
Abstract
Individuals encounter varying environmental exposures throughout their lifetimes. Some exposures such as smoking are readily observed and have high personal recall; others are more indirect or sporadic and might only be inferred from long occupational histories or lifestyles. We evaluated the utility of using lifetime-long self-reported exposures for identifying differential methylation in an amyotrophic lateral sclerosis cases-control cohort of 855 individuals. Individuals submitted paper-based surveys on exposure and occupational histories as well as whole blood samples. Genome-wide DNA methylation levels were quantified using the Illumina Infinium Human Methylation450 array. We analyzed 15 environmental exposures using the OSCA software linear and MOA models, where we regressed exposures individually by methylation adjusted for batch effects and disease status as well as predicted scores for age, sex, cell count, and smoking status. We also regressed on the first principal components on clustered environmental exposures to detect DNA methylation changes associated with a more generalised definition of environmental exposure. Five DNA methylation probes across three environmental exposures (cadmium, mercury and metalwork) were significantly associated using the MOA models and seven through the linear models, with one additionally across a principal component representing chemical exposures. Methylome-wide significance for four of these markers was driven by extreme hyper/hypo-methylation in small numbers of individuals. The results indicate the potential for using self-reported exposure histories in detecting DNA methylation changes in response to the environment, but also highlight the confounded nature of environmental exposure in cohort studies.
Similar content being viewed by others

Introduction
DNA methylation (DNAm) is the addition of a methyl group to DNA, most commonly at CpG dineculotide sites in mammals. DNAm provides an epigenetic mechanism for the regulation of transcription, playing a vital role in cellular differentiation, X chromosome inactivation and maintaining genomic stability1. Furthermore, DNAm is influenced by both innate effects (i.e., ageing and cell type) as well as environmental exposures which further interact with each other2.
If predictable DNAm changes occur as a consequence of environmental exposure then DNAm measures can be used to construct a proxy measure for exposure in the absence of direct measurement of the exposure. For example, cigarette smoking results in substantial changes in DNAm at hundreds of sites across the genome3, with the changes in a proportion of these sites remaining long after smoking cessation4. The methylation at these CpG sites can be used to predict both current and prior smoking status, with the potential to validate or replace inaccurate measures such as self-reporting5. Individuals whose exposure to cigarette smoking is secondary (e.g., through living with a spouse who smokes or being raised in a smoking household) can be identified from DNA methylation measures, whereas they would otherwise self-report as a non-smoker6. This approach of identifying historical exposures from their effects on DNAm has a range of potential uses, particularly in disease epidemiology, because relevant exposures may be difficult to quantify or are unknown to the study participant.
Environmental toxicants are suspected to have a role in the etiology of some neurodegenerative disorders7,8. For example, lead and aluminium exposure accelerates the accumulation of amyloid-peptide deposits related to Alzheimer’s disease8,9 and manganese can trigger Parkinsonism10. Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by upper and lower motor neuron dysfunction with little variation being explained by known heritable genetic factors, and is the most prevalent form of all motor neurone diseases (MND)11. ALS has been reported to be associated with a large range of environmental factors, including heavy metals such as lead7,12, cycad genotoxins13, and unknown causative agents associated with military service14. However, the replication of these association studies has been poor15, mostly due to difficulties in quantifying individual exposure due to a lack of standardised methodology.
In the current study, methylation-wide association studies (MWAS) were performed on a range of self-reported environmental exposures as potential ALS risk factors. While the study participants were ALS patients and non-ALS controls, the focus was on the exposures using ALS status as a covariate. Several associations between DNA methylation and metal exposures were identified, which highlights the confounded nature of assessing many exposures.
Materials
Sample collection and participant inclusion
ALS patients (cases) and controls were recruited between April 2000 to June 2011 as part of The Australian MND DNA Bank from the University of Sydney16. Cases were white Australians older than 25 years recruited from around Australia via state-based MND association newsletters, which were classified as having ALS based on verification by a neurologist17. Controls had no history of neuromuscular disease, and were recruited as friends or spouses of case participants, or community volunteers. Exclusion criteria were any family history of ALS and slowly progressive disease, defined as survival for greater than 10 years after diagnosis. Participants provided a blood sample for DNA analysis.
Participants were asked to complete a 6-page paper questionnaire. This questionnaire included short-form, multiple-choice and yes/no questions under the following categories: General, lifetime residences, lifetime travel, family illnesses, lifetime employment, exposure to chemicals or toxins, injuries, physical exercise, personal habits, past illnesses, and medication. A total of 855 genetically confirmed unrelated individuals (438 ALS cases and 417 controls) were included in the analysis. The age and sex distributions of the groups are reported in Table S1, with mean age and percentage female being 59 years and 54% for controls, 62 years and 38% for cases, respectively.
Collection of DNA and questionnaire information was approved by the Human Research Ethics Committee of the Sydney South West Area Health Service. All DNA data generation and analyses were approved by the University of Queensland Human Research Ethics Committee. All research was performed in accordance with relevant regulations and guidelines, and informed consent was obtained from all participants.
Evaluation of environmental exposures reported in questionnaire
Figure S1 delineates the steps of the analysis as a flowchart. A total of 15 binary-response (yes/no) questions relating to environmental exposures were selected from the questionnaire. Unmarked fields were interpreted as an implied ‘no’, since participants often responded only in the case of exposure18. To consolidate highly-related exposures, we used the hierarchical clustering algorithm within the R Clustovar package19. Cluster numbers from 2 to 15 were tested, with the cluster stability (Figure S2) and the variance explained by the first principal component of the clusters (Figure S3) used to determine the optimal grouping. The principal component analyses of the final clusters are reported in Figure S4. All 15 individual binary variables were exported as outcome variables for the methylation profiling, as well as the first principal component of each of the four final clusters (Fig. 1). Self-reported current smoking status was used as a validation variable due to its established status as an exposure that causes methylation changes.

Correlation network of tested environmental phenotypes. Edges shown have \(p < 0.05\) after Holm correction for multiple testing. Edge weight and repulsion are based on strength of correlation, which are all positive. Nodes are colored based on final cluster allocation: Mine (blue), Chem (orange), Farm (green) and 'blue collar' Work (purple) exposures. HP: Herbicide/pesticide. Chem/Solv: Other chemicals or solvents. Cu: Copper. Cd: Cadmium. Hg: Mercury. Pb: Lead. For variables suffixed with n mo., they qualify living in that locale within the last n months.
Array and DNA methylation processing
Measurement and normalisation of DNAm is described elsewhere20. Briefly, typing for \(468,521\) DNAmsites was conducted using the Infinium HumanMethylation450 BeadChip array, following the standard Infinium HD Methylation protocol. The R Bioconductor package meffil21 was used for initial processing, normalization and quality control following filtering thresholds, as detailed previously20. The mean and minimum bisulfite II conversion efficiencies were 12.47 and 11.51, respectively, falling above the suggested manufacturer threshold of 1 for successful conversion and higher than the majority of values reported in a metanalysis of 80 datasets22. Cell-type proportions were estimated based on the GSE35069 blood methylome profile through the meffil implementation of the Houseman algorithm23. Probes which were sex-chromosome linked, non-uniquely hybridized, or with internal SNPs adjacent to the 3’ end were removed according to previously recommended criteria24. In addition, probes with low variability (SD < 0.02) were removed. After quality control, a total of \(m = 156,874\) DNAm sites remained.
Probe measures were precorrected with predicted scores for sex, age25, smoking26 (omitted for the smoking analysis), and for predicted cell types B, CD4-T, CD8-T, NK-T, monocytes and neutrophils to address bias in cell type composition in DNAm23. To account for batch effects, row and column position were added to the models, with slide (beadchip) as the sole random effect. The residuals of these linear mixed-effects models, generated through the R lme4 package27, were taken forward for the analyses.
Methylome-wide association analysis
Omics-data-based Complex Trait Analysis (OSCA) software28 was used to conduct the methylome-wide association analysis for the exposure variables. The effects of environmental exposures on DNA methylation were tested using several approaches. First, simple linear regressions with each exposure or exposure PC considered as the dependent variable regressed on a single probe fitting ALS status (a binary value denoting control or case) as a covariate. Secondly, we used OSCA’s mixed model approach MLM-based omic association, or MOA using ALS as a covariate, but with a random effect added to the linear model representing genome-wide DNAm level of an individual. The variance/covariance structure of the random effect is described by the omic-data-based relationship matrix (ORM), in which off-diagonal elements capture the genome-wide similarity of DNA methylation values between pairs of individuals. This approach has been shown in the original publication to be robust to confounding of probes across the the genome. When possible, we also ran the highly conservative multi-component MLM-based omic association excluding the target (MOMENT) method which used two random effects in the model constructed from two sets of probes grouped based on genome-wide significance, omitting those within 50 kb of the tested probe. Applying the MOMENT method is only possible or relevant when a set or locally high correlated probe associations is detected29. We stringently accounted for multiple testing across CpGs by using a Bonferroni correction for epigenome-wide significance threshold (\(0.05/m = 3.19* 10^{ - 7}\) for the m DNAm sites tested). No additional multiple testing corrections were made for the 15 exposures due to their extensive correlation structure.
To verify the robustness of the significance tests, 1000 permutations of the MOA model were performed for phenotypes with epigenome-wide significant probes, by random reassignment of the phenotype (with or without replacement) to the DNAm data. The number of epigenome-wide significant probes for each permutation, as well as the p-values of previously detected probes, was extracted from each model.
A differentially methylated region (DMR) analysis was applied to the complete results of all MOA models using comb-p to determine whether the epigenome-wide differentially methylated probes belong to a DMR of probes methylated in the same direction. This software’s full pipeline aggregates the p-values of adjacent probes to calculate an autocorrelation factor, combines p-values through the modified Stouffer-Liptak-Kechris (SLK) correction and finally returns region p-values with and without the multiple testing Sidak correction30. The parameters for seed threshold was set to 0.05 and window distance to 750 base pairs as the best performing settings according to a previously published benchmark study31.
Results
Endorsement counts of environmental exposures included in the analyses are reported in Table 1. Across these 15 variables, the median response rate was 96% with a minimum of 71%. These variables all had high proportions of nonresponse which were interpreted as ‘no’ responses, given that other parts of the questionnaire had been fully completed.
Clustering of environmental exposures
The clustering analysis of the questionaire response items generated four partitions, with this choice being justified by cluster stability analyses, and the variance explained by the principal components of the partitions (Figures S1-3). The correlation network (Fig. 1) and the clustering dendrogram (Fig. 2) illustrate these final four partitions. The cluster with the largest height or homogeneity, with 5 constituent exposures, represents questions that can be broadly summarized as metal exposures. These relate to either exposure to named metals (cadmium, lead, copper or mercury) or working in occupations that involve metallurgy, either handling metallic ores or creating metals. We labelled this partition ‘Metal’. The second largest partition represents indicators for living or working in rural/farm areas. In addition to survey questions relating to residence outside of urban areas, the exposure to and use of bore water was included in this cluster which we refer to as ‘Farm’. The third partition consists of a grouping of herbicide/pesticide (HP) exposures (Intermittent domestic, Regular domestic or Industrial use) as well as exposure to other chemicals or solvents. The fourth partition was “blue collar” or “manual” work, with the inclusion of diesel exposure, or manual labor roles obtained from occupational reporting. We refer to these two groups as Chemical (abbreviated Chem) and Work, respectively.

Hierarchical clustering of exposure measures. Exposures were separated into \(k = 4\) clusters (coloured) based on cluster stability over 100 bootstrap samples.
The grouping of manual labor and exposure to diesel fuel in the fourth partition may reflect the use of occupation-reporting fields to identify diesel exposure. Diesel exposure was not explicitly indicated on the questionnaire, but we interpreted likely diesel exposure as having worked within a set of ANZCO/ISCO occupation codes (Table S2) formerly established as high users of diesel fuel through this cohort by Pamphlett and Rikard-Bell32. The classification of former or current work in manual labor was directly indicated for each listed occupation on the questionnaire form.
The correlation network (Fig. 1) shows a degree of correlation between all exposures. In the case of the metal exposures, the proportion of cases and controls who have reported exposure is relatively small (\(\le 17.8\%\), Table 1). The first principal component generated from each of the four partitions was then used as a continuous variable representative of its correlated constituents, which are otherwise binary. The proportion of variance explained by each are 51% for Mine, 46% for Chem, 50% for Farm, and 72% for Work (Figures S2-3).
Methylome-wide association analyses
We benchmarked our dataset for the ability to detect effects on DNAm from environmental exposures by using the exposure variable of self-reported smoking. Within our cohort of 855 participants, 50 of 438 ALS cases and 32 of 417 controls report themselves as current smokers (Table 1). Linear and MOA analyses identify \(m = 212\) and \(m = 74\) smoking-associated probes, respectively (minimum association p-value of 3.19e − 7). The Pearson correlation of smoking probe effect sizes compared to those published previously33 (for the current versus never-smoker contrast) was 0.64 and 0.68 for the linear and MOA and models respectively, comparing probes FDR < 0.05 in the reference data (\(m = 18,760\)). The MOMENT analysis identified two probes associated with AHRR that have been shown to explain the largest proportion of variation in smoking status and lung cancer risk34, as well as an unannotated intergenic region. These AHRR probes (cg05575921 and cg26703534) are hypomethylated in smokers, while the significance of the unannotated region appears to be driven by hypomethylated outliers in the nonsmoking group (Figure S5). The stringency of MOMENT compared to the linear and MOA methods is visualized within Figures S5-6. The top 20 probes detected in the MOA analysis are reported in Figure S4. These results demonstrate that our cohort size is well-powered to detect these known effects, with a smoking rate of ~ 10%.
In contrast to self-reported smoking status, there were fewer associations detected across the 15 individual binary environmental exposure variables. A total of 7 epigenome-wide significant sig- natures were detected by linear regression across exposures for cadmium, mercury, and metallurgy (Table 2). Of these associated CpG sites, cg10071091 (annotated to KSR2) for metallurgy was the only one that did not pass the significance threshold via MOA. Likewise, the Chem PC1 continuous variable was associated with cg09369863 (annotated to LINGO1) through the linear method, though not MOA. The peaks of co-located inflated loci as seen in the smoking MWAS using MOA (Figure S7b) are not present in any of the environmental MWAS with associated CpGs (Figure S8) and as such the gain in stringency from MOMENT28 is not needed. The Q-Q plots of each of these models are reported in Figure S9, with little difference between the linear and MOA methods. We consider the small deflation in significance from the linear to the MOA models as indicative that the observed associations in the linear models were not solely influenced by the confounding structure within local and distal probes, and regard probes detected by both methods as less likely to be false positives.
The direction of differential methylation at these CpG sites was mixed (Fig. 3, Table 2). In response to cadmium exposure, cg03085637 (in proximity to GNRHR2 and within PEX11B) and cg16655883 (annotated to ZFR2) were hypermethylated, while cg21124714 (P2R76) was hypomethylated. For Metallurgy, cg07503918 (PRKG1-AS1) was hypermethylated and the cg10071091 site hypomethylated. Mercury exposure replicated a similar effect for cg03085637 as cadmium exposure, while cg16845679 (an intergenic CpG site on on chromosome 12) was hypomethylated.
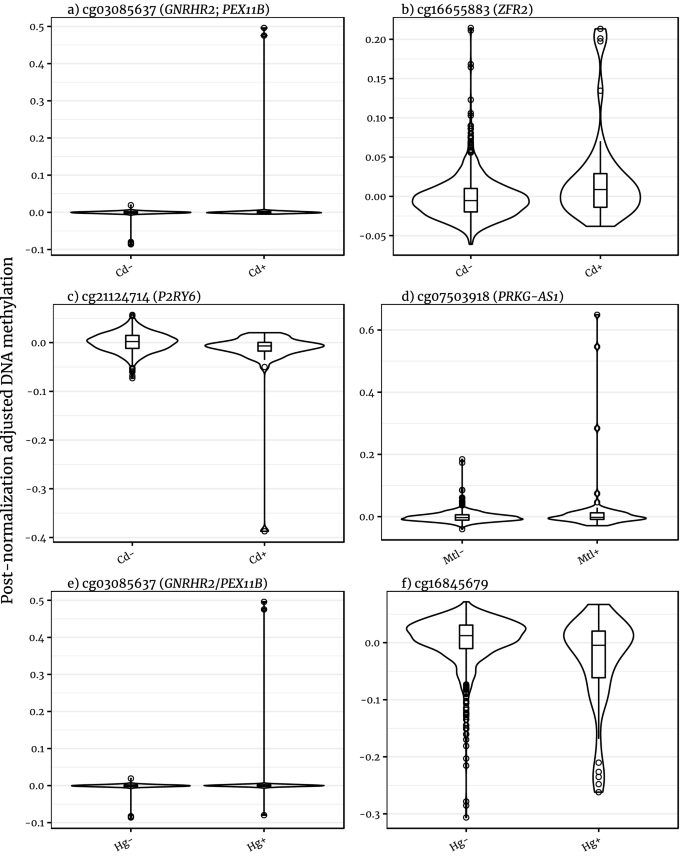
Violin plots of the distribution of normalised DNA methylation values for significant probe-exposure associations. Cd: cadmium; Mtl: metallurgy; Hg: mercury; -: no exposure + : exposure.
The association of the cg03085637 site to both cadmium and mercury exposure highlights the correlation between the two exposures and abnormal hypermethylation. For example, of 855 participants, 19 reported exposure to cadmium and not mercury, 30 to mercury and not cadmium, and 21 to both. Figure 3 shows the DNAm values for these epigenome-wide significant sites. Figure 3a shows cg03085637 DNAm values plotted by the cadmium exposure, while Fig. 3e shows the sites separated by the mercury exposure. Notably, the significance of this GNRHR2/PEX11B probe is driven by two cadmium and mercury-exposed individuals with extreme hypermethylation (\(v > {\text{Q}}_{3} + 100 \times {\text{IQR}}\), where IQR is the interquantile range, Q is the quantile and \(v\) is the adjusted methylation value), which illustrates the difficulty in differentiating the exposures.
This pattern of few extreme methylation values skewing the distributions in the exposure groups is reflected in other MOA-significant DNAm sites. The association between DNAm values of cg21124714 and exposure to cadmium (Fig. 3c) was driven by one individual (\(v \approx {\text{Q}}_{1} - 15 \times {\text{IQR}}\)). Of the 22 individuals reporting having worked with metals, 6 showed extreme methylation in the cg07503918 site, as evident in Fig. 3d (\(v \le {\text{Q}}_{3} + 9 \times {\text{IQR}}\)). Both cadmium and mercury exposures are associated with DNAm levels which are not principally driven by a few outliers, cg16655883 and cg16845679, respectively (Fig. 3b,f). Associations at individual CpG sites were not supported by differences at nearby methylation loci, although this is common in MWAS due to lack of correlation between co-located probes on the array35.
Figures S9 and S10 show results of the permutation analyses of the MOA models for cadmium, mercury and metallurgical exposures. The six epigenome-wide significant loci remain significant under permutation mode as visualized in Figure S10. This indicates that the reported associations based on hyper- or hypomethylated outliers are unlikely to be incidentally captured by chance. However, genome-wide permutations given in Figure S11 show the number of epigenome-wide significant loci in this study was not more than expected by chance. Hence, these results should be treated with caution until future studies are able to provide validation data.
Our DMR analysis of the MOA results indicate that none of the previously detected significant probes fall within DMRs and exist as singular hyper- or hypomethylated points. However, the DMR analyses were conducted on the subset of probes passing the 0.02 SD threshold on the already limited 450 k probe array which covers a small fraction of CpG sites across the genome. Figure S12 is a histogram displaying the number of probes in a given 750 bp window, with 43.4% of the probes in the analysis having no others within their own window (Figure S12a). There is little evidence of artificial inflation of the previous results due to spatially correlated probes; however, this does not discount that methylation can occur in the flanking region. Of the 15 phenotypes tested through comb-p, 13 reported DMRs of more than 1 probe with significant Sidak-corrected regional p-values, with the highest number (\(n = 16\)) of DMRs associated with bore water exposure. All DMRs comprised probes with the same direction of effect. These significant DMRs are reported for each tested phenotype in the supplementary file Table S4.
Discussion
We performed MWAS with a range of environmental exposures potentially associated with ALS, identifying several associations between DNAm and exposure to heavy metals in analyses that fitted ALS case/control status as a covariate. DNAm near the PEX11B gene was found to be associated with cadmium exposure. PEX11B is expressed widely across tissue types, and its aggregation on the perixosomal membrane initiates the elongation and then constriction required in the replication pathway of peroxisomes36. This gene family has been measured to respond to cadmium exposure in Arabidopsis in targeted time-course analyses, with increased expression of PEX11 isoforms (PEX11A and PEX11E)37.
We replicated previously published observations of methylation changes in response to smoking status. Unlike smoking, the effects of the other exposures on DNAm were more limited, possibly attributable to reporting and recall. Exposures were variably reported, with cadmium exposure as the least frequent (5%). Exposures were self-reported or derived in the case of diesel, with severity, duration and temporal period unreported for all exposures excepting smoking. We acknowledge that exposures should be regarded as continuous and not binary as encoded in the MWAS analysis, specifically those which are able to be physiologically measured through established biomarkers. For example, the long half-life of 13.6 years of Cadmium in urine38 would enable it to be a tested as a continuous outcome variable in MWAS if measured in this cohort. Including biological verification of self-reported exposures would be an advantage for future study designs of the epigenetic effects for environmental factors. Additionally, while extreme variations in methylation values (epivariations) are principally associated with epigenetic remodelling, others are a secondary consequence of cis-linked DNA mutations that can be verified through targeted sequencing 39.
There are several possibilities for the differences in epigenome-wide significant sites of methylation as a result of environmental exposures. In particular, cigarette smoke is a ubiquitous and highly frequent exposure with evidence of bioaccumulation in most of the population40 and highly associated tobacco-related biomarkers in active smokers41. Biomarkers for other exposures may have short half-lives in the span of weeks or be rapidly metabolized, as well as having low chemical concentrations, vulnerability to handling contamination42, and variability in measurements within and between laboratories43. Measurable toxicants emitted per cigarette dose are comparatively large, with tar levels between \(7.4 - 19.4\) mg44. In comparison, ambient air cadmium concentrations measured at a cadmium pigment factory ranged from \(0.02 - 1.22\) mg/m3 in the first year, to \(0.01 - 0.52\) mg/m3 in the second, depending on the tasks performed45. Finally, the measurement method (self-reported exposure as a binary trait) limits the detection of the effect. Our permutation analyses highlighted that while the individual significant probes have robust significance, we did not observe more significant probes epigenome-wide than expected by chance. These limitations indicate the need for substantially larger sample sizes to robustly detect associations with such exposures.
Correlation between exposures reported by participants introduces additional difficulties in interpreting the relationship between DNAm and exposures. This is illustrated by the association with PEX11B and cadmium, where the two individuals who drove the significance of this effect both shared self-reported cadmium and mercury exposures. Whether this effect is related to either of the two reported metal-based variables, or to another unknown shared exposure or cause remains indecipherable with the available data and the limitation of it being observed in only two individuals. Replication in other data sets is needed to verify these results. The first principal component of all metal-related exposures, which accounted for 51% of their variance, was used as a proxy for this invisible “lifestyle type”. However, no associations were identified with metal-PC1 at the methylome-wide significance threshold.
In summary, we attempted to predict a set of self-reported environmental exposures through an MWAS consisting of 855 ALS cases and controls. We illustrate the utility of capturing historical exposures as a way of elucidating the epigenome, with the qualifier that larger study cohorts will be required to differentiate these effects. While smoking may be detected in a small cohort owing to its effect size, other exposures will require more individuals to distinguish small effects or to correctly differentiate outliers in methylation. We present the summary statistics of associations to allow future replication and meta-analyses.
Data availability
The DNA methylation array data are available in the NCBI dbGaP repository with accession number phs002068.v1.p1.
References
Allis, C. D. & Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17, 487–500 (2016).
Christensen, B. C. et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, e1000602 (2009).
Wan, E. S. et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum. Mol. 21, 3073–3082 (2012).
Wilson, R. et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics 18, 805 (2017).
Maas, S. C. E. et al. Validated inference of smoking habits from blood with a finite DNA methylation marker set. Eur. J. Epidemiol. 34, 1055–1074 (2019).
Reynolds, L. M. et al. Secondhand tobacco smoke exposure associations with DNA methylation of the aryl hydrocarbon receptor repressor. Nicotine Tob. Res. 19, 442–451 (2016).
Wang, M.-D., Gomes, J., Cashman, N. R., Little, J. & Krewski, D. A meta-analysis of observational studies of the association between chronic occupational exposure to lead and amyotrophic lateral sclerosis. J. Occup. Environ. Med. 56, 1235–1242 (2014).
Chin-Chan, M., Navarro-Yepes, J. & Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front. Cell .Neurosci. 9, (2015).
Cheignon, C. et al. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 14, 450–464 (2018).
Guilarte, T. R. & Gonzales, K. K. Manganese-induced parkinsonism is not idiopathic Parkinson’s disease: Environmental and genetic evidence. Toxicol. Sci. 146, 204–212 (2015).
Mathis, S., Goizet, C., Soulages, A., Vallat, J.-M. & Masson, G. L. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 399, 217–226 (2019).
Wang, M.-D., Little, J., Gomes, J., Cashman, N. R. & Krewski, D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology 61, 101–130 (2017).
Spencer, P. S., Palmer, V. S. & Kisby, G. E. Cycad β-N-methylamino-L-alanine (BMAA), methylazoxymethanol, genotoxicity, and neurodegeneration. Toxicon 155, 49–50 (2018).
Beard, J. D. et al. Military service, deployments, and exposures in relation to amyotrophic lateral sclerosis etiology. Environ. Int. 91, 104–115 (2016).
Belbasis, L., Bellou, V. & Evangelou, E. Environmental risk factors and amyotrophic lateral sclerosis: An umbrella review and critical assessment of current evidence from systematic reviews and meta-analyses of observational studies. Neuroepidemiology 46, 96–105 (2016).
Morahan, J. M. & Pamphlett, R. Amyotrophic lateral sclerosis and exposure to environmental toxins: An Australian case-control study. Neuroepidemiology 27, 130–135 (2006).
Stoyanov, A. & Pamphlett, R. Is the Risk of Motor Neuron Disease Increased or Decreased after Cancer? An Australian Case-Control Study. PLoS ONE 9, e103572 (2014).
Pamphlett, R. Exposure to environmental toxins and the risk of sporadic motor neuron disease: an expanded Australian case-control study. Eur. J. Neurol. 19, 1343–1348 (2012).
Chavent, M., Kuentz-Simonet, V., Liquet, B. & Saracco, J. ClustOfVar : An R Package for the Clustering of Variables. J. Stat. Soft 50, (2012).
Nabais, M. F. et al. Significant out-of-sample classification from methylation profile scoring for amyotrophic lateral sclerosis. NPJ Genom. Med .(2020).
Min, J. L., Hemani, G., Davey Smith, G., Relton, C. & Suderman, M. Meffil: efficient normalization and analysis of very large DNA methylation datasets. Bioinformatics https://doi.org/10.1093/bioinformatics/bty476 (2018).
Heiss, J. A. & Just, A. C. Identifying mislabeled and contaminated DNA methylation microarray data: An extended quality control toolset with examples from GEO. Clin. Epigenet. 10, 73 (2018).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf. 13, 86 (2012).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. https://doi.org/10.1093/nar/gkw967 (2016).
Zhang, Q. et al. Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Med. 11, 54 (2019).
McCartney, D. L. et al. Epigenetic prediction of complex traits and death. Genome Biol. 19, 136 (2018).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Soft 67, (2015).
Zhang, F. et al. OSCA: A tool for omic-data-based complex trait analysis. Genome Biol. 20, 107 (2019).
Zhang, F. et al. OSCA: A tool for omic-data-based complex trait analysis. http://biorxiv.org/lookup/doi/https://doi.org/10.1101/445163 (2018) https://doi.org/10.1101/445163.
Pedersen, B. S., Schwartz, D. A., Yang, I. V. & Kechris, K. J. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics 28, 2986–2988 (2012).
Mallik, S. et al. An evaluation of supervised methods for identifying differentially methylated regions in Illumina methylation arrays. Brief. Bioinform. 20, 2224–2235 (2019).
Pamphlett, R. & Rikard-Bell, A. Different occupations associated with amyotrophic lateral sclerosis: Is diesel exhaust the link?. PLoS ONE 8, e80993 (2013).
Joehanes, R. et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 9, 436–447 (2016).
Fasanelli, F. et al. Hypomethylation of smoking-related genes is associated with future lung cancer in four prospective cohorts. Nat. Commun. 6, 10192 (2015).
Zhang, W., Spector, T. D., Deloukas, P., Bell, J. T. & Engelhardt, B. E. Predicting genome-wide DNA methylation using methylation marks, genomic position, and DNA regulatory elements. Genome Biol. 16, 14 (2015).
Honsho, M., Yamashita, S. & Fujiki, Y. Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. BBA – Mol. Cell Res. 1863, 984–991 (2016).
Rodríguez-Serrano, M., Romero-Puertas, M. C., Sanz-Fernández, M., Hu, J. & Sandalio, L. M. Peroxisomes extend peroxules in a fast response to stress via a reactive oxygen species-mediated induction of the peroxin PEX11a. Plant Physiol. 171, 1665–1674 (2016).
Suwazono, Y. et al. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 14, 77–81 (2009).
Barbosa, M. et al. Identification of rare de novo epigenetic variations in congenital disorders. Nat. Commun. 9, 2064 (2018).
Wei, B., Blount, B. C., Xia, B. & Wang, L. Assessing exposure to tobacco-specific carcinogen NNK using its urinary metabolite NNAL measured in US population: 2011–2012. J. Expo. Sci. Environ. Epidemiol. 26, 249–256 (2016).
Roethig, H. J. et al. Population estimates for biomarkers of exposure to cigarette smoke in adult U.S. cigarette smokers. Nicotine Tob. Res. 11, 1216–1225 (2009).
Esteban, M. & Castaño, A. Non-invasive matrices in human biomonitoring: A review. Environ. Int. 35, 438–449 (2009).
Snoj Tratnik, J., Mazej, D. & Horvat, M. Analytical quality requirements in human biomonitoring programs: Trace elements in human blood. Int. J. Environ. Res. Public Health 16, 2287 (2019).
Melikian, A. A., Djordjevic, M. V., Chen, S., Richie, J. & Stellman, S. D. Effect of delivered dosage of cigarette smoke toxins on the levels of urinary biomarkers of exposure. Cancer Epidemiol. Biomarkers Prev. 16, 1408–1415 (2007).
Kawasaki, T. et al. Markers of cadmium exposure in workers in a cadmium pigment factory after changes in the exposure conditions. Toxicol. Ind. Health 20, 51–56 (2004).
Acknowledgements
We thank the participants who contributed to this research project through providing biological samples, as well as releasing clinical information and completing surveys and the laboratory researchers for processing samples to generate the DNAm data. The Australian MND DNA Bank was funded by an Australian National Health and Medical Research Council Enabling Grant (402783). This work was funded by the National Health and Medical Research Council (1078901, 1083187, 1113400, 1121962). DNAm array and phenotypic data will be accessioned onto NCBI dbGaP.
Author information
Authors and Affiliations
Contributions
R.P., I.P.B., A.F.M. and N.R.W. conceived and designed the study. R.P. coordinated the initial study and oversaw blood collection and developed the questionnaire. K.L.W. and R.P. assisted in phenotypic data access and blood sample availability. A.K.H. and L.W. coordinated sample processing and laboratory QC for generating methylation data. A.F. analyzed the data. M.F.N. and T.L. performed QC and pipeline development of the DNA methylation data. A.F., A.F.M., and N.R.W. wrote the paper. All authors contributed in editing and approving the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Freydenzon, A., Nabais, M.F., Lin, T. et al. Association between DNA methylation variability and self-reported exposure to heavy metals. Sci Rep 12, 10582 (2022). https://doi.org/10.1038/s41598-022-13892-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13892-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
