decision-making
Apoptosis (2023)
-
Role of androgen receptor signaling pathway-related lncRNAs in the prognosis and immune infiltration of breast cancer
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
