
Abstract
Genetics play an important role in late-onset Alzheimer’s Disease (AD) etiology and dozens of genetic variants have been implicated in AD risk through large-scale GWAS meta-analyses. However, the precise mechanistic effects of most of these variants have yet to be determined. Deeply phenotyped cohort data can reveal physiological changes associated with genetic risk for AD across an age spectrum that may provide clues to the biology of the disease. We utilized over 2000 high-quality quantitative measurements obtained from blood of 2831 cognitively normal adult clients of a consumer-based scientific wellness company, each with CLIA-certified whole-genome sequencing data. Measurements included: clinical laboratory blood tests, targeted chip-based proteomics, and metabolomics. We performed a phenome-wide association study utilizing this diverse blood marker data and 25 known AD genetic variants and an AD-specific polygenic risk score (PGRS), adjusting for sex, age, vendor (for clinical labs), and the first four genetic principal components; sex-SNP interactions were also assessed. We observed statistically significant SNP-analyte associations for five genetic variants after correction for multiple testing (for SNPs in or near NYAP1, ABCA7, INPP5D, and APOE), with effects detectable from early adulthood. The ABCA7 SNP and the APOE2 and APOE4 encoding alleles were associated with lipid variability, as seen in previous studies; in addition, six novel proteins were associated with the e2 allele. The most statistically significant finding was between the NYAP1 variant and PILRA and PILRB protein levels, supporting previous functional genomic studies in the identification of a putative causal variant within the PILRA gene. We did not observe associations between the PGRS and any analyte. Sex modified the effects of four genetic variants, with multiple interrelated immune-modulating effects associated with the PICALM variant. In post-hoc analysis, sex-stratified GWAS results from an independent AD case–control meta-analysis supported sex-specific disease effects of the PICALM variant, highlighting the importance of sex as a biological variable. Known AD genetic variation influenced lipid metabolism and immune response systems in a population of non-AD individuals, with associations observed from early adulthood onward. Further research is needed to determine whether and how these effects are implicated in early-stage biological pathways to AD. These analyses aim to complement ongoing work on the functional interpretation of AD-associated genetic variants.
Similar content being viewed by others

Introduction
The rapidly decreasing cost of genomics paired with technological advances in the generation of multi-omic data has resulted in multiple datasets of deeply phenotyped individuals with a variety of health outcomes1,2,3. The data collected in these studies have the potential to yield important insights into potential molecular drivers of health observable in the blood periphery. The present study seeks to leverage a unique and relatively large set of multi-omic, deep-phenotyping data to shed light on genetic pathways to late-onset Alzheimer’s disease (AD) by assessing differences in ~ 2000 analytes in the blood that show association with known genetic risk variants for AD. Coupled with high-dimensional data sets, this approach has the potential to yield clues into gene pleiotropy, disease processes, and possible early-intervention strategies, which are critically important given the essentially untreatable nature of late-stage Alzheimer’s disease once significant brain deterioration has occurred.
Genetic variation plays a substantial role in AD risk, with twin studies estimating AD heritability at 58–79%4. While the emergence of recent large-scale consortia efforts has facilitated well-powered meta-analyses of genome-wide association studies (GWAS) to identify multiple common variants with small effect sizes5,6, the research community is still untangling exactly how this genetic variation influences disease risk. Functional genomics studies are beginning to identify likely genetic pathways to disease with the aid of transcriptomic, epigenomic, and endophenotypic data7,8,9,10. So far, genetic and multi-omic studies of AD studies have largely focused on older individuals with either clinically diagnosed AD or milder symptoms of cognitive decline, despite research pointing to highly variable AD pathobiology that occurs on a spectrum, and begins decades before clinical symptoms onset11.
In this study, we leveraged the results from a large-scale GWAS meta-analysis5 alongside data from a deeply phenotyped wellness cohort to investigate the physiological periphery effects of genetic risk for AD in individuals without known cognitive impairment, at all ages. We undertook an agnostic approach by adopting a phenome-wide association study (PheWAS) design12. By examining how genetic variation impacts 2008 analytes in the blood of 2831 individuals, we sought to complement previous functional genomics studies as well as potentially reveal new testable hypotheses for future studies. In addition, we tested for associations between a polygenic risk score (PGRS) for AD and blood analytes to determine if a relative burden of genetic risk might impact observable changes in the blood, and we assessed for effect modification of genetic risk by sex.
Results
Summary of population and study design
Sixty-one percent of Arivale participants were female, 22% were of non-white self-reported ethnicity, and 28% were obese (Table 1). The mean age at blood draw was 47 years, with a range of 18 to 89+. In general, individuals who joined Arivale had somewhat higher levels of cardiovascular risk markers compared to the US population, and slightly lower rates of obesity and pre-diabetes3 (these rates were consistent with rates in the geographies and ethnicities of the population, mostly from the west coast region of the United States).
Phenome-wide association study results
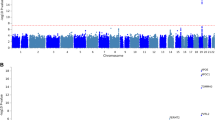
We observed 33 SNP-analyte associations that were statistically significant at FDR-adjusted p-value < 0.05, with most of the associations observed for the APOE SNPs (rs7412, or the e2-defining allele, and rs429358, or the e4-defining allele). The other SNPs showing significant associations with at least one clinical chemistry, protein, or metabolite were rs10933431, rs12539172, and rs3752246 (Fig. 1, Table S2). Complete PheWAS results, including beta coefficients, sample sizes, minor allele frequencies, Hardy–Weinberg Equilibrium p-values, and raw and adjusted p-values for each SNP are in Supplementary Excel File 1. Sample sizes varied among analytes collected (particularly among protein analytes, as a small subset of the population (N = 354) had samples submitted for the full range of protein panels, as described in “Methods” section).

Statistically significant SNP-analyte associations after correcting for multiple testing (threshold FDR-adjusted p-value = 0.05), by SNP. Top panel: log-transformed beta-coefficient from the linear regression model adjusted for sex, age, and genetic principal components 1–4; markers above the zero line (orange) indicate analytes that increased in value with the minor allele, while markers below the line indicate markers that decreased in value. Second panel: FDR-adjusted − log10 p-value; orange line at FDR-p = 0.05. Proteins = red, metabolites = blue, clinical chemistries = purple. Metabolite codes: DG diacylglycerol, LC lactosylceramide, o oleoyl; a arachidonoyl, g glycerol, l linoleoyl, p palmitoyl. Third panel: minor allele frequency (MAF). Bottom panel: Total sample size for each analyte-SNP regression.
NYAP1
The most robust SNP-analyte associations we observed were between rs12539712 in the 3’ region of NYAP1 (Neuronal Tyrosine Phosphorylated Phosphoinositide-3-Kinase Adaptor 1), and two co-regulated proteins, paired immunoglobulin-like type 2 receptors beta and alpha (PILRB and PILRA) (Fig. 2). Carriage of the minor allele (AD risk odds ratio (OR) = 0.92) was associated with significant reduction in normalized protein expression (NPX) of PILRB and PILRA compared to individuals homozygous for the major allele (FDR-adjusted p-values = 2.2 × 10–33 and 2.3 × 10–17, respectively), while the overall level of NPX increased with age among all genotypes. The reduction in protein levels appears roughly dose-dependent with the number of minor alleles and was observed in all but the oldest and youngest age groups (likely due to small numbers of the minor allele in these groups (Table S3A). These observations led us to previous studies pointing to variation in PILRA as the causal gene at this locus, with a missense SNP as a leading candidate (G78R, rs1859788)13,14,15,16. In post-hoc analysis, we repeated the PheWAS with this putative causal SNP (which was in LD with our index SNP rs12539172, R2 = 0.77), and the associations became stronger (FDR-adjusted p-value for PILRB = 3.6 × 10–52; for PILRA = 1.4 × 10–22) (Fig. 2), with genotype differences observed in all age groups (Table S3A).

Unadjusted box plots of normalized protein expression (NPX) levels of PILRA and PILRB by genotype and age group. White boxplots = individuals who are homozygous for the major allele, gray boxplots = heterozygotes, black boxplots = minor allele homozygotes. Box plot midline = median value, lower/upper hinges = 25th and 75th percentiles, respectively; lower whisker ends/upper whisker ends no further than 1.5× interquartile range from the hinge. Data beyond whiskers are outlying points. Top panel: NPX of PILRA and PILRB by rs12539172 (NYAP1) genotype; Bottom panel: NPX of PILRA and PILRB by rs1859788 genotype.
APOE4
We observed significant associations between rs429358 (which encodes the e4 allele) and multiple related clinical measures of cholesterol (Fig. S1). Differences by genotype were less pronounced in older age groups likely due to statin use (Table S3B); exploratory analyses visualizing only individuals who did not report use of statin-lowering medications showed more consistent genotype-dependent differences between rs429358 and the top cholesterol marker, low-density lipoprotein (LDL) particle number (Fig. S2, Table S3B). The concentration of two proteins in the blood were associated with the e4 allele: PLA2G7 (Platelet Activating Factor Acetylhydrolase) and CD28 (T-Cell-Specific Surface Glycoprotein CD28). Selected lipid metabolites in the blood were positively associated with e4: two diacylglycerol (DG) metabolites (one of which was measured twice in the Metabolon panel) were higher in e4 carriers compared to non-carriers.
APOE2
We observed significantly lower levels of multiple clinical measures of LDL cholesterol associated with carriage of the e2 allele (Fig. S3). As the unadjusted plots show, e2 homozygotes are dramatically different than other genotypes, though it should be noted that few e2 homozygotes were present in the population (n = 16) and were within a limited age range (30–59 years). Selected lipid metabolites in the blood were positively associated with e2: a monoglyceride (MG) and four diacylglycerol (DG) metabolites (one of which was a replicate) were higher in e2 carriers compared to non-carriers. We observed six e2-protein associations (Fig. 3), such that each of the following proteins were observed at higher levels in e2 carriers: low density lipoprotein receptor (LDLR), heme oxygenase-1 (HMOX-1), SLAM family member 8 (SLAMF8), ring finger protein 31 (RNF31), contactin associated protein 2 (CNTNAP2), and signal recognition particle 14 (SRP14).

Unadjusted box plots of normalized protein expression levels (NPX) of six proteins significantly associated with APOE2 genotype, by age group. White boxplots = individuals who are homozygous for the major allele, gray boxplots = heterozygotes, black boxplots = minor allele homozygotes. Box plot midline = median value, lower/upper hinges = 25th and 75th percentiles, respectively; lower whisker ends/upper whisker ends no further than 1.5× interquartile range from the hinge. Data beyond whiskers are outlying points. LDLR low-density lipoprotein receptor, HMOX1 heme oxygenase-1, SLAMF8 SLAM family member 8, RNF31 E3 ubiquitin-protein ligase RNF31, CNTNAP2 contactin-associated protein-like 2, SRP14 signal recognition particle 14 kDa protein.
ABCA7
The ABCA7 (ATP Binding Cassette Subfamily A Member 7) variant (rs3752246), which has been associated with increased risk of AD (OR 1.15, Table S1), was associated with lower levels of two lactosylceramide (LC) metabolites in the sphingolipid family. These differences were evident starting in the youngest age groups (Fig. S4, Table S3A). The minor allele of rs3752246 was also associated with higher levels of DEFA1 (Defensin Alpha 1), an antimicrobial peptide.
INPP5D
An intronic SNP in INPP5D (Inositol Polyphosphate-5-Phosphatase D) (rs10933431), which was associated with a lowered risk of AD in meta-analyses, was associated with lower levels of the protein IDUA (alpha-l-iduronidase) (Fig. S4).
Polygenic risk score
No associations were observed between the APOE-free PGRS and any analyte after FDR correction for multiple testing, either in primary analyses or in analyses adjusted for e4 status, or among non-e4 individuals only. No effect modification by sex or APOE4 status was observed.
Sex-specific findings
We observed a SNP × sex interaction involving the AD-protective PICALM variant, such that the minor allele was associated with higher levels of 30 proteins in men and lower levels of the proteins in women (Fig. 4, Fig. S5, Table S4). These proteins were highly correlated with one another (mean pairwise spearman’s rho = 0.49); thus, it is unclear whether the associations are independently biologically meaningful, or whether there is a passenger effect, in which one or a few proteins are driving the sex-differential association with genotype observed in the data. In addition, the PICALM variant is associated with a sex-specific effect on five highly correlated long-chain fatty acid (LCFA) metabolites and one polyunsaturated fatty acid (PFA) metabolite (Docosahexaenoic acid) (Fig. 4). To investigate further, we conducted a post-hoc analysis examining the impact of this variant on AD risk stratified by sex, in a meta-analysis of clinically diagnosed late-onset AD (18,812 individuals, Table S5). While AD risk was reduced in both men and women among carriers of the minor allele, the effect was stronger among men (Table 2, Table S6), which was consistent with the sex-stratified SNP-analyte analyses (data not shown).

Heatmap of statistically significant genotype × sex interaction terms at FDR-adjusted p-value < 0.1. Beta coefficients obtained from sex-stratified analyses, middle-column p-values from interaction term in the full model. SL sphingolipid, LCFA long-chain fatty acid, PFA polyunsaturated fatty acid.
Other observed sex-specific effects were more modest. The SNP near CD2AP (CD2 Associated Protein) interacted with sex to affect three highly correlated sphingomyelins and three plasmologens, while the SNP in SPI1 (Transcription Factor PU.1) interacted with sex to affect SPARC related modular calcium binding 2 (SMOC2). Lastly, the missense ABCA7 SNP interacted with sex to affect levels of Ubiquitin conjugating enzyme E2f (UBE2F).
Stratification by self-identified race/ethnicity
Unfortunately, due to vanishingly small numbers in individual self-identified groups (Table 1), we were not able to assess genetic risk effects in individual groups with statistical rigor. As expected, analyses restricted to white individuals recapitulated results of the overall analysis (Fig. S6). In the nonwhite group overall, we observed effect sizes that were consistent with the overall results and white-only results (Fig. S7).
Discussion
Our study examines associations between known genetic risk factors for AD and blood markers (clinical labs, proteins, and metabolites). It provides insight into the manifestation of AD-related genetic risk in blood-borne analytes from cognitively normal individuals and demonstrated how AD-related genetic variation manifests in the blood across adulthood. Our results contribute to the growing literature highlighting a potential causal variant (missense SNP in PILRA), point to potential new mechanisms of protection among APOE2 carriers, and suggest a role for infectious diseases as AD risk factors, alongside lipid metabolism, immune response, and endocytosis. We also uncovered intriguing differences between men and women in how genetic risk manifests in the blood. These analyses not only add to the existing literature on functional genomics in AD, but also offer up multiple potential new hypotheses to catalyze future studies.
The strongest associations in the study were between the NYAP1 SNP (rs12539172) and the PILRB/PILRA proteins. This locus was originally identified by rs1476679 near ZCWCP16. NYAP1 and ZCWPW1 are located near PILRA and PILRB on chromosome 7, within a linkage disequilibrium (LD) block. In previous gene expression studies, the initial index SNP for ZWCWP1 has been associated with expression of multiple PILRB and PILRA transcripts in brain9,17. PILRA and PILRB are paired, co-regulated inhibiting/activating receptors, respectively, that are expressed on innate immune cells, recognize certain O-glycosylated proteins, and have an important role in regulating acute inflammatory reactions18. The R78 substitution in PILRA (rs1859788) has been shown to reduce the binding capacity of endogenous ligands and thereby potentially increase microglial activity16. In addition, while controversial, work from our group and others19,20,21 has suggested a potential viral role in AD risk. Notably, the R78 variant has been implicated in HSV-1 (Herpes Simplex Virus type 1) infectivity16 and differences in HSV-1 antibody titer levels13. While previous studies have hypothesized that reduced activity of PILRA was due to steric conformational changes in the protein leading to reduced binding of key ligands (including HSV-1 glycoprotein B), our results suggest that reduced levels of circulating PILRA protein in R78 carriers could also be a factor in the overall protective effect of this genetic variant.
Statistically significant associations were observed between multiple lipid analytes and the SNPs encoding both APOE4 (rs429358) and APOE2 (rs7412). APOE normally plays a key role in lipid transport, including shuttling cholesterol to neurons in healthy brains. Notably, APOE has a role in beta-amyloid (Aβ) metabolism, and while the exact mechanism is unknown, the e4 variant appears to accelerate neurotoxic Aβ accumulation, aggregation, and deposition in the brain22. The associations we observed between the e4 variant and increased levels of total cholesterol and LDL cholesterol, along with lower levels of high-density lipoprotein (HDL), were consistent with previous cardiovascular disease cohort studies that included young, middle-aged, and older adults23,24,25,26. The e4 allele was associated with increased NPX of two inflammatory proteins. PLA2G7 is a known cardiovascular risk marker with pro-inflammatory and oxidative activities27 which has previously been associated with APOE genotypes28 and implicated in AD and cognitive decline27,29. To our knowledge, CD28 protein levels have not previously been associated with e4 status, though this relatively weak association may be a downstream result of APOE isoform-specific effects on inflammation30.
Blood cholesterol levels among APOE2 carriers were also largely consistent with a body of existing data24; the e2 variant was associated with lower levels of multiple measures of LDL cholesterol. It should be noted that while 5–10% of e2 homozygotes develop type III hyperlipoproteinemia (typically in the presence of an existing metabolic disorder31) resulting in elevated cholesterol levels, all e2 homozygotes in the study had significantly decreased levels of LDL cholesterol compared to other genotypes. In contrast, the e2 variant was associated with higher levels of six lipid metabolites in the diacylglycerol and monoacylglycerol family; interestingly, both the e4 variant and e2 variants were associated with increased levels of the same two lipid metabolites in the diacylglycerol family, despite the opposite effects of these two variants on circulating blood cholesterol. Diacylglycerol is a precursor to triacylglyceride (TG), which is typically higher in APOE2 carriers26. The effects of high DGs and TGs remains unclear. DG-rich diets fed to diabetic APOE-knockout mice had reduced atherosclerosis and lower plasma cholesterol than mice fed TG-rich or western diets32,33; however, non-targeted metabolomics studies have shown elevated levels of DGs and MGs in AD and mild cognitive impairment (MCI) patient brains and blood compared to cognitively intact individuals34,35.
We observed six proteins that were significantly upregulated in APOE2 carriers (Fig. 3). The LDLR protein had higher levels of NPX in e2 carriers, particularly in e2 homozygotes. Though APOE2 is known to bind poorly to LDLR (~ 2% of e3 or e4 binding activity)36, APOE2 was associated with lower levels of LDL cholesterol across age groups as noted previously, perhaps due to compensatory up-regulation of LDLR26. Greater understanding of the compensatory mechanism leading to upregulated LDLR and lower circulating LDL cholesterol is needed. The e2 variant was associated with increased levels of the highly inducible HMOX-1, which has antioxidant properties and has been associated with both neuroprotection and neurodegeneration37. SLAMF8 may be another link to an antioxidant effect of APOE2, as it has been implicated in modulation of reactive oxygen species and inflammation via negative regulation of NOX activity38. APOE2 carriers displayed higher levels of RNF31 protein (aka HOIP). HOIP is the catalytic component of the linear ubiquitin chain assembly complex (LUBAC), which was shown to have a role in the recognition and degradation of misfolded proteins39. Variation in CNTNAP2, a member of the neurexin superfamily of proteins involved in cell–cell interactions in the nervous system, has been associated with neurodevelopmental disorders40, and has been implicated in AD-related dementia41. Lastly, SRP14, which has a role in targeting secretory proteins to the rough endoplasmic reticulum (ER) membrane, has been identified as one of many tau-associated ER proteins in AD brains42. To our knowledge, the APOE2-protein associations described here are novel and may help point to the mechanisms of protection associated with the e2 variant.
ABCA7 is involved in lipid efflux from cells into lipoprotein particles, plays a role in lipid homeostasis43, and has also been implicated in Aβ processing and deposition in the brain44. Our results support ABCA7’s lipid-related function by showing lower levels of two LC metabolites among individuals carrying the AD-risk allele of rs3752246. In contrast, we observed higher NPX of DEFA1 protein in carriers of the ABCA7 variant, which is consistent with previous studies showing higher levels of this protein in cerebral spinal fluid (CSF) and sera of AD patients compared to controls45,46, potentially linking ABCA7 with an inflammatory response pathway to AD. Lastly, lower NPX of IDUA was associated with the INPP5D SNP. INPP5D, which encodes the lipid phosphatase SHIP1, is a negative regulator of immune signaling and is expressed in microglia47. To our knowledge, this association has not been previously observed.
Genetic variation likely affects men and women differentially, pointing to mechanisms that contribute to known differences in AD pathology between the sexes48. The set of proteins that were differentially affected by sex and PICALM genotype are primarily implicated in immune processes, cell adhesion, and regulatory processes, with widely overlapping functions (Fig. S8). Our results highlight an interaction between the AD-risk variant in PICALM and multiple proteins implicated in immune response in a sex-specific manner, and support emerging research showing sex differences in the neuroimmune response that impact microglia function49. We also observed a sex-differential effect of the variant on multiple LCFA metabolites and one PFA metabolite (DHA). A potential link between PICALM function, lipids, and AD is feasible: fatty acids, and DHA in particular, have long been known to have a role in maintaining brain health and cognition50, while PICALM expression has been shown to influence cholesterol homeostasis through multiple mechanisms51. This multi-analyte interaction was supported by results from sex stratified GWAS meta-analyses, which showed differing effect sizes of the variant on men vs. women.
In addition to these sex-specific PICALM effects, the SNP near CD2AP, a scaffolding protein, interacted with sex to affect three highly correlated sphingomyelins and three plasmologens, while the SNP in SPI1, a transcription factor associated with microglial activation52, interacted with sex to affect SMOC2, a protein involved in microgliosis that has been previously associated with Aβ positivity in CSF53.
We also examined an AD-specific polygenic risk score. While the PGRS is predictive of disease in case/control studies54, it was not associated with any blood analytes in the all-ages AD-free Arivale cohort. Combining genetic effects into a single score for AD likely served to dilute any individual genetic effect on the manifestation of genetic risk in the blood. In addition, the relative youth and cognitive health of this cohort should be considered. The PGRS may be more likely to detect perturbation in analytes that are markers of systemic inflammation or immune dysfunction in later life and among cohorts experiencing cognitive impairment.
The results presented here are novel and we believe will be of interest to the AD-related functional genomics community, though several limitations should be noted. The study population was not a random sample but was self-selected. The population is largely self-identified non-hispanic white, was mostly located on the west coast, and likely has higher than average socio-economic status (though these data were not captured). Thus, results may not be generalizable to a broader population. At this time, we were not aware of a suitable replication cohort that would contain parallel-omics panels in an all-ages health-heterogeneous cohort. Future studies will be needed to assess generality of the findings to other populations, not only for the sake of replicability of the findings, but due to the relative ancestral homogeneity of this data set. Previous studies have shown genetic heterogeneity between white and non-white individuals, particularly with regard to African Americans and risk of cognitive outcomes among carriers of APOE and ABCA7 variants55,56. Given known wide-ranging racial/ethnic disparities in dementia incidence57, it is imperative that future deep-phenotyping studies are far more inclusive than the study presented here.
Another limitation to the interpretation of results concerns the issue of pleiotropy; we cannot discern pleiotropic, non-AD-related effects from true causal effects that are implicated in AD pathogenesis. However, even if the associations described here are purely the result of pleiotropy and are unrelated to causal mechanisms of AD, the novel associations we described may provide clues to the function of several genes that are highly interesting to the AD community. Related, we only obtained peripheral plasma, and are unable to examine effects in AD-relevant compartments such as brain or CSF. We had high-coverage WGS available and did not interrogate other types of genetic variation such as copy number variants, indels, and short tandem repeats. Lastly, data harmonization with other studies will be a challenge. For instance, most previous metabolomics studies used metabolomics data that lacked complete speciation, and more work is needed with newer technologies that yield high fidelity data to determine the biological effects of specific serum metabolites.
This study also has multiple strengths. While most studies focused on AD-related genetic variation consist of case/control cohorts in older adults, the Arivale data offered an unprecedented look into how genetic variation perturbs physiological pathways in the blood long before disease onset, in health-heterogeneous individuals of all ages. This feature allowed us to observe subtle changes in blood associated with genetic variation, due to the relatively large sample size (2831 individuals with WGS) and the high quality of the blood analytes collected. Our results are from a “real-world” cohort, which promises to be an increasing source of large-scale data in the community going forward, with its accompanying advantages and disadvantages. Some results were previously unobserved and need to be replicated (such as the associations between APOE2 and multiple proteins), while other results agree with previous findings and serve to reinforce confidence that the results are reasonably representative and not simply spurious.
Conclusions
Due to a unified world-wide effort, dozens of genetic variants have been robustly implicated in the development of AD, though we are still in the early stages of understanding exactly how genetic variation contributes to disease. Our study showed that AD-related genetic variation manifests in the blood, from early adulthood onward, and highlights known targets for prevention in early and mid-life, such as cholesterol monitoring, mitigation of inflammation, and possibly, HSV-1 prevention and/or viral load management. Importantly, as well as yielding new insight into the pathobiology of AD through adulthood, these results may provide a significant number of new drug targets that are highly novel and biologically plausible or may serve as biomarkers if confirmed to have a consistent influence on AD pathophysiology. Lastly, these results highlight the need to assess for sex differences in future studies. Taken together, these results not only illustrate previously unobserved biological phenomenon as a result of AD-associated genetic variation, but also serve as an important resource for the generation of hypotheses for future functional genomics studies and emphasize the potential insight that can be gleaned from deeply phenotyped individuals.
Methods
Population
The Institute for Systems Biology, through partnership with their spin-out company Arivale, has access to a wealth of data collected from subscribers in the commercially available (now closed) Arivale Scientific Wellness program3,58, from July 2015 to May 2019. In brief, participants in the Arivale program were assigned a health coach upon joining the program, who then utilized data from clinical blood assays and detailed health-history and behavioral questionnaires to personalize health advice and management of health goals.
All research was conducted in accordance with regulations and guidelines for observational research in human subjects. Informed consent was obtained from all participants for the use of their anonymized data in research. The study was reviewed and approved by the Western International Review Board (Study Number 1178906 at Arivale and Study Number 20170658 at the Institute for Systems Biology, in Seattle, WA).
Blood-derived clinical laboratory tests and whole genome sequencing
We identified 2831 individuals with whole genome sequencing (WGS) and at least one class of blood-derived analyte, described as follows. For each participant, fasting clinical blood laboratory tests were measured upon joining the program. Blood samples were collected at either local facilities hosted by LabCorp (North Carolina, USA) or Quest Diagnostics (New Jersey, USA). Whole genome sequencing was performed on DNA extracted from whole blood with library preparation using the Illumina TruSeq Nano Library prep kit and sequenced using Illumina HiSeq X, PE-150, target 30× coverage at a single Clinical Laboratory Improvement Amendmnets (CLIA)-approved sequencing laboratory. Only values with < 20% missing were included, and no imputation was performed. At the baseline blood draw, 2827 of the 2831 individuals with sequenced whole genomes had up to 63 fasting clinical blood lab tests. Clinical blood tests included standard markers for cardiometabolic health (lipid levels), diabetes, inflammation, kidney and liver function, nutrition (vitamins and minerals), and blood cell counts. All clinical lab tests included, with descriptions and units where available, are in Supplementary Excel File 2.
Proteomics: Frozen plasma samples (aliquots of the initial blood draw) were also sent to Olink (Olink Bioscience, Sweden) for targeted proteomics assays based on Olink’s proximity extension assay (PEA) technique59, which is a dual-recognition, DNA-coupled methodology that is quantified by quantitative real-time PCR and enables high multiplex, high throughput proteomics that are both sensitive and specific (for further details, see https://www.olink.com/our-platform/our-pea-technology/). Full details of normalization and batch effect adjustment have been described previously60. For analysis, only proteins with < 20% missing were included and no imputation was performed. Up to 2694 of these participants had quantitative proteomic data on 274 proteins from three Olink panels (Cardiovascular II, Cardiovascular III, and Inflammation panels). An additional 919 proteins (from 10 additional panels available at Olink at the time) were obtained from a subsample of 354 individuals, in which Apolipoprotein E (APOE) e2/e2 and APOE e4/e4 genotypes were overrepresented. Since multiple batches were performed, previously generated pooled control samples were run with each batch and used for batch correction and multiple control samples were included on each plate.
Metabolomics
Aliquots of frozen plasma samples were sent to Metabolon, Inc. (North Carolina) to conduct metabolomics assays using the Metabolon HD4 discovery platform. In brief, Metabolon conducted their Global Metabolomics high-performance liquid chromatography (HPLC)-mass spectrometry assays on the plasma samples. Full details of sample handling, quality control, biochemical identification, data curation, and quantification and normalization has been described previously60,61. For analysis, only metabolites with < 20% missing (or detectable) were included and no imputation was performed. Up to 1909 of the participants had data from 754 metabolites, though due to technical variability and variation in detection rates of rare metabolites, sample sizes ranged from 1539 to 1909 after pruning metabolites with < 20% missing. Relative concentration values were reported for each metabolite. Full biochemical annotation for each metabolite (when available), as provided by Metabolon at the time of quantification, can be found in Supplementary Excel File 2.
SNP selection
We selected 25 common and somewhat-rare (> 1% allele frequency) single nucleotide polymorphisms (SNPs) that were significantly associated with AD in a large-scale meta-analysis based on updated data from the International Genomics of Alzheimer’s Project (IGAP)5. In addition to these variants, we also included the SNP coding for APOE e2 (rs7412). The 25 SNPs were linked to 24 genes (two SNPs in APOE), as detailed in Table S1.
Polygenic risk score calculation for AD
PGRS for age-associated AD risk was computed using summary statistics from the initial IGAP-driven GWAS meta-analysis6. Briefly, the set of SNPs included in the PGS was determined as follows. The Benjamini–Hochberg62 procedure was applied to the p-values for all SNPs tested in the GWAS to account for multiple testing by controlling the false discovery rate (FDR) at a 5% level. This FDR-filtered set of SNPs was then further pruned using linkage disequilibrium (LD): pairs of SNPs in close proximity capturing highly correlated information (r2 > 0.2) were identified, and the SNP with the smaller p-value in the pair was kept; this was repeated until all remaining SNPs were mutually uncorrelated (r2 < 0.2 for all pairs). The PGRS for each individual was then calculated by summing up the published effect size for each selected SNP multiplied by the number of effect alleles the individual carried for that SNP, across all of the selected SNPs. Missing genotypes were mean imputed using the effect allele frequency.
Statistical analysis
Following a phenome-wide association study approach (PheWAS)12,63, the primary model for each SNP used linear regression, with genotype (0, 1, or 2, with 0 indicating homozygosity for the major allele and 2 indicating homozygosity for the minor allele) as the predictor, and each continuous quantitative analyte as the dependent variable. Clinical lab and metabolite values were natural log transformed to account for right skewness and outliers, with + 1 added to each natural log transformation to prevent zero values. Proteomic quantities were presented as normalized protein expression (NPX), Olink’s arbitrary unit, which is in log2 scale. Genetic ancestry was represented by principal components (PCs) 1–4, calculated using previously described methods64. All SNP models were adjusted for age, sex, genetic ancestry PCs 1–4, and vendor identification for the clinical labs. Secondary models tested effect modification by sex by including a gene x sex interaction term in the models. We accounted for multiple comparisons by applying the Benjamini–Hochberg method62 at alpha = 0.05 on a per-SNP basis and applied to the main effect of genotype in the primary models, while we set B-H alpha = 0.1 of the sex-SNP interaction term as the threshold for the gene x sex interaction models. The FDR rate took into account testing for all 2008 possible analytes, with the understanding that this adjustment was highly conservative given a high degree of correlation among multiple groups of analytes, and the fact that some analytes were sampled in only a subset of individuals. Both raw and adjusted p-values are reported.
We also repeated the primary PheWAS approach with participants stratified by self-identified race, due to evidence for variable genetic risk for cognitive outcomes between non-Hispanic white (hereafter referred to as “white”) and non-white populations55,56. Unfortunately, due to small numbers of individuals in specific non-white racial and ethnic groups, which become vanishingly small when accounting for allele frequency and numbers of available samples (Table 1), we were not able to assess genetic risk effects in individual groups with statistical rigor and had to group all non-white participants into one stratum for analysis. The stratified white and non-white group analyses serve as an investigation into whether our primary results reflected the majority-white makeup of the Arivale population. PheWAS was applied as described above, with FDR to account for multiple comparisons.
To visualize genotype-analyte associations across adulthood, we created boxplots of the log-transformed analyte values by genotype, stratified by age group (by decade, from 18–29 to 70 and over). One-way analysis of variance (ANOVA) was used to test whether there was an overall difference between genotypes within each age group. All statistical analyses were performed in R v3.5.1 (https://www.R-project.org/).
In post-hoc exploratory analysis focused on the SNP in the PICALM (Phosphatidylinositol Binding Clathrin Assembly Protein) locus (rs3851179), sex-stratified and sex-interaction analyses was performed on 12,324 cases (57.7% female) and 11,453 controls (59.9% female) of European ancestry from the Alzheimer’s Disease Genetics Consortium (ADGC) (see Supplementary Table 4 for dataset details). Datasets were imputed to the Haplotype Reference Consortium (HRC)65 panel using the Michigan Imputation Server (https://imputationserver.sph.umich.edu/index.html#!). Standard pre-imputation quality control was performed on all datasets individually, including exclusion of individuals with low call rate, individuals with a high degree of relatedness, and variants with low call rate66. Individuals with non-European ancestry according to principal components analysis of ancestry-informative markers were excluded from the further analysis. Detailed descriptions of individual ADGC datasets can be found in Kunkle et al.5 and Table S5. Study-specific logistic regression analyses employed Plink67 for sex-interaction analysis and SNPTest68 for sex-stratified analysis. Sex-interaction, which analyzed the sex × variant interaction, and sex-stratified analysis of males and females separately, were performed for two separate models per analysis, one adjusting for age, sex and PCs (model 1) and a second adjusting for age, sex, PCs and APOE (model 2). Results were meta-analyzed with METAL using inverse variance-based analysis69. In order to explore the relationships among the proteins associated with the PICALM variant, we input the list of sex-interacting proteins into Cytoscape software, utilizing the CLUEGO plug-in70,71, which drew a network linking proteins through their known GO Biological processes.
Data availability
The datasets generated and/or analysed during the current study are not publicly available because the data was generated by a private investment firm under legal terms that mandate researchers to sign a data access agreement permitting the use of these data for non-proift research purposes. Upon reasonable request, researchers can access the Arivale deidentified dataset supporting the findings in this study for research purposes from ISB. Requests should be sent to data-access@isbscience.org. The data are available to qualified researchers on submission and approval of a research plan.
Code availability
Code used for PheWAS statistical analysis is available through the Sage Bionetworks Github (https://github.com/Sage-Bionetworks/ADsnps_PheWAS.git).
References
Price, N. D. et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat. Biotechnol. https://doi.org/10.1038/nbt.3870 (2017).
Schüssler-Fiorenza Rose, S. M. et al. A longitudinal big data approach for precision health. Nat. Med. 25, 792–804 (2019).
Zubair, N. et al. Genetic predisposition impacts clinical changes in a lifestyle coaching program. Sci. Rep. https://doi.org/10.1038/s41598-019-43058-0 (2019).
Gatz, M. et al. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174 (2006).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019).
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Karch, C. M. & Goate, A. M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51 (2015).
Pimenova, A. A., Raj, T. & Goate, A. M. Untangling genetic risk for Alzheimer’s disease. Biol. Psychiatry 83, 300–310 (2018).
Allen, M. et al. Late-onset Alzheimer disease risk variants mark brain regulatory loci. Neurol. Genet. 1, e15 (2015).
Ertekin-Taner, N. Gene expression endophenotypes: A novel approach for gene discovery in Alzheimer’s disease. Mol. Neurodegener. 6, 31 (2011).
Atri, A. The Alzheimer’s disease clinical spectrum: Diagnosis and management. Med. Clin. N. Am. 103, 263–293 (2019).
Pendergrass, S. A. & Ritchie, M. D. Phenome-wide association studies: Leveraging comprehensive phenotypic and genotypic data for discovery. Curr. Genet. Med. Rep. 3, 92–100 (2015).
Agostini, S. et al. The PILRA G78R variant correlates with higher HSV-1-specific IgG titers in Alzheimer’s disease. Cell. Mol. Neurobiol. 39, 1217–1221 (2019).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Patel, T. et al. Whole-exome sequencing of the BDR cohort: Evidence to support the role of the PILRA gene in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 44, 506–521 (2018).
Rathore, N. et al. Paired Immunoglobulin-like type 2 receptor alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet. 14, e1007427 (2018).
Karch, C. M., Ezerskiy, L. A., Bertelsen, S., Alzheimer’s Disease Genetics Consortium (ADGC) & Goate, A. M. Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 Loci. PLoS ONE 11, e0148717 (2016).
Wang, J., Shiratori, I., Uehori, J., Ikawa, M. & Arase, H. Neutrophil infiltration during inflammation is regulated by PILRα via modulation of integrin activation. Nat. Immunol. 14, 34–40 (2013).
Eimer, W. A. et al. Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 99, 56–63 (2018).
Itzhaki, R. F. & Wozniak, M. A. Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J. Alzheimers Dis. 13, 393–405 (2008).
Readhead, B. et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99, 64–82 (2018).
Liu, C.-C., Liu, C.-C., Kanekiyo, T., Xu, H. & Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013).
Karjalainen, J.-P. et al. The effect of apolipoprotein E polymorphism on serum metabolome—A population-based 10-year follow-up study. Sci. Rep. 9, 458 (2019).
Downer, B., Estus, S., Katsumata, Y. & Fardo, D. W. Longitudinal trajectories of cholesterol from midlife through late life according to apolipoprotein E allele status. Int. J. Environ. Res. Public Health 11, 10663–10693 (2014).
Grönroos, P. et al. Influence of apolipoprotein E polymorphism on serum lipid and lipoprotein changes: A 21-year follow-up study from childhood to adulthood. The Cardiovascular Risk in Young Finns Study. Clin. Chem. Lab. Med. 45, 592–598 (2007).
Bennet, A. M. et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA 298, 1300–1311 (2007).
Davidson, J. E. et al. Plasma lipoprotein-associated phospholipase A2 activity in Alzheimer’s disease, amnestic mild cognitive impairment, and cognitively healthy elderly subjects: A cross-sectional study. Alzheimers Res. Ther. 4, 51 (2012).
Drenos, F. et al. Integrated associations of genotypes with multiple blood biomarkers linked to coronary heart disease risk. Hum. Mol. Genet. 18, 2305–2316 (2009).
van Oijen, M. et al. Lipoprotein-associated phospholipase A2 is associated with risk of dementia. Ann. Neurol. 59, 139–144 (2006).
Zhang, H., Wu, L.-M. & Wu, J. Cross-talk between apolipoprotein E and cytokines. Mediat. Inflamm. 2011, 949072 (2011).
Mahley, R. W., Huang, Y. & Rall, S. C. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J. Lipid Res. 40, 1933–1949 (1999).
Fujii, A., Allen, T. J. & Nestel, P. J. A 1,3-diacylglycerol-rich oil induces less atherosclerosis and lowers plasma cholesterol in diabetic apoE-deficient mice. Atherosclerosis 193, 55–61 (2007).
Ijiri, Y. et al. Dietary diacylglycerol extenuates arterial thrombosis in apoE and LDLR deficient mice. Thromb. Res. 117, 411–417 (2006).
Wood, P. L., Barnette, B. L., Kaye, J. A., Quinn, J. F. & Woltjer, R. L. Non-targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer’s disease subjects. Acta Neuropsychiatr. 27, 270–278 (2015).
Wood, P. L. et al. Targeted lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer’s disease: Validation of DAG accumulation early in the pathophysiology of Alzheimer’s disease. J. Alzheimers Dis. 48, 537–546 (2015).
Mahley, R. W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. Berl. Ger. 94, 739–746 (2016).
Barone, E., Di Domenico, F., Mancuso, C. & Butterfield, D. A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 62, 144–159 (2014).
Wang, G. et al. Cutting edge: Slamf8 is a negative regulator of Nox2 activity in macrophages. J. Immunol. Baltim. Md. 1950(188), 5829–5832 (2012).
van Well, E. M. et al. A protein quality control pathway regulated by linear ubiquitination. EMBO J. https://doi.org/10.15252/embj.2018100730 (2019).
Rodenas-Cuadrado, P., Ho, J. & Vernes, S. C. Shining a light on CNTNAP2: Complex functions to complex disorders. Eur. J. Hum. Genet. EJHG 22, 171–178 (2014).
van Abel, D. et al. Direct downregulation of CNTNAP2 by STOX1A is associated with Alzheimer’s disease. J. Alzheimers Dis. 31, 793–800 (2012).
Meier, S. et al. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer’s disease brain. J. Alzheimers Dis. 48, 687–702 (2015).
De Roeck, A., Van Broeckhoven, C. & Sleegers, K. The role of ABCA7 in Alzheimer’s disease: Evidence from genomics, transcriptomics and methylomics. Acta Neuropathol. (Berl.) 138, 201–220 (2019).
Sakae, N. et al. ABCA7 deficiency accelerates amyloid-β generation and Alzheimer’s neuronal pathology. J. Neurosci. Off. J. Soc. Neurosci. 36, 3848–3859 (2016).
Szekeres, M. et al. Relevance of defensin β-2 and α defensins (HNP1-3) in Alzheimer’s disease. Psychiatry Res. 239, 342–345 (2016).
Watt, A. D. et al. Peripheral α-defensins 1 and 2 are elevated in Alzheimer’s disease. J. Alzheimers Dis. 44, 1131–1143 (2015).
Efthymiou, A. G. & Goate, A. M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 12, 43 (2017).
Dumitrescu, L., Mayeda, E. R., Sharman, K., Moore, A. M. & Hohman, T. J. Sex differences in the genetic architecture of Alzheimer’s disease. Curr. Genet. Med. Rep. 7, 13–21 (2019).
Osborne, B. F., Turano, A. & Schwarz, J. M. Sex differences in the neuroimmune system. Curr. Opin. Behav. Sci. 23, 118–123 (2018).
Fotuhi, M., Mohassel, P. & Yaffe, K. Fish consumption, long-chain omega-3 fatty acids and risk of cognitive decline or Alzheimer disease: A complex association. Nat. Clin. Pract. Neurol. 5, 140–152 (2009).
Mercer, J. L. et al. Modulation of PICALM levels perturbs cellular cholesterol homeostasis. PLoS ONE 10, e0129776 (2015).
Verheijen, J. & Sleegers, K. Understanding Alzheimer disease at the interface between genetics and transcriptomics. Trends Genet. 34, 434–447 (2018).
Whelan, C. D. et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol. Commun. 7, 169 (2019).
Harrison, J. R., Mistry, S., Muskett, N. & Escott-Price, V. From polygenic scores to precision medicine in Alzheimer’s disease: A systematic review. J. Alzheimers Dis. 74, 1271–1283 (2020).
N’Songo, A. et al. African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol. Genet. 3, e141 (2017).
Berg, C. N., Sinha, N. & Gluck, M. A. The effects of APOE and ABCA7 on cognitive function and Alzheimer’s disease risk in African Americans: A focused mini review. Front. Hum. Neurosci. 13, 387 (2019).
Mayeda, E. R., Glymour, M. M., Quesenberry, C. P. & Whitmer, R. A. Inequalities in dementia incidence between six racial and ethnic groups over 14 years. Alzheimers Dement. J. Alzheimers Assoc. 12, 216–224 (2016).
Xu, X. et al. Habitual sleep duration and sleep duration variation are independently associated with body mass index. Int. J. Obes. 2005(42), 794–800 (2018).
Assarsson, E. et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192 (2014).
Manor, O. et al. A multi-omic association study of trimethylamine N-oxide. Cell Rep. 24, 935–946 (2018).
Wittmann, B. M. et al. Bladder cancer biomarker discovery using global metabolomic profiling of urine. PLoS ONE 9, e115870 (2014).
Hochberg, Y. & Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med. 9, 811–818 (1990).
Hall, M. A. et al. Detection of pleiotropy through a phenome-wide association study (PheWAS) of epidemiologic data as part of the environmental architecture for genes linked to environment (EAGLE) study. PLoS Genet. 10, e1004678 (2014).
Conomos, M. P. et al. Genetic diversity and association studies in US Hispanic/Latino populations: Applications in the hispanic community health study/study of latinos. Am. J. Hum. Genet. 98, 165–184 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Sims, R. et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet. 49, 1373–1384 (2017).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Marchini, J., Howie, B., Myers, S., McVean, G. & Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39, 906–913 (2007).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinform. Oxf. Engl. 26, 2190–2191 (2010).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Bindea, G. et al. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009).
Funding
This study was supported by the National Institutes of Health, National Institute on Aging Grants U01 AG046139 (N.E-T, T.E.G, N.D.P), R01 AG061796 (N.E-T), RF1 AG051504 (N.E-T), R01-AG062634-01 (B.W.K, E.R.M), and U19 AG023122 (N.R). ADGC. The National Institutes of Health, National Institute on Aging (NIH-NIA) supported this work through the following Grants: ADGC, U01 AG032984, RC2 AG036528; Samples from the National Cell Repository for Alzheimer’s Disease (NCRAD), which receives government support under a cooperative agreement Grant (U24 AG21886) awarded by the National Institute on Aging (NIA), were used in this study. We thank contributors who collected samples used in this study, as well as patients and their families, whose help and participation made this work possible; Data for this study were prepared, archived, and distributed by the National Institute on Aging Alzheimer’s Disease Data Storage Site (NIAGADS) at the University of Pennsylvania (U24-AG041689-01); NACC, U01 AG016976; NIA LOAD (Columbia University), U24 AG026395, U24 AG026390, R01AG041797; Banner Sun Health Research Institute P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01 AG048927, R01AG33193, R01 AG009029; Columbia University, P50 AG008702, R37 AG015473, R01 AG037212, R01 AG028786; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG006781, UO1 HG004610, UO1 HG006375, U01 HG008657; Indiana University, P30 AG10133, R01 AG009956, RC2 AG036650; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG016574, R01 AG032990, KL2 RR024151; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, UL1 RR029893, 5R01AG012101, 5R01AG022374, 5R01AG013616, 1RC2AG036502, 1R01AG035137; North Carolina A&T University, P20 MD000546, R01 AG28786-01A1; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG030146, R01 AG01101, RC2 AG036650, R01 AG22018; TGen, R01 NS059873; University of Alabama at Birmingham, P50 AG016582; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653, AG041718, AG07562, AG02365; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136, R01 AG042437; University of Wisconsin, P50 AG033514; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991, P01 AG026276. The Kathleen Price Bryan Brain Bank at Duke University Medical Center is funded by NINDS Grant # NS39764, NIMH MH60451 and by Glaxo Smith Kline. Support was also from the Alzheimer’s Association (LAF, IIRG-08-89720; MP-V, IIRG-05-14147), the US Department of Veterans Affairs Administration, Office of Research and Development, Biomedical Laboratory Research Program, and BrightFocus Foundation (MP-V, A2111048). P.S.G.-H. is supported by Wellcome Trust, Howard Hughes Medical Institute, and the Canadian Institute of Health Research. Genotyping of the TGEN2 cohort was supported by Kronos Science. The TGen series was also funded by NIA Grant AG041232 to AJM and MJH, The Banner Alzheimer’s Foundation, The Johnnie B. Byrd Sr. Alzheimer’s Institute, the Medical Research Council, and the state of Arizona and also includes samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts and Newcastle University), MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council),South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England (HEFCE), Alzheimer’s Research Trust (ART), BRACE as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN), The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stiching Alzheimer Onderzoek), Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona. ADNI data collection and sharing was funded by the National Institutes of Health Grant U01 AG024904 and Department of Defense award number W81XWH-12-2-0012. ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. We thank Drs. D. Stephen Snyder and Marilyn Miller from NIA who are ex-officio ADGC members.
Author information
Authors and Affiliations
Consortia
Contributions
L.Heath and N.D.P. conceived and designed the study; L.Heath, J.C.E., B.W.K., A.C.N., and E.R.M. performed data analyses and figure generation; A.T.M., J.C.L., B.W.K., E.R.M., N.E.-T., L.Hood, N.R., and N.D.P. acquired the data; L.Heath, J.C.E., N.R., A.T.M., S.A.K., J.C.L., C.C.F., B.A.L., L.M.M., N.E.-T., T.E.G., L.Hood, and N.D.P. made substantial contributions to interpretation of the results. L.Heath and N.D.P. were primary authors of the manuscript. All authors read and approved final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heath, L., Earls, J.C., Magis, A.T. et al. Manifestations of Alzheimer’s disease genetic risk in the blood are evident in a multiomic analysis in healthy adults aged 18 to 90. Sci Rep 12, 6117 (2022). https://doi.org/10.1038/s41598-022-09825-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-09825-2
This article is cited by
-
The relationship between 11 different polygenic longevity scores, parental lifespan, and disease diagnosis in the UK Biobank
GeroScience (2024)
-
A global view of the genetic basis of Alzheimer disease
Nature Reviews Neurology (2023)
-
ABCA7-Associated Clinical Features and Molecular Mechanisms in Alzheimer’s Disease
Molecular Neurobiology (2023)
-
Common factors among three types of cells aged in mice
Biogerontology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
