
Abstract
Phosphorus (P) is an essential plant nutrient. Most rice growing lands lack adequate P, requiring multiple P fertiliser applications to obtain expected yields. However, P fertiliser is environmentally damaging, and already unaffordable to the marginal farmers. This warrants developing P-efficient rice varieties that require less P to produce the expected yield. However, genetic factors underlying P-use efficiency (PUE) in rice remain elusive. Here, we conducted comparative transcriptome analysis using two rice varieties with contrasting PUE; a P-efficient landrace DJ123 and a P-inefficient modern cultivar IR64. We aimed to understand the transcriptomic responses in DJ123 that allow it to achieve a high PUE under low P conditions. Our results showed that both DJ123 and IR64 had replete tissue P concentrations after 48 h of P deprivation. Yet, DJ123 strongly responded to the external low P availability by inducing P starvation-inducible genes that included SPX2, PHO1, PAPs and SQDs, while these genes were not significantly induced in IR64. We envisage that the ability of DJ123 to rapidly respond to low P conditions might be the key to its high PUE. Our findings lay a valuable foundation in elucidating PUE mechanism in rice, thus will potentially contribute to developing P-efficient modern rice variety.
Similar content being viewed by others

Introduction
Phosphorus (P) is an essential macronutrient for plant growth, development, and yield. It is a critical component in various biochemical processes like photosynthesis, respiration, and lipid metabolism. It is also a pivotal part of nucleic acids and the energy currency of the cell, ATP1. Under normal physiological conditions, plants take up P from soil as inorganic phosphate (Pi), H2PO4−. Soil Pi concentration across the globe ranges from < 0.6 to 11 µM with an average 3 µM2. However, plants require a far greater concentration of Pi at the intracellular level, ranging from 5 to 20 mM1,3,4. Therefore, most soils need to be supplemented by external P for optimal plant growth and development.
About half of the world-wide rice growing soils are deficient in P5. As such, rice farming heavily relies on external P fertiliser applications for achieving desired yield6. This P fertiliser dependence is greater in the economically poor regions, which are characterised by upland and rainfed rice farms7. However, most farmers in these regions, where rice demand is generally higher, already cannot afford the fertiliser costs or do not have access to P fertilisers7,8,9. This situation is bound to be aggravated as the demand for rice production is on the rise to feed an alarmingly increasing world population10,11. Furthermore, the application of P-fertilisers in agricultural systems has already caused some profound concerns. For example, depletion of rock phosphate reserve within next 50–100 years12,13 that is used as a raw material for manufacturing P fertiliser14,15 and negative environmental impacts from excess fertiliser application, including eutrophication16. This situation warrants developing rice varieties that can thrive on low P soils, with minimal fertiliser and still achieve the desired yield17,18.
There is a world-wide effort to breed P-efficient rice cultivars19. Rice breeders cross modern high yielding varieties with P-efficient rice varieties, which are generally lower-yielding; to effectively transfer P-efficient traits to the modern varieties20. The P-efficient rice breeding programs generally aim for two traits, namely (1) P acquisition efficiency (PAE) that accounts for greater P uptake from soil and (2i) P utilisation efficiency (PUE) that produces greater yield and biomass per unit P21.
DJ123 and IR64 are two cultivars contrasting in P use efficiency22 that are often used in rice breeding programs as a donor and recipient of P-efficiency traits, respectively. DJ123 is an aus-type rice variety that thrives in low P soil23,24 by operating at a greater PAE24,25 and PUE22,26. On the other hand, IR64 is a popular high-yielding indica variety27 but poorly performs in low-P conditions23. However, the basic question remains to be elucidated as to how DJ123 differs from other genotypes that constitutes its remarkable tolerance to low P. This knowledge will likely provide the breeders with specific targets in P-efficient rice breeding.
Here, we aimed to carry out a comparative transcriptomics analysis between DJ123 and IR64 as it provides an wholistic approach for dissecting genotypic differences28,29. Our goal was to gain insights on the earlier events that potentially lead to low-P tolerance in DJ123. In accordance, we designed the experiment in a way that allowed us to capture the immediate transcriptomic responses to low P (see “Materials and methods” section). We included IR64 as a contrasting genotype. Then, we followed the genotypic differences in the transcriptomes through the differential gene expression analysis.
Results
Our experimental design (Supplementary Fig. S1) attempted to distinguish long-term from short-term responses to P starvation. The long-term response is expected to be captured in the contrast between the HPC and LPC treatments, which supplied either high P continuously (HPC) or low P continuously (LPC) throughout the experimental period. Short-term P starvation was induced in both high and low-P treatments by stopping P supply 48 h prior to harvesting the plants and labelled as HPS and LPS, respectively. Thus, plants in the HPS treatment were supplied with P in excess of demand for 40 days and should have maintained tissue P concentrations well above deficiency levels but were expected to have reacted to a shift to a P-free growth medium 48 h prior to sampling. We adopted this transfer treatment approach to ensure that plants were exposed to only short-term external P deficiency, thus allowing us to capture the genotypic differences in short-term responses to P starvation.
Genotypic variation in P-dependent growth and tissue P concentrations
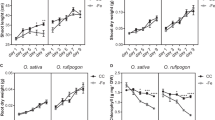
Low P supply over 40 days after germination reduced shoot biomass by around 65–70% in both DJ123 and IR64 (Fig. 1A,B) but had the opposite effect on root biomass, which significantly increased (140%) in genotype DJ123 relative to the high P treatments. Root biomass of IR64 remained non-responsive to P deficiency (Fig. 1C). Both genotypes increased root-to-shoot biomass ratio under LPC compared to that in HPC (Fig. 1D). However, this increase was twice as pronounced in DJ123 because of its significantly higher root biomass in both low-P treatments (Fig. 1D).

Whole plant, shoot and root biomass production and root-to-shoot biomass ratio of DJ123 and IR64 grown under varying phosphorus supply. Data are the mean ± SE of three to four independent replicates. ‘HP’ and ‘LP’ stand for high- and low-phosphorus supply, respectively. Plants were grown on HP and LP treatments for 40 days after germination. Then, both groups were splitted into two sub-groups. P supply was continued as usual for one sub-group of each treatment (labelled as ‘C’) and stopped for the other group (labelled as ‘S’). After approximately 24 and 48 h, LP and HP sub-groups were harvested, respectively. Significant differences among treatments were determined by ‘generalised least square’ model, separated by Tukey’s test (P < 0.05), and are indicated by different letters.
Maximum root length increased in DJ123 in response to low availability of P but remained constant in IR64 (Fig. 2A). DJ123 was consistently taller compared to IR64 but this difference in plant height decreased under P deficiency (Fig. 2B).

Maximum root length and plant height of DJ123 and IR64 under varying phosphorus supply. Data are the mean ± SE of four independent replicates. ‘HP’ and ‘LP’ stand for high- and low-phosphorus supply, respectively. Plants were grown on HP and LP treatments for 40 days after germination. Then, both groups were splitted into two sub-groups. Phosphorus supply was continued as usual for one sub-group of each treatment (labelled as ‘C’) and stopped for the other group (labelled as ‘S’). After approximately 24 and 48 h, LP and HP sub-groups were harvested, respectively. Significant differences among treatments were determined by ‘generalised least square’ model, separated by Tukey’s test (P < 0.05), and are indicated by different letters.
In the high P treatments leaf P concentrations of the top three youngest leaves remained within or even slightly above optimum tissue concentration range of 2.0–4.0 mg g−130 (Fig. 3A–C), whereas leaf P concentrations dropped far below this optimum range in the low P treatments. Tissue P concentrations did not differ between genotypes under P deficiency but DJ123 tended to have slightly higher P concentrations under high P supply (Fig. 3A–E). The total plant P content was at least fourfold higher in the high-P treatment and DJ123 took up significantly more P than IR64 in the HPC but not the HPS treatment (Fig. 4A–C). In the low-P treatments, genotypic differences for P uptake were not significant, which matched the intended experimental design of supplying each genotype with an equal (low) amount of P that should have been taken up entirely. A clear difference in the distribution of P between root and shoot was observed with DJ123 having allocated more P to roots compared to IR64 (Fig. 4C).

Phosphorus concentration in different tissues of DJ123 and IR64 grown under varying phosphorus supply. Data are the mean ± SE of four independent replicates. ‘HP’ and ‘LP’ stand for high- and low-phosphorus supply, respectively. Plants were grown on HP and LP treatments for 40 days after germination. Then, both groups were splitted into two sub-groups. Phosphorus supply was continued as usual for one sub-group of each treatment (labelled as ‘C’) and stopped for the other group (labelled as ‘S’). After approximately 24 and 48 h, LP and HP sub-groups were harvested, respectively. Significant differences among treatments were determined by ‘generalised least square’ model, separated by Tukey’s test (P < 0.05), and are indicated by different letters.

Phosphorus content in whole plant, shoot and root of DJ123 and IR64 grown under varying phosphorus supply. Data are the mean ± SE of four independent replicates. ‘HP’ and ‘LP’ stand for high- and low-phosphorus supply, respectively. Plants were grown on HP and LP treatments for 40 days after germination. Then, both groups were splitted into two sub-groups. Phosphorus supply was continued as usual for one sub-group of each treatment (labelled as ‘C’) and stopped for the other group (labelled as ‘S’). After approximately 24 and 48 h, LP and HP sub-groups were harvested, respectively. Significant differences among treatments were determined by ‘generalised least square’ model, separated by Tukey’s test (P < 0.05), and are indicated by different letters.
Global gene expression analysis by RNA-sequencing
RNA-seq of leaf and root tissues emanated into an average of 40 million raw reads per sample (Table 1). A set of about 20 million adapter-free and high-quality reads were filtered for each sample and mapped to the rice reference genome (Oryza sativa L. ssp. japonica cv. Nipponbare)31. More than 80% filtered reads were mapped uniquely while approximately 4% mapped multiple times and 10% remained unmapped. The overall alignment of the reads to the reference genome was 95% (Table 1).
A first global analysis of the RNA-seq data through Principal Component Analysis (PCA) attributed 89% of the variance in the data to the difference between root and leaf tissues (Fig. 5A), indicating that these tissues should be analyzed separately. Subsequent separate analyses of root and shoot samples revealed that the factor genotype accounted for most of the variation whereas P treatment effects were less dominant (Fig. 5B,E). A further PCA analysis of root samples within an individual genotype suggested that different P supply treatments explained 46% and 45% of the variance in DJ123 and IR64, respectively (Fig. 5C,D). Very similar results were seen for the global gene expression patterns in leaf samples, where P treatments explained 47–49% (Fig. 5F,G) of the variance within each genotype. The main difference between expression patterns in root and shoot samples is the very clear separation by high versus low P supply in shoots (Fig. 5E,F), whereas this separation was less distinct in roots (Fig. 5B), especially in DJ123 (Fig. 5C) where HPS-treated roots appeared to be in between HPC and both low-P treatments. The shift of the HPS-treated root transcriptomes towards the LP-treated ones might suggest that the former has already responded to the 48 h of P deficiency in the growing medium, especially in DJ123. Based on these observations, we hypothesized that DJ123 responded faster to the short-term P deficiency (in high-P plants) than IR64, and in order to elucidate this fast response, we focused our further analyses on the HPC versus HPS comparison in root tissue.

Principal component analysis of the RNA-seq results.
Differentially expressed genes (DEGs) by short-time deprivation of P in root samples
Consistent with the PCA analysis, HPS-treated roots of DJ123 had more differentially expressed genes (DEGs) than those of IR64 and twice as many DEGs were upregulated in DJ123 compared to IR64 (Fig. 6A,B). Only 56 DEGs were commonly detected in both genotypes (Fig. 6C, Supplementary Table S1), thus most DEGs were genotype-specific, suggesting that IR64 and DJ123 have distinct response patterns to a short-term P deprivation.

Volcano plots showing significantly up- or down-regulated genes in different tissues between DJ123 and IR64. The logarithm of fold change (log2) of each transcript is presented on x-axis, and the log10 of the P value are on y-axis. Thresholds are ‘pCutoff = 10e−16’ and ‘log2FoldChange > 1’. Venn diagram of the differentially expressed transcripts (DETs) in roots of DJ123 and IR64. The numbers of the DETs represent the DETs in HPS treatment against HPC treatment (HPS vs HPC).
Gene Ontology analysis of the differentially expressed genes
Gene Ontology (GO) enrichment analysis was carried out to functionally annotate the HPS-induced DEGs that were DJ123- and IR64-specific as well as common in both genotypes. For each group, we analysed the up- and down-regulated genes separately for biological process (BP), molecular function (MF), cellular component, pathway and protein class (Protein) categories in PANTHER32,33. The GO-term enrichment analysis showed that DJ123 and IR64 had a common set of GO terms enriched as well as a genotype-specific set under HPS treatment (Fig. 7). The common GO terms were related with lipid metabolism and metabolite interconversion. DJ123-specific GO terms were related to responses to external stimuli, dephosphorylation and phosphatase activities. In addition, one GO term (PC00262:metabolite interconversion enzyme) from the common set of terms was further enriched by the DJ123 specific genes (Fig. 7). On the other hand, IR64-specific GO terms were all related to nucleotide binding. Interestingly, all the enriched GO terms were enriched by the up-regulated genes only except for the GO term ‘PC00176:oxidoreductase’, which was enriched by the IR64 down-regulated genes (Fig. 7). However, a single DEG was associated with multiple GO terms (Supplementary Table S1) as expected32. Accordingly, there were only 29 and 34 DEGs in the DJ123- and IR64-specific GO terms, respectively (Supplementary Table S2).

Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) in roots of rice seedlings. The Y-axis represents the enriched GO terms with their identification numbers. The X-axis represents the negative log10 of P values indicating the significance level of term-enrichment. The purple dotted-line represents P = 0.05. The up- and down-regulated genes under HPS treatment (compared to HPC treatment) were annotated separately for biological process (BP), molecular function (MF), cellular component, pathway and protein class (Protein) categories in PANTHER32,33. Different colors represent different categories. The categories that are absent in the graph were not significantly enriched. The bars on the right-hand side of (0,0) intercept indicate the GO-terms enriched by the up-regulated genes while the one on the left-hand side by the down-regulated genes. The numbers on top of the bars are the numbers of mapped genes. Three panels distinguish the GO-terms that were enriched in both genotypes (Both), only in DJ123 (DJ) and only in IR64 (IR).
Differentially expressed genes that enriched the GO terms under short-term P deprivation
We hypothesized that the genes specifically induced in DJ123 by HPS treatment and/or molecular pathways enriched specifically in DJ123 are key components that characterize the response pattern of DJ123 to a short-term P deprivation. Accordingly, we compared the expression pattern of the genes from the enriched GO-terms (Supplementary TableS S1, S2) between DJ123 and IR64 genotypes under the HPS treatment (Fig. 8, Supplementary Fig S2). Considering together, our findings showed that the following genes were up-regulated to a greater extent in DJ123 compared to those in IR64 under the HPS treatment: Os01g0110100 (PHO1;1), Os02g0168800, Os02g0202200 (SPX2), Os04g0326201, Os09g0506000 (PAP27A), Os11g0151700 (PAP21B), Os11g0439100 and Os12g0189300 (Fig. 8); PAP10a, SQD2.1 and SQD2.2 (Supplementary Fig S2).

Fold change of the genes that enriched P-stress related GO terms in DJ123 under HPS treatment. Fold change was calculated by dividing the transcript abundance under HPS treatment by that under the HPC treatment in genotype-wise. The red line represents two-fold up-regulation of the corresponding gene in HPS compared to that in HPC. The red rectangular boxes highlight the genes, which are overdriven by DJ123 compared to IR64.
Validation
To confirm the accuracy and reproducibility of the Illumina RNA-Seq results, we carried out qPCR assays for a panel of 6 differentially expressed genes under HPS: SPX2, PHO1;1, PAP21b, PAP10a, SQD2.1, SQD2.2, and two house-keeping genes ELF1 and UBI (Supplementary Table S3). The ‘Fold change’ was calculated in the same way as in the transcriptome analysis, i.e., the transcript abundance under HPS treatment was divided by that under the HPC treatment in each genotype (Fig. 8). The expression pattern of the DEGs in the comparative transcriptome analysis under the HPS treatment (SPX2, PHO1;1, PAP21b, PAP10a, SQD2.1 and SQD2.2) (Fig. 8) corroborated with that in the qPCR assay (Supplementary Fig. S2). The level of expression of both house-keeping genes (ELF1 and UBI) remained the same between DJ123 and IR64, as expected (Supplementary Fig. S2).
Discussion
Characteristic response pattern of DJ123 to low P stress
DJ123 had a greater root biomass under low P treatments compared to that in high P, consistent with the increased maximum root length (Figs. 1, 2). In sharp contrast, root biomass, as well as maximum root length in IR64, remained non-responsive to the variation in P supply. Reflecting these contrasts, root to shoot ratio increased drastically in DJ123 by the low P treatment, indicating that DJ123 invested more resources to root growth than to shoot growth as compared with IR64. These results suggest that the low-P treatments in the experiment created P stress to the plants and triggered genotype-specific responses. Preferential resource allocation of DJ123 to root growth under P deficient conditions, as reported in previous studies22,24,34, is associated with its greater ability to explore larger soil volume, thus allowing efficient acquisition of scarcely available P24. In root, DJ123 significantly decreased P concentration by 48 h of P deprivation, while IR64 did not. Similar trend was observed for shoot and leaf 1 samples (Fig. 3). These observations show that DJ123 more drastically responds to low P stress than IR64, which could be a strategy to cope with low P stress more effectively.
DJ123 induces P starvation responsive genes more strongly than IR64
Comparative transcriptomics have effectively been used to dissect underlying genetic mechanisms of a trait in rice28,29. To reveal genetic components that characterizes the response patterns of DJ123, we obtained transcriptome datasets from 48 leaf and 24 root samples across high- and low-P treatments from DJ123 and IR64. PCA analysis (Fig. 5) demonstrated that root- and leaf transcriptomes are tissue-specific explaining 89% variation, thus warranted a separate analysis. We focused our comparative analysis on the root tissues and between the HPC- and HPS-treatments. Because our PCA analysis showed that the 48 h P deficiency (HPS treatment) had induced transcriptional changes in the DJ123 root tissues that separate the transcriptome from that of the HPC (control) and towards the low-P treatments (Fig. 5C). Thus, the comparison between DJ123 and IR64 root transcriptome under HPS treatment might reveal the early events of low-P tolerance mechanisms in DJ123.
The GO enrichment analysis highlighted the genotypic differences between DJ123 and IR64 under low P conditions (Fig. 7). Unlike IR64, DJ123 root transcriptomes were specifically enriched with 14 P-starvation related GO terms under the HPS treatment (Fig. 7). This suggests that DJ123 employed different strategies to cope with low P supply. The DJ123-specific GO term ‘response to extracellular stimulus or nutrient levels’ could suggest that DJ123 responds to low P availability more strongly or earlier than IR64. The phosphatase and hydrolase GO terms suggest that DJ123 is more efficient in internal P scavenging activities to obtain more P from otherwise inaccessible sources35,36. The enrichment of the GO term ‘dephosphorylation’ may indicate activating P stress responsive mechanism37. The enrichment of these DJ123-specific GO terms suggests that DJ123 initiated several adaptive mechanisms in response to low P conditions.
We analysed the expression pattern of the genes that enriched the above GO terms in DJ123 under the HPS treatment. We compared this pattern between HPC and HPS treatments and between DJ123 and IR64 genotypes (Fig. 8). Our findings showed that the following genes were expressed to a greater extent in DJ123 in comparison to those in IR64 under the HPS treatment: (1) Os01g0110100 (PHO1;1), which encodes a protein involved in transferring Pi from root to shoot38 and maintaining Pi homeostasis39,40, (2) Os02g0168800 encodes a porphobilinogen deaminase41. Porphobilinogen deaminase has been reported to promote vegetative and reproductive development42, (3) Os04g0326201 encodes a glycosyltransferase43. Glycosyltransferase is involved in synthesizing non-phosphorus lipid under phosphorus deprivation44, (4) Os09g0506000 and Os11g0151700 encode purple acid phosphatases31,45 and might play a role in Pi mobilization under low P condition46, (5) Os11g0439100 encodes an oxidoreductase47 that is exuded by roots for degrading organic matter48, (6) Os12g0189300 encodes a phosphoenolpyruvate carboxylase47. This enzyme, classified as an isomerase, is a major player for organic acid synthesis that is liberated in soil to scavenge Pi from insoluble P-complex49, (7) Os02g0202200 (SPX2) is one of the six SPX genes in rice50. SPX2 modulates the activity of PHR2, the master protein that controls the expression of Phosphate Starvation Induced (PSI) genes in rice50,51,52. In summary, the above genes are reported in literature as P starvation inducible (PSI) genes. The greater expression of these PSI genes in DJ123 compared to that in IR64 under short-term P deprivation suggests that DJ123 strongly responds to P deprivation than IR64.
Possible mechanisms for differential response patterns between DJ123 and IR64
Consistent with the change in tissue P concentration, DJ123 responded to a short-term P deprivation more drastically than IR64, in terms of the genome-wide gene expression pattern in root. Recent investigations revealed the mechanisms for induction of P starvation responses. Under P deficient conditions, the master regulators for P starvation response, PHR transcription factors, bind to the PHR1-binding sequence (P1BS) elements on the promoters of an array of PSI genes and induce their expression. On the other hand, under the P-replete conditions, SPX family proteins, including SPX2, bind to PHR transcription factors and inhibit their binding to P1BS elements or nuclear localization52,53,54,55. Since the interaction of SPX proteins and PHR proteins is mediated by inositol phosphate28,56,57, reduction in its concentration is a key for triggering of P starvation responses. Since the genes that are specifically induced in DJ123 in response to 48 h P deprivation are mostly PSI genes, it is likely that DJ123 more strongly or more promptly triggers P deficiency response pathways by reducing inositol phosphate and releasing PHR transcription factors upon short-term P deprivation compared with IR64. Alternatively, even though DJ123 maintains a relatively high P concentration in the whole root system, it is possible that the P concentration in specific root cells may be reduced, inducing low-P responsive genes in these cells. This is supported by the fact that the strengths of P deficiency responses are heterogenous among cells58. However, the potential for reduction in root inositol phosphate concentration or differential root cell P concentrations and consequently inducing PSI genes which may drive whole-plant responses to low P conditions certainly warrants further investigation.
Conclusion
Considering together the entirety of our findings, our results demonstrate that DJ123 rapidly responded to the low-P availability in the growing medium, despite having abundant internal P to support growth and development. In response, DJ123 swiftly employed an array of the PSI genes. These PSI genes are reported in literature to be associated with low P adaptive mechanisms that included (1) increasing Pi uptake (PHO1), (2) scavenging P from metabolite interconversion (PAPs), (3) reducing P consumption by switching to non-phosphorus lipid synthesis (SQD2.1, SQD2.2), and (4) increasing organic acid synthesis that is liberated by roots for releasing P from the insoluble P-complexes (Os12g0189300) (Fig. 9). The ability to promptly deploy adaptive mechanisms in response to low-P in the growing media might underlie the remarkable low-P tolerance in DJ123. However, further investigations are required to determine what triggers these prompt responses in DJ123.

Diagram depicting the rapid response of DJ123 to low-P condition. High P and low-P conditions in the growing medium were represented by the green and red colors in the solid arrow, respectively. A. Plants were supplied with high P for the entire experimental period (42 days). B. High P supply was stopped after 40 days, then plants were harvested within 24–48 h. The response of DJ123 to low-P condition was determined by analyzing the differential gene expression in the root transcriptomes between A and B.
Materials and methods
Biological materials
Experimental research and field studies on plants complies with all relevant institutional, national, and international guidelines and legislation.
Plant materials and treatment
Rice (Oryza sativa L.) genotypes DJ123 and IR64 were used in this study. DJ123 develops a larger root system earlier than IR64 under low-P conditions. This early root vigor allows DJ123 to explore a greater soil volume and improve P acquisition. In contrast, IR64 is a high-yielding modern cultivar but lacks early root vigor and is comparatively less tolerant to low P availability. DJ123 and IR64 seeds were washed with 70% ethanol once, followed by three washes with deionized water and incubated in petri dishes at 30 °C. Germinated seeds were transferred to mesh floating in a 6 L tray filled with deionized water. Nutrients except P and N were added in the tray as follows: 0.4 mL of 0.5 M K2SO4 and 1 M MgSO4·7H2O + 0.2 mL of 2000× Micronutrients (1× Micronutrients = 9 µM MnCl2·4H2O + 0.5 µM (NH4)6MO7O24·4H2O + 18.5 µM H3BO3 + 0.16 µM CuSO4·5H2O + 0.3 µM ZnSO4·7H2O) + 0.8 mL of 100 µM Ca + 1.12 mL of 10 µM Fe. pH in the trays was 5.4. Six days after germination (DAG), the trays were replenished with 0.2× Yoshida solution without P and N. The full-strength Yoshida solution (1×) is composed of: N, 2.86 mM (as NH4NO3); P, 0.05 mM (as NaH2PO4·2H2O); K, 1 mM (as K2SO4); Ca, 1 mM (as CaCl2); Mg, 1 mM (as MgSO4·7H2O); Mn, 9 μM as (MnCl2·4H2O); Mo, 0.5 μM (as (NH4)6MO7O24·4H2O); B, 18.5 μM (as H3BO3); Cu, 0.16 μM as (CuSO4·5H2O); Fe, 36 μM (as FeCl3·6H2O); Zn, 0.15 μM (as ZnSO4·7H2O)59. At 10 DAG, plants were transferred to 1.1 L black bottles containing low P (0.2 × Yoshida + 2 µM Na2HPO4) and high-P (0.2 × Yoshida + 50 µM Na2HPO4) solutions. Nutrient solutions in the bottles were completely replaced by fresh low- and high-P solution on every alternative day. Each bottle had two seedlings (technical replicates) and represented one biological replicate. Each treatment had three biological replicates. Plants were grown between September and November 2019, in a naturally lit temperature-controlled glass house at the Japan International Research Center for Agricultural Sciences, Tsukuba, Japan; at a mean temperature of 27 °C (23–33 °C) and a mean relative humidity of 40% (10–78%). Plants were regularly monitored for P-deficiency symptoms and harvested when the low-P treated plants appeared to be P-deficient. All low-P treatment replicates were harvested on DAG 41 and the high-P treatment ones on DAG 42. For transcriptomic study, first and third youngest leaves and roots were collected from both genotypes of all treatments and frozen immediately in liquid nitrogen and stored at − 80 °C until RNA extraction.
Phosphorus concentration and content
An aliquot of approximately 100 mg dried sample was pre-digested in 8 mL of a mixture of HNO3, HClO4 (3:1) in 50 mL of acid digestion tubes overnight, followed by digestion at 105 °C for 1 h and removal of cHNO3 at 140˚C for 1 h in a block digester. The block digester was heated up to a maximum of 170˚C to dehydrate any silica present in the digest. After cooling, the digested solution was diluted with DI water to a volume of 50 mL. The digested solution was centrifuged at 12,000 rpm for 5 min and the supernatant was used for P concentration measurement using the molybdenum blue method60. The P content in leaves and roots was calculated as P concentration in the corresponding organ times its dry biomass.
RNA extraction
Total RNA was extracted from rice leaf and root tissues using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions including DNase treatment. The quantity and purity of the total RNA in each sample were determined using a NanoDrop spectrophotometer (Thermo Scientific). The RNA integrity was checked using an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA). The samples used for sequencing had a RIN value ranging from 6.2 to 8.6.
cDNA library construction and illumina sequencing
The cDNA libraries were individually prepared from each sample using the “TruSeq Stranded mRNA Low Throughput (LT) Sample Preparation Kit (Illumina, San Diego, CA, USA)” following the manufacturer’s instructions (TruSeq Stranded mRNA Sample Preparation Guide, Part # 15031047 Rev. E). The DNA libraries were purified from the PCR reactions using AMPure XP Beads, and quantified using Agilent Technologies 2100 Bioanalyzer. The sequencing libraries were sequenced on an Illumina NovaSeq6000 sequencing platform by Macrogen Japan (Kyoto, Japan).
Quality control
The quality control and reference mapping of the RNA-Seq data were carried out using open-source tools in Ubuntu command-line interface. First, the quality of the raw reads was evaluated using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) commands. Then, the raw reads were processed using ‘trimmomatic’ package for trimming the Illumina PE adapters (TruSeq3-PE-2.fa:2:30:10), removing the low-quality or poly-N bases from the ends that were below quality score 3, removing the sequences when the average quality per base dropped below 15 in a 4-base sliding window, and dropping the reads with less than 25 bases in length. This led to a set of clean and high-quality RNA-seq reads for subsequent analyses.
Reference mapping
Rice reference genome (fasta) and genome annotation (GTF) files were downloaded from ‘EnsemblPlants’ website (Oryza sativa Japonica Group, https://plants.ensembl.org/info/data/ftp/index.html). The pre-processed paired-end RNA-Seq reads were mapped to the reference genome using HISAT261. First, the splice and exon information from the rice GTF file were extracted using two python scripts—‘extract_splice_sites.py’ and ‘extract_exons.py’—provided with the HISAT2 package (https://cloud.biohpc.swmed.edu/index.php/s/hisat2-220-source/download). Then, this information together with the reference genome in ‘fasta’ format were used as input to build a reference genome index followed by aligning the reads to these indexes. The alignment was produced in ‘sam’ format. ‘samtools sort’ function was used to convert the ‘sam’ format to ‘bam’, and sort the bam files62. The sorted bam files were passed to StringTie63 for assembling transcripts and quantifying expressed genes and transcripts. However, some samples might have partial read coverage for some transcripts resulting in partial assembly of these transcripts. The ‘StringTie merge’ function was used to merge all the gene structures found in any of the samples and re-estimate the transcript abundances using the merged structures. This created a consistent set of transcripts across the samples making them comparable in the downstream analysis.
StringTie uses MSTRG gene ids. We ran a Python postprocessing script, ‘mstrg_prep.py’ (Pertea, https://gist.github.com/gpertea/4207fa9cb30fe7fec0eb52bd29b9a976) that appends reference gene ids to the MSTRG gene ids used in StringTie. However, we used ‘stringtie-e-B’ function to estimate the transcript abundance and create count tables for differentially expressed genes. We processed the row count table using a Python script ‘prepDE.py’ (Pertea M, http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual#de) for performing differential gene expression analysis using the DESeq2 package.
Differential gene expression analysis
We used DESeq2 R package for analyzing the differentially expressed genes (DEGs) using the ‘design = ~ Genotypes + Treatments + Genotypes:Treatments’ Generalized Linear Model64. We filtered out the genes with less than 10 counts. The ‘relevel’ function was used to manually set ‘IR64’ and ‘HPC’ as a reference for genotype and treatment, respectively. By default, the ‘DESeq’ function normalized the raw read counts for library size, estimated the dispersion of counts for each DEG, and calculated the significance of coefficients using ‘nbinomWaldTest’. Then, we estimated the treatment effects on the DEGs using the ‘results’ function with a threshold for False Discovery Rate set at 0.005. We used ‘lfcShrink’ function to perform shrinkage on log2 fold change. All ‘log2 fold change’ values presented in this manuscript are shrinkage estimated value. The thresholds to define significantly DEGs across the genotypes, treatments and tissues were ‘padj < 0.001’ and ‘log2FoldChange < −1|log2FoldChange > 1’. To determine the global pattern of the DEGs in the samples, we performed Principal Components Analysis (PCA) on expression values using ‘plotPCA’. Using ‘EnhancedVolcano’65, we determined the DEGs that were treatment- or genotype-specific.
Gene Ontology (GO) term enrichment and pathway analysis
We refined a list of DEGs in DJ123 roots compared with IR64 under low-P treatment. We functionally annotated these DEGs against a series of ‘Annotation Data Set’ viz. ‘GO biological process complete’, ‘GO molecular function complete’, ‘GO cellular component complete’, ‘PANTHER Pathways’, ‘PANTHER GO-Slim Biological Process’, ‘PANTHER GO-Slim Molecular Function’, ‘PANTHER GO-Slim Cellular Component’ and ‘PANTHER Protein Class’ that are implemented in PANTHER classification system (http://pantherdb.org/tools/compareToRefList.jsp or http://www.pantherdb.org)66. We used ‘Oryza sativa all genes in the database’ as a reference list. The annotation used ‘Fisher’s Exact’ test with ‘No correction’.
Validation of the RNA-seq data
We validated our RNA-seq data using quantitative real-time PCR (qRT-PCR) assay on a CFX96 Touch Real-Time PCR system (BioRad, USA). The qRT-PCR assay was carried out on a panel of eight genes including two house-keeping ones, ELF1 (Elongation factor) and UBI (Ubiquitin) (Supplementary Table S3). Gene specific primers were designed using the Primer3 software keeping the default parameters67. Total RNA from three independent biological replicates was reverse transcribed (RT) into cDNA using the PrimeScript RT Enzyme Mix I (Takara, Japan). qRT-PCR reaction volume was 10μL containing 5 µL TB Green® Premix Ex Taq™ II (Takara, Japan), 1 µL cDNA, 0.04 µL of each of the primers (100 μM) and 3.92 μL RNase-free water.
qRT-PCR cycling conditions were as follows: 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s, 60 °C for 30 s. For melt curve analysis, the denaturation temperature was incrementally increased from 65.0 to 95.0 °C by 0.5 °C per cycle. Relative gene expression level was calculated using the standard-curve method and expressed as fold-change.
Data availability
RNA-seq data generated in the study have been deposited in the National Centre for Biotechnology Information (NCBI) under BioProject ID PRJNA823747, BioSample ID SUB11348183 and Sequence Read Archive (SRA) submission ID SUB11348231.
References
Vance, C. P., Uhde-Stone, C. & Allan, D. L. Phosphorus acquisition and use: Critical adaptations by plants for securing a nonrenewable resource. New Phytol. 157, 423–447 (2003).
Pierre, W. H. & Parker, F. W. Soil phosphorus studies: II. The concentration of organic and inorganic phosphorus in the soil solution and soil extracts and the availability of the organic phosphorus to plants. Soil Sci. 24, 119–128 (1927).
Fang, Z., Shao, C., Meng, Y., Wu, P. & Chen, M. Phosphate signalling in Arabidopsis and Oryza sativa. Plant Sci. 176, 170–180 (2009).
Lambers, H. & Plaxton, W. C. Phosphorus: Back to the roots. In Annual Plant Reviews Vol 48 1–22 (Wiley, 2015). https://doi.org/10.1002/9781118958841.ch1.
Ismail, A. M., Heuer, S., Thomson, M. J. & Wissuwa, M. Genetic and genomic approaches to develop rice germplasm for problem soils. Plant Mol. Biol. 65, 547–570 (2007).
Vandamme, E., Wissuwa, M., Rose, T., Ahouanton, K. & Saito, K. Strategic phosphorus (P) application to the nursery bed increases seedling growth and yield of transplanted rice at low P supply. Field Crop Res. 186, 10–17 (2016).
Kirk, G. J. D., George, T., Courtois, B. & Senadhira, D. Opportunities to improve phosphorus efficiency and soil fertility in rainfed lowland and upland rice ecosystems. Field Crop Res. 56, 73–92 (1998).
Saito, K. et al. Yield-limiting macronutrients for rice in sub-Saharan Africa. Geoderma 338, 546–554 (2019).
Andrianary, B. H. et al. Phosphorus application affects lowland rice yields by changing phenological development and cold stress degrees in the central highlands of Madagascar. Field Crop Res. 271, 108256 (2021).
Van Nguyen, N. & Ferrero, A. Meeting the challenges of global rice production. Paddy Water Environ. 4, 1–9 (2006).
Prasad, R., Shivay, Y. S. & Kumar, D. Current status, challenges, and opportunities in rice production. In Rice Production Worldwide (eds Chauhan, B. S. et al.) 1–32 (Springer, 2017). https://doi.org/10.1007/978-3-319-47516-5_1.
Fixen, P. E. & Johnston, A. M. World fertilizer nutrient reserves: A view to the future. J. Sci. Food Agric. 92, 1001–1005 (2012).
Johnston, A. E., Poulton, P. R., Fixen, P. E. & Curtin, D. Phosphorus: Its Efficient use in agriculture. In Advances in Agronomy Vol 123 (ed. Sparks, D. L.) 177–228 (Academic Press, 2014).
Gilbert, N. Environment: The disappearing nutrient. Nature 461, 716–718 (2009).
Scholz, R. W. & Wellmer, F.-W. Approaching a dynamic view on the availability of mineral resources: What we may learn from the case of phosphorus?. Glob. Environ. Chang. 23, 11–27 (2013).
Stoner, D. J. Industrial by-Product Characterization for Phosphorus Removal in Environmental Contaminant Filters (Oklahoma State University, 2011).
Gamuyao, R. et al. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 488, 535–539 (2012).
Tanaka, R. et al. From gene banks to farmer’s fields: Using genomic selection to identify donors for a breeding program in rice to close the yield gap on smallholder farms. Theor. Appl. Genet. 134, 3397–3410 (2021).
Heuer, S. et al. Improving phosphorus use efficiency: A complex trait with emerging opportunities. Plant J. 90, 868–885 (2017).
Chithrameenal, K. et al. Genetic enhancement of phosphorus starvation tolerance through marker assisted introgression of OsPSTOL1 gene in rice genotypes harbouring bacterial blight and blast resistance. PLoS One 13, e0204144 (2018).
Adem, G. D., Ueda, Y., Hayes, P. E. & Wissuwa, M. Genetic and physiological traits for internal phosphorus utilization efficiency in rice. PLoS One 15, e0241842 (2020).
Hayes, P. E., Adem, G. D., Pariasca-Tanaka, J. & Wissuwa, M. Leaf phosphorus fractionation in rice to understand internal phosphorus-use efficiency. Ann. Bot. 129, 287–302 (2022).
Vandamme, E. et al. Genotypic variation in grain P loading across diverse rice growing environments and implications for field P balances. Front. Plant Sci. 7, 1435 (2016).
Mori, A. et al. The role of root size versus root efficiency in phosphorus acquisition in rice. J. Exp. Bot. 67, 1179–1189 (2016).
Wissuwa, M., Gonzalez, D. & Watts-Williams, S. J. The contribution of plant traits and soil microbes to phosphorus uptake from low-phosphorus soil in upland rice varieties. Plant Soil 448, 523–537 (2020).
Rose, T. J., Mori, A., Julia, C. C. & Wissuwa, M. Screening for internal phosphorus utilisation efficiency: Comparison of genotypes at equal shoot P content is critical. Plant Soil 401, 79–91 (2016).
Mackill, D. J. & Khush, G. S. IR64: A high-quality and high-yielding mega variety. Rice 11, 18 (2018).
Deng, Q.-W. et al. Transcriptome analysis of phosphorus stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). Biol. Res. 51, 7 (2018).
Jeong, K. et al. Transcriptional response of rice flag leaves to restricted external phosphorus supply during grain filling in rice cv IR64. PLoS One 13, e0203654 (2018).
Dobermann, A. & Fairhurst, T. Rice: Nutrient Disorders and Nutrient Management (Potash & Phosphate Institute (PPI), Potash & Phosphate Institute of Canada (PPIC) and International Rice Research Institute, 2000).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4 (2013).
Ashburner, M. et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
The Gene Ontology Consortium. The gene ontology resource: 20 years and still going strong. Nucleic Acids Res. 47, D330–D338 (2019).
Rakotoson, T., Holz, M. & Wissuwa, M. Phosphorus deficiency tolerance in Oryza sativa: Root and rhizosphere traits. Rhizosphere 14, 100198 (2020).
Hu, B. & Chu, C. Phosphate starvation signaling in rice. Plant Signal Behav. 6, 927–929 (2011).
Balemi, T. & Negisho, K. Management of soil phosphorus and plant adaptation mechanisms to phosphorus stress for sustainable crop production: A review. J. Soil Sci. Plant Nutr. 12, 547–562 (2012).
Lan, P., Li, W. & Schmidt, W. ‘Omics’ approaches towards understanding plant phosphorus acquisition and use. In Annual Plant Reviews online 65–97 (Wiley, 2017). https://doi.org/10.1002/9781119312994.apr0518.
Poirier, Y., Thoma, S., Somerville, C. & Schiefelbein, J. Mutant of Arabidopsis deficient in xylem loading of phosphate1. Plant Physiol. 97, 1087–1093 (1991).
Che, J. et al. Node-localized transporters of phosphorus essential for seed development in rice. Plant Cell Physiol. 61, 1387–1398 (2020).
Secco, D., Baumann, A. & Poirier, Y. Characterization of the rice pho1 gene family reveals a key role for ospho1;2 in phosphate homeostasis and the evolution of a distinct clade in dicotyledons1[C][W][OA]. Plant Physiol. 152, 1693–1704 (2010).
Song, T. et al. Transcriptomic analysis of photosynthesis-related genes regulated by alternate wetting and drying irrigation in flag leaves of rice. Food Energy Secur. 9, e221 (2020).
Quesada, V. et al. Porphobilinogen deaminase deficiency alters vegetative and reproductive development and causes lesions in Arabidopsis. PLoS One 8, e53378 (2013).
Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 44, D7–D19 (2016).
Devers, E. A., Wewer, V., Dombrink, I., Dörmann, P. & Hölzl, G. A processive glycosyltransferase involved in glycolipid synthesis during phosphate deprivation in Mesorhizobium loti. J. Bacteriol. 193, 1377–1384 (2011).
Sakai, H. et al. Rice Annotation Project Database (RAP-DB): An integrative and interactive database for rice genomics. Plant Cell Physiol. 54, e6 (2013).
Zhang, Q., Wang, C., Tian, J., Li, K. & Shou, H. Identification of rice purple acid phosphatases related to posphate starvation signalling. Plant Biol. 13, 7–15 (2011).
Mi, H., Muruganujan, A., Ebert, D., Huang, X. & Thomas, P. D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019).
Gramss, G., Voigt, K.-D. & Kirsche, B. Oxidoreductase enzymes liberated by plant roots and their effects on soil humic material. Chemosphere 38, 1481–1494 (1999).
Peñaloza, E., Muñoz, G., Salvo-Garrido, H., Silva, H. & Corcuera, L. J. Phosphate deficiency regulates phosphoenolpyruvate carboxylase expression in proteoid root clusters of white lupin. J. Exp. Bot. 56, 145–153 (2005).
Liu, N. et al. Evolution of the SPX gene family in plants and its role in the response mechanism to phosphorus stress. Open Biol. 8, 170231 (2018).
Guan, Z. et al. Mechanistic insights into the regulation of plant phosphate homeostasis by the rice SPX2–PHR2 complex. Nat. Commun. 13, 1581 (2022).
Wang, Z. et al. Rice SPX1 and SPX2 inhibit phosphate starvation responses through interacting with PHR2 in a phosphate-dependent manner. Proc. Natl. Acad. Sci. 111, 14953–14958 (2014).
Zhong, Y. et al. Rice SPX6 negatively regulates the phosphate starvation response through suppression of the transcription factor PHR2. New Phytol. 219, 135–148 (2018).
Qi, W., Manfield, I. W., Muench, S. P. & Baker, A. AtSPX1 affects the AtPHR1–DNA-binding equilibrium by binding monomeric AtPHR1 in solution. Biochem. J. 474, 3675–3687 (2017).
Lv, Q. et al. SPX4 negatively regulates phosphate signalling and homeostasis through its interaction with phr2 in rice. Plant Cell 26, 1586–1597 (2014).
Zhu, J. et al. Two bifunctional inositol pyrophosphate kinases/phosphatases control plant phosphate homeostasis. Elife 8, e43582 (2019).
Ried, M. K. et al. Inositol pyrophosphates promote the interaction of SPX domains with the coiled-coil motif of PHR transcription factors to regulate plant phosphate homeostasis. Nat. Commun. 12, 384 (2021).
Hani, S. et al. Live single-cell transcriptional dynamics via RNA labelling during the phosphate response in plants. Nat. Plants 7, 1050–1064 (2021).
Yoshida, S., Forno, D. A., Cock, J. H. & Gomez, K. A. Laboratory Manual for Physiological Studies of Rice (The International Rice Research Institute, 1976).
Murphy, J. & Riley, J. P. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 27, 31–36 (1962).
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Blighe, K., Rana S, & Lewis M. EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labelling. https://github.com/kevinblighe/EnhancedVolcano. Accessed 28 Aug 2020 (2020).
Mi, H. et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721 (2019).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012).
Acknowledgements
We thank Taro Matsuda, Mari Yonemoto and all the technical staffs at the Japan International Research Center for Agricultural Sciences (JIRCAS) for their technical assistance. This study was funded by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship awarded to MAP. We thank the Australian Academy of Science for the JSPS Postdoctoral Fellowship nomination, and the Pawsey Supercomputer Centre (https://pawsey.org.au/) for the computing resources.
Author information
Authors and Affiliations
Contributions
M.A.P., J.P.-T., P.E.H., and M.W. conceived the experiment. M.A.P. and P.E.H. set up the experiment. M.A.P. conducted the experiment, extracted RNA, carried out the entire bioinformatics workflow for RNA-seq analysis, and interpreted the outputs. P.E.H. performed acid digestion for tissue phosphorus assay. M.A.P., J.P.-T., Y.U. and M.W. critically discussed the results. M.A.P. wrote the manuscript. J.P.-T., Y.U., P.E.H. and M.W. provided editorial comments. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prodhan, M.A., Pariasca-Tanaka, J., Ueda, Y. et al. Comparative transcriptome analysis reveals a rapid response to phosphorus deficiency in a phosphorus-efficient rice genotype. Sci Rep 12, 9460 (2022). https://doi.org/10.1038/s41598-022-13709-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-13709-w
This article is cited by
-
Novel QTL for Lateral Root Density and Length Improve Phosphorus Uptake in Rice (Oryza sativa L.)
Rice (2023)
-
Physiological evidence that nitrate use positively correlates with internal phosphorus utilization efficiency and phosphorus uptake efficiency in rice (Oryza sativa L.)
Plant and Soil (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
