
Abstract
Loquat (Eriobotrya japonica) have originated in southeastern China and spread as a cultivated plant worldwide. Many of the loquat genetic resources collected internationally are of unknown origin, and their genetic background requires clarification. This study analyzed the genetic diversity of 95 accessions by using Rad-Seq SNP markers. Data analysis broadly classified loquat into three groups: (1) Japanese and Chinese cultivars and some Japanese strains (wild plants that are not used for commercial cultivation), (2) Vietnamese, Israeli, Greek, USA, and Mexican cultivars and strains, and (3) other Japanese strains. Group 2 is cultivated mostly outside of East Asia and was clearly distinct from the other groups, indicating that varieties of unknown origin with genetic backgrounds different from those of Japanese and Chinese cultivars may have been introduced to Mediterranean countries and North America. Because Japanese and Chinese cultivars belong to group 1, the current Japanese cultivars are derived from genetic resources brought from China. Some of group 1 may have been introduced to Japan before excellent varieties were developed in China, while group 3 may have been indigenous to Japan that have not been introduced by human activities, or may have been brought to Japan by human activities from China.
Similar content being viewed by others


Introduction
Loquat (Eriobotrya japonica (Thunb.) Lindl.) is a diploid species (2n = 34)1 in the family Rosaceae, subfamily Spiraeoideae, tribe Pyreae, subtribe Pyrinae2,3. These fruit trees are cultivated in temperate and subtropical zones worldwide. Among the 20 species in the genus Eriobotrya, E. japonica is the only species used commercially for fruit production4. The origin of loquat is reportedly southeastern China5,6. In 1784, loquat was transferred from China to the National Garden in Paris, France, and in 1787 from China to the Royal Botanic Gardens at Kew, England7. Subsequently, loquat was grown on the Riviera, in Malta, French North Africa, and the Near East, where the fruit began to appear in local markets8. There is also a record that it was introduced to California from Japan in 1899 (Fig. 1A), but the details are unknown9.

The spread of loquat from its origins in China to other parts of the world (A: Propagation by citation) (B: Propagation proposed by this study). The base map was produced using SimpleMappr (https://www.simplemappr.net/). Dark green circles indicate Group 1, dark blue circles indicate Group 2, and orange circles indicate Group 3.
In Japan, loquat seeds have been discovered in excavations of strata from the Yayoi period (300 B.C.–300 A.D.), when rice cultivation technology was introduced from the continent. Loquat was first described in Japan in 762 in the Shōsōin archives, and was also recorded in the Nihon Sandai Jitsuroku (the English translation is “The True History of Three Reigns of Japan”) in 90110. Because the current Japanese cultivars were most likely introduced from China about 150–400 years ago (see below), the descendants of the Japanese loquat that predated this introduction are now likely growing as wild strains (wild plants that are not used for commercial cultivation are defined as “strains”). Two possibilities (not necessarily mutually exclusive) have been proposed for the origin of these wild strains11: they may have been introduced from China in ancient times by human activity, or they may have been originally indigenous to Japan. The first possibility is supported by the fact that loquat has no native Japanese name, but only a Sino-Japanese name (a Japanese word of Chinese origin)10. On the other hand, because some wild strains have been found mixed with native plants throughout Japan and are scattered without forming colonies, and cultivars have not been documented to become wild, wild strains are likely indigenous in Japan12. Loquat that grew wild in Japan may not have been actively cultivated for a long time because of the small size and thin flesh of its fruit12, which may explain the lack of a native Japanese name.
Although loquat has existed in Japan for more than 1000 years, the current Japanese cultivars were probably bred and spread from seeds introduced from China in the Edo period (1600–1868)11. Later, the number of cultivars increased through crossbreeding and bud sport mutations, mainly from ‘Mogi’, ‘Tanaka’, and ‘Kusunoki’.
There are still many unknowns in the transmission of loquat, and elucidation of genetic relationships within this species is important and interesting in loquat breeding research. Loquat genetic diversity has been analyzed with SSR markers13,14 and SNP markers15, but the number of markers was small. Genome-wide analysis is crucial for accurate evaluation of genetic diversity. Compared to these conventional methods, whole genome resequencing is the most effective method for studying genetic diversity. However, the cost has not decreased sufficiently to analyze a large number of individuals with whole genome resequencing. Restriction site–associated DNA sequencing (RAD-Seq) is an inexpensive method that can analyze a large number of individuals, and it can analyze a large number of markers compared to conventional methods. We have used RAD-Seq to study the genetic diversity of citrus16,17, firefly18, Japanese pepper19, and razor clam20, to construct the linkage map of bronze loquat21, and to study the phylogenetic relationships among Aurantioideae (citrus and its relatives)22. In this study, we used genome-wide SNP markers obtained by RAD-Seq to analyze the genetic diversity and structure of wild loquat strains from various parts of Japan; Japanese cultivars; and cultivars and strains introduced from China, Vietnam, Israel, Greece, the United States, and Mexico.
Results
Variant detection by mapping of RAD‑Seq data
The double-digest RAD-Seq (ddRAD-Seq) of the 95 samples (Table 1) generated over 2.8 gigabases with 54.6 million single-ended 51-bp reads. Quality-based filtering yielded 95 samples with an average of 0.57 million reads (maximum, 1.1 million reads; minimum, 0.22 million reads) (Supplementary Table S1). The Stacks program constructed new loci with an average coverage depth of 14 times (Supplementary Table S2). The following analysis was performed using data from 1,822 variant sites.
Genetic structure
In principal component analysis (PCA), the contributions of the first and second principal components were 17.1% and 9.47%, respectively (Fig. 2). The first principal component separated the accessions into three major groups: (1) Japanese and Chinese cultivars and some Japanese wild strains, (2) cultivars and strains from Vietnam, Israel, Greece, USA, and Mexico, and (3) other Japanese wild strains. The second principal component separated group 3 from the other groups. Wild strains from different parts of Japan were divided into two groups: some formed a separate group (group 3) and some belonged to group 1. Multidimensional scaling (MDS) analysis also divided the accessions into three major groups (Fig. 3) and supported the grouping by PCA.

Principal component analysis (PCA) of Eriobotrya japonica accessions in the first two components based on 1822 SNP markers. The color of the sample number indicates the country or region where the sample was collected. Cyan represents germplasm resources and cultivars from China, darkgreen represents cultivars from Japan, orange represents germplasm resources from Japan, dark blue represents germplasm resources from Vietnam, purple represents cultivars from Israel, pink represents germplasm resources from Greece, black represents germplasm resources and cultivars from North America. Figure was generated using R software (version 4.1.1).

Multidimensional scaling of Eriobotrya japonica accessions using 2-dimensional data based on 1822 SNP markers. The color scheme is the same as in Fig. 1. Figure was generated using R software (version 4.1.1).
PCA and MDS analyses indicated the presence of subgroups. In group 1, most of the Japanese cultivars were in the upper right part, and the Chinese cultivars and some of the Japanese cultivars (22–24, 28, 38, and 48) were in the lower left part. In group 2, with some exceptions, country-specific subgroups could be discerned: Greek subgroup (86, 87, 89, and 90), Israeli subgroup (78, 79, 82, 84, and 85), and Vietnamese subgroup (72–77). Accessions from the USA and Mexico (80, 91 and 93–95) seem to be close to the Vietnamese subgroup. One of the US cultivars (60) belonged to group 1.
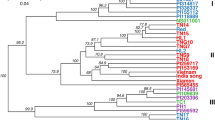
Cluster analysis was performed using a pairwise distance matrix. The cluster analysis identified the two clades (A and B) (Fig. 4). Group 2 was placed in clade A, and group 1 and group 3 were placed in clade B.

Cluster analysis of Eriobotrya japonica accessions. The color scheme is the same as in Fig. 2. Figure was generated using R software (version 4.1.1).
Admixture analysis performed with the number of ancestral populations (K) ranging from 1 to 10 (Fig. 5, Supplementary Fig. S1) clearly divided the accessions into three groups, which strongly supports the PCA and MDS results. Reflecting the presence of subgroups as described above, the most likely number of ancestral populations was 7 (Supplementary Fig. S1). No gene flow from other groups was detected in group 3, which consisted of Japanese wild strains.

Admixture analysis of Eriobotrya japonica accessions. Admixture plots at K = 3 to 8 are shown. The colors for the sample numbers are the same as in Fig. 2. The figure was generated using R software (version 4.1.1).
Population genetic statistics
Estimation of the degree of genetic diversity among the three groups using the Fst values (Table 2) showed that groups 1 and 3 were the genetically closest pair, as indicated by the lowest Fst value, and groups 2 and 3 were the genetically most distant pair. The mean values of nucleotide diversity (π) and mean expected heterozygosity (He) were highest in group 2, followed by group 1 (Table 3, Supplementary Table S3). The mean value of inbreeding coefficient (Fis) was lowest in group 3, whereas the Fis values for groups 2 and 3 were similar to each other.
The heterozygosity of each individual was calculated (Supplementary Table S4). Twenty-five individuals exhibited relatively low heterozygosity with a value below 0.15. All 11 individuals (61–71) in group 3 and 6 Japanese wild strains (52–54, and 57–59) in group 2 were in this low heterozygosity group.
Elucidation of the process of asexual reproduction
Several cultivars have been generated through asexual reproduction, as in the case of a bud sport mutation. To examine whether the parent cultivar and its possible child cultivar were derived from a single tree by a somatic mutation, we checked the conservation of heterozygosity using pairwise alignments (Table 4, Supplementary Fig. S2). The conservation ratio of heterozygous sites between technical replicates was above 90%, which is the criterion17 for determining the combinations of the parent–child cultivars derived by asexual reproduction. Consistent with the available records23, ‘Tanaka’ (46) was a parent of ‘Morimoto’ (37). Heterozygous sites were conserved among ‘Amakusagokuwase’ (22), ‘Amakusawase’ (23), and ‘Moriowase’ (38). According to oral tradition, ‘Moriowase’ is the oldest, so ‘Amakusagokuwase’ and ‘Amakusawase’ are its descendants. Although the records10 claim that ‘Moriowase’ (38) is a bud sport mutant of ‘Mogi’ (32), our analysis rejected this possibility because the conservation ratio of heterozygous sites between these two cultivars was only 19.7%.
Discussion
Our study analyzed the genetic diversity of 95 cultivars and strains of loquat collected from all over the world. On the basis of the analysis of the population structure of 19 Chinese loquat cultivars and strains by RAD-Seq, Hubei Province in China has been suggested as the center of origin of loquat cultivation6. However, the authors have not analyzed the movement of loquats following their introduction from China to other countries and have not verified whether the hypothesis that the cultivation of loquat started in a small area in China24 is correct or not. On the basis of the analysis of samples from around the world, we here propose and discuss a more complex history of loquat cultivation (Fig. 1B).
Although genetic diversity analysis using SSR markers revealed no country-specific cultivar clusters14, the RAD-Seq analysis used in this study allowed for such clusters to be detected. This was possible because more markers are available in RAD-Seq analysis than in conventional SSR marker analysis. RAD-Seq analysis classified the loquat genetic resources into three groups: (1) Japanese and Chinese cultivars and some Japanese wild strains, (2) Vietnamese, Israeli, Greek, US, and Mexican cultivars and strains, and (3) other Japanese wild strains.
Group 2 had the highest mean values of π and He (Table 3). The Fst values (Table 2) showed that group 2 is well separated from groups 1 and 3, which may be due to differences in their place of origin, as discussed below. The Fis value of group 2 was higher than that of the Japanese wild strains (Table 3), which may reflect the fact that group 2 plants have been grown and crossbred by humans.
Group 2 was genetically separated from the Japanese and Chinese loquats. Blasco et al.25 reported that SSR and S-allele markers can distinguish European cultivars from other cultivars. Our results are in good agreement with the above study. Morton8 reported that loquat genetic resources were introduced to the West from China and Japan, but our study does not agree with this report. When plants are preserved in botanical gardens, records are well kept. However, for commercial use, records are not always kept, because the purpose of plant introduction is different. Group 2 plants may have been introduced to the West for commercial cultivation in a different way than they were introduced to botanical gardens.
The place of origin for group 2 may be in China or Japan, although our analysis failed to find any cultivars or strains in group 2 that originated from China or Japan. Unfortunately, the cultivars and strains from China analyzed in this study are not representative of all loquat cultivars from China, and we were unable to analyze cultivars and strains from southern China, such as Yunnan, Guangdong, and Guangxi. Wang et al.6 reported that ‘Younan’ from Guangdong has a close genetic relationship with cultivars from Spain, Italy, and the USA. This suggests that genetic resources from particular regions of China may have been introduced to the West.
The fact that the Vietnamese strains belong to group 2 suggests two possibilities. One is that group 2 originated from Southeast Asian countries other than China, including Vietnam, in particular because cultivars from the USA and strains from Mexico were genetically similar to strains from Vietnam. Interestingly, when group 2 was analyzed in detail, we detected the presence of an Israeli subgroup and a Greek subgroup. They may have originated in Southeast Asian countries other than Vietnam. The second possibility is that cultivars or stains introduced from China and other countries to the USA or France (the former colonial master of Vietnam) were further introduced to Vietnam. Researchers who collected the strains in Vietnam told us that these strains were not used for edible fruit, even though they grew near houses. These plants may have been cultivated in Vietnam as ornamental trees or to be offered to Westerners. Thus, group 2 cultivars are likely to originate from various places.
The differences in taste preferences between the Greek, Israeli, American, and Mexican people and the Japanese and Chinese people may have influenced cultivar differentiation. Indeed, many cultivars and strains from Greece, Israel, USA, and Mexico have higher acid content than those from Japan and China26,27. However, differences in taste preferences need to be studied in detail in the future. Although group 2 strains grow in Vietnam, the Vietnamese most likely prefer to eat group 1 fruits because they do not eat group 2 fruits. In Vietnam, loquat imported from East Asia is commercially available.
In group 1, the mean values of π and He were lower than in group 2 but higher than in group 3 (Table 3). The Fst values indicated that group 1 is genetically closer to group 3 than to group 2 (Table 2). The Fis value of group 1 was higher than that of the Japanese wild strains (Table 3), which may reflect the fact that group 1 plants, like group 2 plants, have been grown and crossbred by humans.
The PCA and MDS analyses placed Japanese and Chinese cultivars in the center of the group 1 cluster. The present Japanese cultivars originated from excellent Chinese cultivars introduced at the Edo period (1600–1868) and were further improved in Japan11. The detection of group 1 reflects this proposed history. The cultivars were believed to have been improved through crossbreeding and bud sport mutations, using mainly ‘Mogi’, ‘Tanaka’, and ‘Kusunoki’. However, some samples (22–24, 28, 38, and 48) were not related to these three cultivars. Other cultivars introduced at the Edo period or earlier or gene flow from Japanese wild strains may have contributed to the formation of these six cultivars.
With the exception of these six samples, the central part of the cluster in group 1 was divided into a subcluster of Japanese cultivars and a subcluster of Chinese cultivars. This separation may reflect differences in the breeding process between China and Japan, where mainly ‘Mogi’, ‘Tanaka’, and ‘Kusunoki’ were used. The slight difference in taste preferences between the Japanese and Chinese may have influenced the formation of the two subclusters, but further research is needed to address this issue.
The PCA and MDS analyses placed the Japanese wild strains at the periphery of group 1. Because group 1 contains Chinese cultivars, the wild strains in this group are closely related to plants introduced to Japan from China. These wild strains may be related to plants introduced before the development of excellent cultivars in China, as well as being related to plants described in ancient Japanese documents. Although other possibilities exist for the five wild strains (52, 53, and 55–57) as discussed below, two strains (54 and 58) are possible descendants of plants introduced to Japan before the Edo period. Analysis of wild strains in China could help to solve this problem, but unfortunately we were unable to analyze them. Among these wild samples, sample 59 was closer to cultivars such as Mogi (32–36, 43, 44, 47), which may be related to cultivar escape.
PCA and MDS analyses placed the Japanese and Chinese cultivars and the Japanese wild strains at the bottom of the cluster in group 1. Admixture analysis suggested that gene flow from isolated Japanese wild strains in group 3 may have occurred in these plants. The finding that gene flow from group 3 may have occurred in Chinese cultivars suggests that plants belonging to group 3 may be present in China. Alternatively, these Chinese cultivars may have been placed here under the influence of populations not analyzed in this study, rather than group 3. Gene flow from group 3 may have occurred to Japanese cultivars (22–24, 28, 38, and 48) and wild strains (52, 53, and 55–57). It is interesting that genetic resources belonging to group 3 are not suitable for edible use and do not have useful traits that can be used to develop new varieties, such as disease resistance.
There are two possibilities for the origin of group 3 plants growing in Japan. The first possibility is that these plants are indigenous to Japan that have not been introduced by human activities. Genetic diversity analysis using SSR markers has demonstrated that wild strains and cultivars in Japan are genetically different14, and our data support this conclusion. Group 3 plants growing in Japan had the lowest mean values of π and He (Table 3). The Fst values indicated that group 3 was genetically closer to group 1 than to group 2 (Table 2). A notable feature of group 3 was that the lowest Fis value (Table 3), suggesting that humans may not have played a role in the formation of this group. Admixture analysis detected no gene flow from other groups to group 3, even though groups 1 and 3 were genetically close, suggesting that the ancestors of group 3 were not introduced by humans from China, but were indigenous to Japan. Common plants often grow in laurel forest ecoregions in East Asia. Loquat is found in these ecoregions, and may have been growing in Japan since prehistoric times. If these plants are indigenous to Japan, the problem to be elucidated in the future is whether plants belonging to this group are present in China. The second possibility is that the group 3 plants growing in Japan were brought to Japan by human activities from China over the last few thousand years. Although modern people do not use group 3 plants for edible purposes, we cannot rule out the possibility that people in the past used them for edible purposes. In order to clarify these issues, it would be desirable to test and analyze individuals from southern China, such as those from Yunnan, Guangdong, and Guangxi provinces, as well as from the northern parts of Southeast Asian countries.
In this study, we also examined the contribution of breeding through asexual reproduction, as in the case of bud sport mutations (Table 4). We confirmed the available records of the asexual emergence of ‘Morimoto’ from ‘Tanaka’. We also found that ‘Moriowase’, ‘Amakusawase’, and ‘Amakusagokuwase’ were asexually propagated from a single tree. On the other hand, we rejected the possibility that ‘Moriowase’ originated asexually from ‘Mogi’. The above cultivars determined to have been born by asexual reproduction were very similar in fruit traits to their parent cultivars. Thus, our DNA-level analysis allowed to clarify the origin of some cultivars.
This study demonstrates that RAD-Seq analysis is applicable to the genome analysis of loquat, which has relatively low genetic diversity14. The information obtained here can be used for loquat cultivar identification and DNA profiling, and in genetic research and breeding programs.
Methods
Plant materials
The 95 cultivars and strains examined in this study are listed in Table 1. Among them, 24 are Japanese cultivars, including 11 produced by crossbreeding. The Japanese cultivar ‘Morimoto’ is probably bud sport mutant of ‘Tanaka’, and ‘Moriowase’ is probably bud sport mutant of ‘Mogi’. We also examined 21 cultivars from China, 19 Japanese wild strains, 6 strains from Vietnam, 8 cultivars from Israel, 5 strains from Greece, 3 cultivars from the United States of America, and 3 strains from Mexico. All plants were grown at the Nagasaki Prefectural Agricultural and Forestry Technology Development Center, Nagasaki, Japan. These genetic resources were introduced to Japan before 1997. The collectors took the permit, which was required at the time, and obtained the owner's permission. Samples 33–36 were technical replicates of sample 32, and samples 40 and 44 were technical replicates of samples 39 and 43, respectively.
DNA extraction and double-digest restriction site-associated DNA sequencing
Genomic DNA was extracted by the CTAB method28, treated with RNase and then with phenol/chloroform. The concentration of DNA was measured with a Qubit dsDNA BR Assay Kit (Invitrogen, MA, USA) and adjusted to 20 ng/µl. Libraries for ddRAD-Seq were prepared using the method of Sakaguchi et al.29, which is a modification of the original ddRAD-Seq method30. The libraries were sequenced by Macrogen (Seoul, South Korea) in one lane of a HiSeq 2000 (Illumina, San Diego, CA, USA). The reads corresponding to each sample were provided.
Analysis of ddRAD-Seq data
Adapter sequences and low-quality bases were trimmed using fastp v 0.23.0 with default parameters31. The genome sequence of a Chinese cultivar ‘Seventh star’ was used as the reference32. The BWA-MEM algorithm of Burrows-Wheeler Aligner (version 0.7.17 r1188)33 was used for mapping to the reference genome sequence. The ref_map.pl script implemented in the Stacks package (version 2.55)34 was used for SNP identification and genotyping (with-X “populations: − R 0.7—ordered-export—vcf—plink—phylip—phylip-var—write-single-snp—min-maf 0.05”). For the PCA and MDS analyses, genotyping data were created to specify all the samples by assigning one individual per population. Each Obs_Het value from “poples.sumstats_summary.tsv” was used for the heterozygosity of each individual. This program was also used to create pairwise alignments between two individuals by changing the -R option to one.
For statistical analysis, groups separated by PCA and MDS analyses were considered as separate populations in the population map data required for the Stacks package. The denovo_map.pl script was re-performed after modifying the data for the population map (with − r 0.7-X “populations: -k—ordered-export—vcf—plink—phylip—phylip-var—write-single-snp—min-maf 0.05”).
Principal component analysis, multidimensional scaling analyses and cluster analysis
The SNPRelate package35 in the R software environment (version 4.1.1) and the vcf file generated by the denovo_map.pl script were used for PCA and MDS analyses. The program converted the vcf file into a gds (genomic data structure) file and created a PCA diagram. Then, the contribution of each principal component was calculated. At this step, only bi-allelic loci were used. The program also used the gds file to create an MDS diagram. The resulting images were generated with the basic functions of the R software environment. The SNPRelate program plotted the dendrogram by considering the identity by state (IBS) pairwise distance. Images of the results were generated using the basic functions of R software environment.
Admixture analysis
The PLINK 2 program (version 1.90p6.21)36 was used to create the input file for the Admixture program. The admixture program (version 1.3.0)37 was used to create a cross between the history of admixture and a hypothetical run with K (number of ancestral populations) = 1 to 10. The cross-validation error plots were drawn using the obtained log data. The basic functions of R software environment (version 4.1.1) and the Q estimate file created by the admixture program were used to draw the admixture plots.
Data availability
Sequences are available at the DNA Data Bank of Japan Sequence Read Archive (https://www.ddbj.nig.ac.jp/dra/index-e.html; Accession no. DRA013114).
References
Chevreau, E., Lespinasse, Y. & Gallet, M. Inheritance of pollen enzymes and polyploid origin of apple (Malus × domestica Borkh.). Theor. Appl. Genet. 71, 268–277. https://doi.org/10.1007/BF00252066 (1985).
Potter, D. et al. Phylogeny and classification of Rosaceae. PIant Syst. Evol. 266, 5–43 (2007).
Campbell, C. S., Evans, R. C., Morgan, D. R., Dickinson, T. A. & Arsenault, M. P. Phylogeny of subtribe (formerly the Maloideae, Rosaceae): Limited resolution of a complex evolutionary history. PIant Syst. Evol. 266, 119–145. https://doi.org/10.1007/s00606-007-0545-y (2007).
Lin, S. Plant material of loquat in Asian countries. In First International Symposium on Loquat 41–44. https://www.actahort.org/books/403/403_26.htm (2002).
Ding, C., Chen, Q., Sun, T. & Xia, Q. Germplasm resources and breeding of Eriobotrya japonica Lindl. in China. Acta Hort. 403, 121–126 (1995).
Wang, Y., Shahid, M. Q., Lin, S., Chen, C. & Hu, C. Footprints of domestication revealed by RAD-tag resequencing in loquat: SNP data reveals a non-significant domestication bottleneck and a single domestication event. BMC Genomics 18, 354. https://doi.org/10.1186/s12864-017-3738-y (2017).
Bagot, L. W. Letter and notes. Trans. Hort. Soc. Lond. 3, 299–304 (1820).
Morton J. F. Loquat. In: Fruits of warm climates. Creative Resource Systems, Inc., Winterville, 103–108. https://www.hort.purdue.edu/newcrop/morton/loquat.html (1987).
Condit, I. J. The loquat. Calif. Agr. Expt. Sta. Bul. 250, 251–284 (1915).
Ichinose, I. The origin and development of loquat (in Japanese), Series of Agricultural Technology, 4: 1–8. Nousangyosonbunkakyoukai, Tokyo. (1983).
Nesumi, H. Loquat (Biwa). Japan. Soc. Hort. Sci. (ed.). Horticulture in Japan 2006. Shoukadoh Publication, Kyoto. 85–95. (2006).
Kikuchi, A. Kajuengeigaku (in Japanese). Yokendo, Tokyo. (1948).
He, Q. et al. Genetic diversity and identity of Chinese loquat cultivars/accessions (Eriobotrya japonica) using apple SSR markers. Plant Mol. Biol. Rep. 29, 197–208. https://doi.org/10.1007/s11105-010-0218-9 (2011).
Fukuda, S. et al. Genetic diversity of loquat accessions in Japan as assessed by SSR markers. J. Jpn. Soc. Hort. 82, 131–137. https://doi.org/10.2503/jjshs1.82.131 (2013).
Li, X., Xu, H., Feng, J., Zhou, X. & Chen, J. Mining of genic SNPs and diversity evaluation of landraces in loquat. Sci. Hortic. 195, 82–88. https://doi.org/10.1016/j.scienta.2015.08.040 (2015).
Penjor, T., Mimura, T., Matsumoto, R., Yamamoto, M. & Nagano, Y. Characterization of limes (Citrus aurantifolia) grown in Bhutan and Indonesia using high-throughput sequencing. Sci. Rep. 4, 1–9. https://doi.org/10.1038/srep04853 (2014).
Penjor, T. et al. RAD-Seq analysis of typical and minor Citrus accessions, including Bhutanese varieties. Breed. Sci. 66, 797–807. https://doi.org/10.1270/jsbbs.16059 (2016).
Kato, D. et al. Evaluation of the population structure and phylogeography of the Japanese Genji firefly, Luciola cruciata, at the nuclear DNA level using RAD-Seq analysis. Sci. Rep. 10, 1533. https://doi.org/10.1038/s41598-020-58324-9 (2020).
Premarathne, M. D. G. P. et al. Elucidation of Japanese pepper (Zanthoxylum piperitum De Candolle) domestication using RAD-Seq. Sci. Rep. 11, 6464. https://doi.org/10.1038/s41598-021-85909-9 (2021).
Orita, R., Nagano, Y., Kawamura, Y., Kimura, K. & Kobayashi, G. Genetic diversity and population structure of razor clam Sinonovacula constricta in Ariake Bay, Japan, revealed using RAD-Seq SNP markers. Sci. Rep. 11, 7761. https://doi.org/10.1038/s41598-021-87395-5 (2021).
Fukuda, S. et al. Construction of a high-density linkage map for bronze loquat using RAD-Seq. Sci. Hortic. 251, 59–64. https://doi.org/10.1016/j.scienta.2019.02.065 (2019).
Nagano, Y. et al. Phylogenetic relationships of Aurantioideae (Rutaceae) based on RAD-Seq. Tree Genet. Genomes. 14, 6. https://doi.org/10.1007/s11295-017-1223-z (2018).
Hiehata, N., Fukuda, S., Sato, Y., Tominaga, Y. & Terai, O. Quantitative inheritance of resistance to loquat canker (Pseudomonas syringae pv. eriobotryae, Group C) in loquat progenies from crosses between a resistant cultivar, ‘Champagne’, and susceptible cultivars. HortScience 49, 1486–1491. https://doi.org/10.21273/HORTSCI.49.12.1486 (2014).
Ding, C., Chen, Q., Sun, T. & Xia, Q. Germplasm resources and breeding of Eriobotrya japonica Lindl. In china. Acta Hort. 403, 121–126 (1995).
Blasco, M., Naval, M. D. M., Zuriaga, E. & Badenes, M. L. Genetic variation and diversity among loquat accessions. Tree Genet. Genomes 10, 1387–1398. https://doi.org/10.1007/s11295-014-0768-3 (2014).
Nagato, J. et al. Characteristics in generic resources of Eriobotrya spp. Bul. Nagasaki Fruit Tree Expt. Sta. 3, 55–77 (1996).
Nakayama, H. et al. Characteristics in genetic resources of Eriobotrya spp (2nd report). Bul. Nagasaki Agric. For. Tech. Dev. Center 1, 113–134 (2010).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Sakaguchi, S. et al. High-throughput linkage mapping of Australian white cypress pine (Callitris glaucophylla) and map transferability to related species. Tree Genet. Genomes 11, 121. https://doi.org/10.1007/s11295-015-0944-0 (2015).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLOS ONE 7, e37135. https://doi.org/10.1371/journal.pone.0037135 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. https://doi.org/10.1093/bioinformatics/bty560 (2018).
Jiang, S., An, H., Xu, F. & Zhang, X. Chromosome-level genome assembly and annotation of the loquat (Eriobotrya japonica) genome. Giga Sci. 9, 1–9. https://doi.org/10.1093/gigascience/giaa015 (2020).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 1303.3997 (2013).
Rochette, N. C., Rivera-Colón, A. G. & Catchen, J. M. Stacks 2: Analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754. https://doi.org/10.1111/mec.15253 (2019).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328. https://doi.org/10.1093/bioinformatics/bts606 (2012).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. https://doi.org/10.1086/519795 (2007).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. https://doi.org/10.1101/gr.094052.109 (2009).
Acknowledgements
This work was partially supported by a KAKENHI Grant-in-Aid for Scientific Research(C) (18K05624) from the Japan Society for the Promotion of Science (JSPS). We are grateful to Dr. Shao-Hui Zheng for confirming the English transliteration of the Chinese accession names.
Author information
Authors and Affiliations
Contributions
Y.N. and S.F. designed the study. N.H. and S.F. collected samples; H.T. and S.N., performed DNA extraction. Y.N., H.T. and S.N. performed bioinformatics analysis. A.J.N. performed RAD-Seq analysis. Y.N., N.H. and S.F. analyzed the data; Y.N. and S.F. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nagano, Y., Tashiro, H., Nishi, S. et al. Genetic diversity of loquat (Eriobotrya japonica) revealed using RAD-Seq SNP markers. Sci Rep 12, 10200 (2022). https://doi.org/10.1038/s41598-022-14358-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-14358-9
This article is cited by
-
Genetic structure and origin of emu populations in Japanese farms inferred from large-scale SNP genotyping based on double-digest RAD-seq
Scientific Reports (2024)
-
Soil bacterial and protist communities from loquat orchards drive nutrient cycling and fruit yield
Soil Ecology Letters (2024)
-
Single nucleotide polymorphisms reveal the uniqueness of Gushan semi-rock tea in the tea germplasm resources of Fujian, China
Genetic Resources and Crop Evolution (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
