
Abstract
The recognition of the AUG start codon and selection of an open reading frame (ORF) is fundamental to protein biosynthesis. Defect in the fidelity of start codon selection adversely affect proteome and have a pleiotropic effect on cellular function. Using proteomic techniques, we identified differential protein abundance in the translation initiation fidelity defective eIF5G31R mutant that initiates translation using UUG codon in addition to the AUG start codon. Consistently, the eIF5G31R mutant altered proteome involved in protein catabolism, nucleotide biosynthesis, lipid biosynthesis, carbohydrate metabolism, oxidation–reduction pathway, autophagy and re-programs the cellular pathways. The utilization of the upstream UUG codons by the eIF5G31R mutation caused downregulation of uridylate kinase expression, sensitivity to hydroxyurea, and DNA damage. The eIF5G31R mutant cells showed lower glutathione levels, high ROS activity, and sensitivity to H2O2.
Similar content being viewed by others

Introduction
The fidelity of start codon selection is critical for the synthesis of a normal proteome. Defect in the genes involved in the general translation initiation that alters the fidelity of start codon selection could have a pleiotropic effect on cellular function as they change the cellular proteome. However, no studies have been conducted to understand the change in cellular proteome and its effects on cellular physiology caused by the defect in eukaryotic translation initiation factor fidelity. In eukaryotic translation initiation, the AUG start codon is predominantly selected to establish an open reading frame (ORF) to decode the genetic code while scanning the mRNA from 5′ to 3′ direction by the translation initiation machinery. The Saccharomyces cerevisiae translation initiation factors critical for the start codon selection consist of eIF2-GTP-Met-tRNAi Ternary complex (TC), eIF1, eIF1A, eIF5, eIF3, and 40S ribosomal subunit together constitute 43S pre-initiation complex (PIC) that is involve in the selection of AUG start codon on the mRNA1,2. The mRNA binds to the eIF4F complex through its 5′ m7Gppp cap and it is recruited laterally to the PIC by placing the mRNA into the mRNA entry channel results in the formation of 48S initiation complex3,4. The factor eIF1A binds to the A-site of the ribosome and promotes mRNA scanning by its C-terminal tail (CTT)5. The factor eIF1 binds near P-site to monitors anti-codon:codon interaction and prevents non-AUG codon selection1,2. During the scanning for AUG start codon, the N-terminal GTPase activating protein (GAP) domain of eIF5 interacts with the G-domain of eIF2γ subunit to trigger GTP hydrolysis to form GDP and Pi. Once the AUG start codon enters the P-site of the 40S ribosome, the anti-codon:codon interaction causes major rearrangement in the scanning complex. The eIF1A-CTT interacts with the N-terminal domain of eIF5, whereas the eIF1 moves out of the P-site of the ribosome, causing the Met-tRNAi to move from POUT to PIN conformation resulting in the selection of AUG codon and release of Pi from the eIF2-GDP-Pi complex3,6,7.
In yeast, the mitochondrial version of glycyl-tRNA synthetase and alanine-tRNA synthetase is translated using UUG and ACG start codons, respectively8,9. However, the molecular mechanisms involved in the non-AUG codon selection in these cases are unknown. The mRNA features such as cis-acting long structured 5′ untranslated region (UTR) or the short unstructured 5′ UTR and the trans-acting mRNA recruitment and assisting factors such as eIF4B, eIF4G, PABP, or Ded1p plays an important role in the effective translation of a selective mRNAs10,11,12,13,14. However, defects in the assembly of the 48S initiation complex could adversely affect start codon selection and translation efficacy of global mRNA during the scanning process that can alter general proteome. Mutations isolated in some of the initiation factors such as eIF5G31R, eIF2βS264Y and eIF2γN135D cause relax stringency in the recognition of AUG start codon (Suppressor of initiation codon: Sui− phenotype) proposed to be caused due to the premature Pi release, intrinsic GTPase reaction and altered tRNAi binding defect, respectively15,16,17. The Sui− mutants can utilize the in-frame or the out-of-frame UUG, CUG, or GUG codons as a translation start site at the 5′ UTR of the mRNA16. The dominant-negative eIF5G31R hyper GAP mutant is the strongest Sui− mutation identified that preferentially recognizes UUG as a start codon and shows severe slow growth, whereas in the recessive conditions the eIF5G31R mutation is lethal18. This preferential recognition of UUG start from the 5′ regulatory region of the GCN4 transcript causes repression of GCN4 expression (general control non-derepressed: Gcn− phenotype)19. We hypothesized that the aberrant translation initiation at the upstream UUG (upUUG) codon by the Sui− mutant might change the cellular proteome that adversely affects cellular physiology and severely cause growth defects. To gain insights in the underpinning cause of growth defect by the translation initiation fidelity defective eIF5G31R mutation, we used gel-based proteomics approach 2D-PAGE followed by MALDI-TOF/MS and gel-free labeling approach iTRAQ (isobaric tag relative absolute quantification) along with nLC-MALDI-TOF/MS technique to quantitate differential protein abundance. Our results suggest a change in the proteome of the eIF5G31R mutant that has major implications in DNA damage and oxidative stress.
Materials and methods
Yeast strains used in this study
The Saccharomyces cerevisiae yeast strains used in this study were: YP823; Mat α, ura3-52, leu2-3,112, trp1Δ63, GCN2+19. YP824; Mat α, ura3-52, leu2-3,112, trp1Δ63, GCN2+, his4Δ::KanMx620. Ctt1-loxP-Ura3-loxP-Ctt1 cassette was PCR amplified using oligonucleotides oPA1534 and oPA1535 and B4033 vector as a template. This PCR product was transformed into yeast strain YP823 to delete chromosomal CTT1 gene to give rise to yeast strain YP939; MATα leu2-3, -112, ura3-52, trpΔ63, GCN2+, Gal2+, ctt1∆::Ura3. The ctt1∆ in the YP939 strain was confirmed by PCR using oligonucleotides oPA1569 and oPA584.
Plasmids used in this study are listed in Table 1 and oligonucleotides are listed in the Supplementary Table S1.
Cloning
The 2.2 kb eIF5G31R encoding gene was digested from vector A70320 using EcoRI-SalI restriction endonuclease (RE) and cloned into pYCPlac22 to generate pYCPlac22-eIF5G31R (A838) vector. The pYcplac33-Ura6-lacZ reporter construct (A1074) was generated as follows—first, the 534 bp of URA6 promoter and the N-terminal 12 amino acids region was PCR amplified using oligonucleotide combination oPA1005/oPA1006 and cloned into vector pYcplac33 (A309) at HindIII-SalI site. The LacZ region and URA6 3′ UTR region were PCR amplified using oligonucleotides combination oPA1009/oPA1010 and oPA1007/oPA1008 respectively and cloned at SalI-BamHI and BamHI-KpnI sites. The four upstream UUG codons from the URA6 5′ UTR were removed by fusion PCR using oligonucleotides oPA1005, oPA1012, oPA1013, and oPA1011 and cloned at HindIII-SalI site of vector A1074 to generate pYcplac33-upUUGless Ura6-LacZ (A1140) vector. The C-terminal 3xHA tag was introduced into the URA6 gene by PCR amplification using oligonucleotides oPA1041/oPA1042 and cloned into A1074 or A1140 vector by replacing LacZ gene at SalI-BamHI site to generate pYcplac33-Ura6-3xHA (A1141) and pYcplac33-upUUGlessUra6-3xHA (A1142) vectors, respectively. upUUG codon at the − 95 position was put in-frame with the URA6-LacZ reporter by PCR amplification using oligonucleotides oPA1005/oPA1011 followed by a second round of PCR amplification using oPA1005/oPA1467 and cloned at HindIII-SalI site of vector A1074 to generate pYcplac33-upUUG1in-frame_lacZ (A1340) vector. The vector A1340 was modified to insert AUG codon at − 95 position by fusion PCR using oligonucleotides oPA1005, oPA1468, oPA1469, and oPA1470 and cloned at HindIII-SalI site to generate pYcplac33-upAUG1in-frame_lacZ (A1341) vector. The upUUG codon at the − 22 position was put in-frame with the URA6-LacZ reporter by fusion PCR using oligonucleotides oPA1005, oPA1012, oPA1013, and oPA1470 and cloned at HindIII-SalI site of vector A1074 to generate pYcplac33-upUUG3in-frame_lacZ (A1342) vector. The vector A1342 was modified to insert AUG codon at − 25 position by fusion PCR using oligonucleotides oPA1005 and oPA1471 using A1342 as a template and cloned at HindIII-SalI to generate pYcplac33-upAUG3in-frame_lacZ (A1343) vector. A 1636 bp of URA6 gene containing promoter and terminator region from genomic DNA was PCR amplified using oligonucleotide oPA1005/oPA1089 and cloned in pRS425 (B1378) vector at HindIII-SalI site to generate pRS425-Ura6 (A1147) vector.
C-terminal 3xHA tag was introduced into the CTT1 gene as follows—first, the 520 bp CTT1 3′UTR region was PCR amplified using oligonucleotides oPA1036/oPA1037 and cloned at BamHI-KpnI sites of pYcplac33 (A309) vector to generate intermediate vector. Using oligonucleotide oPA1034/oPA1040, a C-terminal 3xHA tag was introduced into the promote-ORF region of the CTT1 gene by PCR amplification and cloned into the intermediate vector at the BamHI-HindIII site to generate pYcplac33-CTT1-3xHA (A1144) vector. The CTT1 overexpression construct pRS425-CTT1 (A1149) was generated by amplifying the 2716 bp CTT1 genomic DNA region using oligonucleotide oPA1108/oPA1109 and cloned into high copy pRS425 vector (B1378) at NotI-BamHI site.
The 1478 bp SOD1 genomic DNA region was PCR amplified using oligonucleotides oPA1173/oPA1174 and the 1722 bp SOD2 genomic DNA region was PCR amplified using oligonucleotides oPA1175/oPA1176, digested with NotI-BamHI-HindIII restriction endonuclease and cloned at the NotI-HindIII site of the high copy pRS425 vector to generate pRS425SOD1-SOD2 (A1170) vector.
The pYCplac111-YAP1_LacZ reporter construct (A1311) was generated as follows- first, the 544 bp YAP1 promoter and the N-terminal 8 amino acids region were PCR amplified using oligonucleotide combination oPA1607/oPA1609 and cloned into vector pYcplac111 (A308) vector at SpeI-BamHI site. The LacZ region and YAP1 3′ UTR region were PCR amplified using oligonucleotides combination oPA1559/oPA1560 and oPA1608/oPA1610 respectively and cloned at BamHI-SalI and SalI-HindIII sites. The six upstream UUG codons from the YAP1 5′ UTR were removed using multiple PCR as follows- First, using A1311 vector template and oligonucleotides combination oPA1607/oPA1640, a PCR product was generated that removed upUUG codons from − 63, − 45, and − 14 position. This PCR product was used as a template to amplify and remove upUUG codon from the − 96 and − 91 position using oligonucleotides combination oPA1609, oPA1638, oPA1639, and oPA1607. Using the last PCR product as a template, the upUUG codon at the − 128 position was removed by PCR amplification using oligonucleotides combination oPA1609, oPA1636, oPA1607, and oPA1637. This PCR product was cloned at the SpeI-BamHI site of the A1311 vector to generate pYCplac111-YAP1-upTTGless_LacZ (A1312) vector. The YAP1 overexpression construct pRS425-YAP1 (A1346) was generated by amplifying 2792 bp YAP1 genomic DNA region using oligonucleotide oPA1607/oPA1608 and cloned into pRS425 vector at SpeI-HindIII site.
2D-PAGE followed by MALDI-TOF/MS proteomics approach
Three biological replicates of wild type (WT) and eIF5G31R mutant cells were grown till OD600 ~ 1.0. The cells were suspended in 2D lysis buffer (7 M Urea, 2 M Thiourea, 65 mM DTT, 2% CHAPS, and a cOmplete Protease Inhibitor tablet) and mechanically lysed by vortexing using acid-washed glass beads (200 μm) at 4 °C. The supernatant containing total cell extract was separated at 13,000×g for 30 min at 4 °C and the total proteins were quantitated by Bradford assay. The total proteins (400 μg) were resolved on 18 cm IPG strip pH 3–11 using Ettan IPGphor 3 isoelectric focusing system (GE) under the following conditions; 12 h (h) at 50 V; 30 min at 250 V; 1 h at 1000 V; 1 h at 2500 V; 1 h at 5000 V; 1 h at 8000 V and held at 8000 V until total Vh reached 50,000 Vh. After IEF, the IPG strips were equilibrated for 30 min in a reduction buffer containing 1% DTT in an equilibration buffer (75 mM Tris–Cl pH 8.8, 6 M urea, 30% v/v glycerol, 2% SDS, 0.002% bromophenol blue), and subsequently alkylated for 30 min in an alkylation buffer containing 2.5% Ioda-acetamide in equilibration buffer. After equilibrium, the strips were placed on 12% polyacrylamide gels and immobilized by overlaying 0.5% agarose on the strips. The second-dimensional separation was carried out in the Bio-Rad Protean II xi Cell and electrophoresed at 80 V for 24 h. The gel was stained with Coomassie brilliant blue (CBB), scanned with GS-800 Calibrated Densitometer (Bio-Rad Laboratories), and analyzed using GE Image Master 2D Platinum v7.0 software. The altered spots were excised, followed by in-gel digestion using trypsin (1 µg/µl trypsin in 1:10 enzyme to protein ratio; AB Sciex #4352157) at 37 °C for 16 h. The resultant digested peptides were subjected to MALDI-TOF/MS. First, it was mixed with α-Cyano-4 hydroxycinnamic acid (CHCA) matrix (1:1) and spotted on MALDI plate and acquired the data as described below.
Isobaric tag relative absolute quantification (iTRAQ) proteomics approach
Three biological replicates of the WT and eIF5G31R mutant cells were suspended in phosphate-buffered saline pH 7.2 (PBS) and mechanically lysed as described above. The supernatant containing total cell extract was separated at 13,000×g for 30 min at 4 °C and the total proteins were quantitated by Bradford assay. Total protein (400 µg) was buffer exchanged with 1 M urea in 0.5 M Triethylammonium bicarbonate (TEAB) solution using 3 kDa Cut-off filter (Millipore # UFC500324) followed by re-quantification using Bradford assay. An aliquot of 75 μg of total protein was treated with 2% SDS and 50 mM of Tris-(2-carboxyethyl) phosphine (TCEP) and incubated at 36 °C for 1 h. The cysteines were blocked with 200 mM Methyl methane thiosulfonate (MMTS) for 10 min at room temperature and then subjected to trypsin digestion (1 µg/µl trypsin in 1:10 enzyme to protein ratio; AB Sciex #4352157) at 37 °C for 16 h. iTRAQ 4-plex reagents (AB Sciex iTRAQ 4-plex kit #4352135) were used to label the resultant peptide digests according to the manufacturer’s instructions. The different isobaric tags labeled peptides were pooled together and subjected to the strong cation exchange chromatogram using the ICAT column (AB Sciex ICAT Kit #4326687). The samples were eluted using 10–350 mM KCl gradient into 12 fractions. These fractions were desalted using C18 tips (Thermo-Pierce #87784) and vacuum dried (Labconco).
NanoLC-MALDI TOF/MS
All the fractions were reconstituted in 0.1% trifluoroacetic acid and chromatogram on reverse phase C18 column (Eksigent, 3 um, 120 Å pores, 0.075 × 150 mm) using Ekspert nanoLC Ultra 2D plus system with 5–90% acetonitrile gradient (2 μl/min flow in 68 min). Eluant from each fraction was deposited with α-CHCA matrix at 15 s/spot and spotted onto the MALDI plate with the help of Ekspot MALDI spotter (AB Sciex). The spotted LC-MALDI plates were subjected to 5800 MALDI TOF/TOF (AB Sciex). Refectron mode mass calibrations were performed externally with a calibration mixture (Cal mix TOF/TOF, AB Sciex #4333604), whereas MS/MS mode mass calibration was executed using the fragments of the Glu-fibrinopeptide B precursor ion. MS and MS/MS data were acquired for all the fractions using defined LC-MALDI parameters. Abundant peptides with a precursor mass of 3568 Da with a mass window ± 100 Da were sent for MS/MS with a 0.8 ppm error. Data Dependent Acquisition (DDA) was activated with automatic Collision Induced Dissociation (CID) and automatic MS/MS acquisition. The MS and MS/MS data of the peptides were used for protein identification and peak areas of the iTRAQ reporter ions suggest the relative abundance of the proteins in the respective samples. The schematics of the iTRAQ and nanoLC-MALDI-TOF/MS approach are outlined in Supplementary Fig. S1a.
Data processing and analysis
Quantification and identification of protein were performed on ProteinPilot 4.0.8085 Software (AB Sciex) integrated with Mascot search engine version:2.3. The Paragon algorithm in the ProteinPilot software was used for the peptide identification. User-defined search parameters were as follows: (i) Sample type, iTRAQ 4-plex (Peptide Labeled); (ii) Cysteine alkylation, MMTS; (iii) Digestion, Trypsin; (iv) Instrument, MALDI TOF/TOF AB Sciex 5800, (iv) Mass tolerance 100 ppm for precursor ion and 0.8 ppm for daughter ions, (v) Database, S. cerevisiae database from Uniprot, SC strain S288C-20160606.fasta and the corresponding reverse sequence (decoy: for false discovery rate (FDR = 2.0 × decoy hit/total_hit) estimation), (vi) Keratin excluded. The peptides were automatically selected by the Pro Group algorithm to calculate the reporter peak area, error factor (EF), and P-value for quantification. Post quantification dataset was auto bias-corrected to eliminate any variation due to unequal mixing while pooling different labeled peptides. The False Discovery Rate (FDR) was estimated using the Proteomics System Performance Evaluation Pipeline (PSPEP) algorithm integrated into the ProteinPilot, and the Global FDR of 5% was used for further analysis. Quantified proteins from both sets were first normalized against the pooled internal control used in both experimental sets. Post normalization, the dataset was filtered based on unused score cut-off 1.3 or more for further analysis from both sets with at least one unique peptide identified. Proteins present in both sets were further tested for statistical significance by performing Student’s t-test, P-value ± 0.05. A cut-off ≥ 1.5-fold and ≤ 0.66-fold were used for differential protein abundance estimation for up-regulation and down-regulation, respectively. The summary of iTRAQ data analysis is outlined in Supplementary Fig. S1b. In addition, the mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE24 partner repository with the data set identifier PXD029460.
Western blotting
Yeast cells were grown till OD600 ~ 0.8. The cell pellet was resuspended in PBS and mechanically lysed as described above. The supernatant containing total cell extract was separated by centrifugation at 13,000×g for 20 min at 4 °C. Total proteins were quantitated by Bradford assay. Total proteins were separated on SDS-PAGE followed by electro-blotting to a PVDF membrane. The blots were probed using an anti-HA antibody (Sigma #H9658, 1:10,000 dilution) and horseradish peroxidase (HRP) conjugated mouse secondary antibody (Sigma, #A9044). The blot was developed using SuperSignal (Thermo Scientific #34075) and quantitated using EvolutionCapt Solo S 17.00 software.
Real-time PCR
Yeast cells were grown till OD600 ~ 0.8 in 30 ml culture. The cell pellet was resuspended in 400 μl TRIzol (Ambion #15596026) and mixed with a 1:1 chloroform-isoamyl alcohol mixture. The supernatant containing total RNA was separated by centrifugation at 13,000×g for 2 min at 4 °C. The total RNA was precipitated by isopropanol and after 75% ethanol wash, the pellet was suspended in distilled water and treated with DNase-I at 37 °C for 30 min. The reaction was terminated by heat inactivation at 70 °C for 10 min. The total RNA (0.5 μg) was subjected to cDNA synthesis using SuperScript III enzyme (Invitrogen #18080044) and oligonucleotide oPA1024, oPA1102, oPA1114, and oPA1642 for ACT1, URA6, CTT1, and YAP1 genes, respectively, and the reaction was carried at 50 °C for 60 min. The reaction was stopped by heat inactivation at 70 °C for 15 min. The cDNA was diluted fivefold and a 3 μl of cDNA was subjected to real-time quantitative PCR using Power SYBR Green PCR Master Mix (Applied Biosystem #1601516) and BioRAD CFX384 Touch Real-Time PCR Detection System. The oligonucleotides combination oPA1023/oPA650, oPA1101/oPA1102, oPA1113/oPA1114, and oPA1641/oPA1642 were used for PCR amplification of ACT1, URA6, CTT1, and YAP1 cDNA, respectively.
ROS assay
Yeast cells were grown till OD600 ~ 0.8, cells were washed with PBS, followed by incubation in PBS containing 100 µM H2DCFDA dye (Sigma # D6883) and 5 µM MitoSOX Red (Invitrogen #M36008) for 30 min in the dark at 37 °C followed by washing with PBS. Slides were mounted and observed under Leica SP8 confocal microscope using a 63× objective lens with the FITC (Em λ529) filter to measure H2DCFDA fluorescence and TRITC (Em λ580) filters to measure MitoSOX Red fluorescence. The images were quantitated using NIH ImageJ software (1.52p).
β-Galactosidase reporter assay
Yeast cells were transformed with appropriate reporter plasmids. Five colonies from each transformant were grown overnight at 30 °C with shaking at 220 rpm in synthetic dextrose (SD) media with appropriate nutrient supplements and grown till OD600 ~ 0.8. Cells were harvested by centrifugation at 13,000×g for 20 min at 4 °C and resuspended in LacZ buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, and 1 mM MgSO4, pH 7.0) and cell lysis was prepared as described above. Clarified cell extract (~ 30 μg) was mixed with LacZ buffer, followed by addition of 180 μl of ONPG (4 mg/ml in LacZ buffer). After 30 min of incubation, absorbance was measured at 420 nm (Molecular Devices Spectra Max). Protein estimation was done using Bradford assay and the β-galactosidase activity (nmol of O-nitrophenyl-β-d-galactopyranoside cleaved per min per mg) was calculated after normalization with total protein.
Estimation of cellular glutathione
Yeast cells were grown till OD600 ~ 0.8. The cells were washed with PBS and treated with 30 μM monochlorobimane (mCBM) dye for 30 min at 30 °C. Fluorescence adduct formation was imaged at Ex λ350 and Em λ460 using a Leica SP8 confocal microscope at × 63 magnification. The images were quantitated using NIH ImageJ software (1.52p).
Tunel assay
Tunel assay was performed using DeadEnd Fluorometric Tunel System (Promega #G3250). Yeast cells OD600 ~ 0.8 (100 μl) was spun down at 4000×g for 2 min, followed by fixation in 3.7% formaldehyde for 30 min at room temperature. Cells were washed with PBS and treated with 10 U of lyticase in PBS at 37 °C for 60 min. The cells were washed with PBS and permeabilized using 0.1% triton-X-100 in 0.1% sodium citrate for 5 min on ice. These cells were equilibrated with rTDT and nucleotide mix in 45 μl of equilibration buffer followed by incubation at 37 °C for 1 h. The cells were washed three times with PBS, followed by DAPI staining for 15 min in the dark. The cells were washed with PBS and imaged using Leica SP8 confocal microscope using DIC, DAPI (Em λ461) filter and FITC (Em λ529) filter. The images were quantitated using NIH ImageJ software (1.52p).
Results
Translation fidelity defective eIF5G31R Sui− mutant alters cellular proteome
Historically suppressor of initiation codon (Sui−) mutations were identified by performing genetic screening to check the growth of Saccharomyces cerevisiae his4-303 mutant (AUG start codon of HIS4 gene was mutated to AUU codon) on histidine minus auxotrophic media. The Sui− mutant could utilize the third in-frame UUG codon as an initiation codon and grow on histidine minus medium15,16. Our bioinformatic analysis suggests ~ 44.6% of Saccharomyces cerevisiae CDS contain at least one UUG codon in the 5′ untranslated region (UTR) within 50 bp of + 1AUG start codon (Supplementary Sheet 1). If Sui− mutants can utilize these upstream UUG codons (upUUG) as translation initiation codon while scanning the mRNA from 5′ to 3′ directions, then that would lead to the formation of either short upORF (ORF with UUG start codon), upORF overlapped out-of-frame with the main ORF or upORF in-frame with main ORF (N-terminal extension) (Fig. 1a). Translation from these out-of-frame upORFs may preclude the translation of main ORF, resulting in the altered proteome in Sui− mutant cells. The eIF5G31R mutation is one of the strongest Sui− mutants that preferentially recognize UUG as a start codon, unlike other Sui− mutants that can also utilize CUG or GUG as a start codon16,18. The eIF5G31R mutation in the recessive condition is lethal, whereas the dominant eIF5G31R mutation represses GCN4 translation (Gcn− phenotype) and shows 3-amino-1,2,4-triazole (3AT) sensitivity due to utilization of upUUG codons from the 5′ regulatory region of GCN4 transcript and show severe slow growth phenotype19,20. To understand how strong utilization of the upUUG codon by the eIF5G31R mutation alters the cell's proteome, we employed a gel-based approach 2D-PAGE followed by MALDI-TOF/MS and a gel-free labeled approach iTRAQ 4-plex coupled with nLC-MALDI-TOF/MS to quantitate the differential abundance of proteins (Fig. 1b). Whole cell extract (WCE) from the WT and eIF5G31R mutant was resolved on 2D-PAGE and differentially abundance proteins spots were analyzed using GE Image Master Platinum software (Fig. 1c). Significantly altered protein spots (total 16) were subjected to LC-MALDI-TOF/MS analysis. Total four proteins could be successfully identified as summarized in Table 2. Four proteins abundance downregulated in the eIF5G31R mutant are involved in the glycolysis pathway and antioxidant activity (Supplementary Table S3). A total of 1098 common proteins (Supplementary Fig. S1b) was successfully identified using a gel-free iTRAQ labeled approach with a significant score in both experimental sets, of which seven proteins abundance were downregulated (< 0.66-fold cut-off), while nineteen proteins abundance were upregulated (> 1.5-fold cut-off) (Fig. 1d, Table 3). Categorizing these proteins for their cellular function and gene ontology indicates involvement in DNA replication stress (VTC4, HSP12, RGI1, HSP31, DUR1,2, TOH1, CTF4)25, micro-autophagy (VTC2, VTC3, VTC4)26, protein oxidation/degradation pathway (PMT2, CIC1, FRA1)27,28,29, cellular stress (MOT3, DCS2, CTT1, HSP31, HSP12), translation repression (CAF20, MOT3, DCS2)30,31,32,33,34, lipid biosynthesis (CHO1, OLE1)35, oxidation–reduction pathway (DLD1, OLE1, DFR1, CTT1)36,37,38, transcription regulation (FRA1, MOT3)28,39, vitamin/co-factor biosynthesis (RIB4, BIO2)40,41, ribosome biogenesis (CIC1)42, protein transport (GOS1, SRP101)43,44, pyrimidine nucleotide biosynthesis gene (URA6)45 and one uncharacterized protein of unknown function (YNL208W) (Supplementary Table S3, Fig. 1e).

Translation fidelity defective eIF5G31R Sui− mutant alters cellular proteome. (a) Schematic of translation of a representative mRNA having upstream ORF initiated from UUG start codon. (b) Schematic of the workflow showing different approaches to estimate differential abundance of proteins in the eIF5G31R mutant cells. (c) Analysis of altered protein expression by 2D-PAGE. The total cell extract (400 μg of protein) of WT or the eIF5G31R mutant was first electrophoresed on an 18 cm 3–11 IEF strip followed by the second dimension run on 12% SDS-PAGE and stained with CBB. Differentially expressed protein spots were analyzed by GE Image Master Platinum software and subjected to MALDI-TOF for identification. The protein molecular weight marker is shown in the middle. (d) Analysis of Isobaric Tag Relative Absolute Quantification (iTRAQ). Total cell extract from WT or eIF5G31R mutant cells was subjected to Isobaric Tag Relative Absolute Quantification (iTRAQ) followed by Mass spectrometric analysis as outlined in Supplementary Fig. S1a. Volcano plot showing − log10 (p-value) versus the log2 (fold change), where the orange and green dots represent down-regulation (< 0.66 fold cut-off) and upregulated (> 1.5-fold cut-off) proteins, respectively. An arrowhead shows a data-point indicating the expression level of URA6 and CTT1 proteins. Heat map showing the expression pattern for all the 26 significantly altered genes in three biological replicates of WT and eIF5G31R mutant cells. (e) Analysis of gene function relationship. The association of genes with different biological functions that are altered in the eIF5G31R mutant cells is represented by Chord diagram64. Statistical differences were determined by the two-tailed Student’s t-test.
Upstream UUG codons repress the URA6 expression in the eIF5G31R mutant and cause DNA damage
URA6 is an essential gene that encodes uridylate kinase and takes part in the de-novo pyrimidine biosynthesis pathway to catalyze the conversion of uridine monophosphate (UMP) to uridine-5′-diphosphate (UDP). The UDP is further converted into UTP and CTP, which are the precursors for DNA and RNA biosynthesis46. Our iTRAQ data analysis showed ~ 0.55-fold less abundant (P-value, 0.0069) uridylate kinase protein (Fig. 1d). To validate this data, we modified the URA6 gene by adding a C-terminal 3xHA tag and checked the plasmid-borne expression of this modified gene in the WT and eIF5G31R mutant yeast strain by Western blot using Anti-HA tag antibody. Consistent with the iTRAQ data analysis, the Western blot showed ~ 0.23-fold less abundance of uridylate kinase protein (Fig. 2a, left). To check if the lower uridylate kinase protein levels were not due to the lower transcription of its mRNA, we performed RT-qPCR using URA6 ORF specific oligonucleotides. To our surprise, we observed ~ 3.5-fold higher URA6 mRNA expression levels in the eIF5G31R mutant, suggesting that the lower levels of uridylate kinase protein indicate translation defect (Fig. 2a, right). It may be possible that the presence of the four upUUG codons at the ~ 160 nucleotides (nt) long 5′ UTR45 has generated an out-of-frame upORFs that prematurely terminate translation before reaching the URA6 main ORF in the eIF5G31R mutant. To test this, we mutated all four upUUG codons (at − 95, − 92, − 22, and − 13 positions) and checked the uridylate kinase protein levels by Western blot. The removal of all upUUG codons caused an overall reduction in uridylate kinase protein levels in the WT cells. However, the relative expression of uridylate kinase protein increased (ratio 0.66) in the eIF5G31R mutant after the removal of upUUG codons from the mRNA as compared to the WT (ratio 0.23) mRNA (Fig. 2a). These results suggest that the eIF5G31R mutant utilizes upUUG codons to translate the out-of-frame upORFs and the ribosomes dissociate before reaching the main ORF causing lower uridylate kinase protein expression.

Upstream UUG codons repress the URA6 expression in eIF5G31R mutant and cause DNA damage. (a) Quantification of URA6 protein expression. YP823 strain transformed with pYcplac33-Ura6-3xHA (A1141) or pYcplac33-upUUGlessUra6-3xHA (A1142) constructs along with empty vector pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) construct were grown overnight on synthetic dextrose (SD) plus leucine medium till OD600 ~ 0.8 at 30 °C. Whole cell extract (WCE) was prepared by mechanical cell breaking using glass beads and 40 µg of WCE was subjected to 12% SDS-PAGE and the URA6p was identified by Western blot using an anti-HA antibody and normalized with blot stained by Coomassie brilliant blue (CBB) (left). YP823 strain carrying empty vector pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown overnight on synthetic dextrose (SD) plus leucine medium till OD600 ~ 0.8 at 30 °C. The whole-cell extract (WCE) was treated with TRIzol, followed by ethanol precipitation. The total RNA isolated was subjected to reverse transcription and quantitative PCR using URA6 ORF specific oligonucleotides oPA1101 and oPA1102 (right). The error represents an average deviation. (b) Analysis of URA6-LacZ expression. Yeast strain YP823 containing empty vector pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were transformed with derivatives of Ura6-LacZ reporter constructs A1074, A1140, A1340, A1341, A1342, and A1343 were grown till OD600 ~ 0.8 in SD plus leucine medium at 30 °C. WCE were prepared, and β-galactosidase activity (nmol of O-nitrophenyl-β-d-galactopyranoside cleaved per min per mg) was measured and plotted. The error represents an average deviation. (c) Growth analysis in the presence of hydroxyurea. High copy (h.c) empty vector (EV) pRS425 (B1378) or high copy pRS425-Ura6 (A1147) constructs were transformed in yeast strain YP828 carrying either EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown overnight, serially diluted, and spotted on SD plus uracil or SD plus uracil plus 25 mM hydroxyurea and incubated at 30 °C for 2–3 days. (d) Analysis of DNA fragmentation by TUNEL assay. Yeast strain YP823 carrying either EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown till OD600 ~ 0.8. The cells were treated with rTDT and nucleotide mix and imaged using Em λ461 (DAPI) and Em λ529 (FITC) filters. The values are average (n = 300) along with the standard error of the mean (SEM). Statistical differences were determined by the two-tailed Student’s t-test. ns non-significant.
The utilization of upUUG codons at the 5′ UTR region by the eIF5G31R mutant creates two upORFs. The upUUG1 (− 95) and upUUG2 (− 92) codons are in the same reading frame and constitute upORF1 that terminates before the main ORF of URA6 gene. The upUUG3 (− 22) and upUUG4 (− 13) codons are in the same reading frame and constitute the second upORF2 that partially overlaps out of frame with the main URA6 ORF (Fig. 2b). To probe which upUUG codons are utilized by the eIF5G31R mutant, we made the URA6-LacZ reporter construct (A1074). This construct was modified to remove all upUUG1-4 codons (A1140), removal of upUUG3-4 and keeping upUUG1 intact in-frame with URA6-LacZ (A1340), removal of upUUG3-4 and changing upUUG1 into AUG codon in-frame with URA6-LacZ (A1341), removal of upUUG1, 2, and 4 and keeping upUUG3 intact in-frame with URA6-LacZ (A1342), and removal of upUUG1, 2 and 4 and changing upUUG3 to AUG codon in frame with URA6-LacZ (A1343). These constructs were transformed into the WT or eIF5G31R mutant yeast cells to check the resultant β-galactosidase activity. Consistent with the results obtained with Western blot, the removal of all the upUUG codons increased the expression of URA6-LacZ reporter in the eIF5G31R mutant. Moreover, whereas the translation from the − 95 position (upUUG1 or AUG) codon severely down-regulated reporter expression, the translation from the − 22 position (upUUG3 or AUG) showed better reporter expression (Fig. 2b). These results suggest that eIF5G31R mutant initiated translation from the second upORF2 that partially overlaps out-of-frame with the main URA6 ORF by utilizing the upUUG3 codon.
If the uridylate kinase protein levels are downregulated in the eIF5G31R mutant strain, it should also affect the catalysis of UMP to UDP. The UDP is further converted into UTP and CTP. Since the UTP and CTP are the precursors for DNA and RNA biosynthesis, we propose that the eIF5G31R mutant affects nucleic acid biosynthesis. Our iTRAQ data analysis showed a significant abundance of proteins previously reported to be associated with DNA replication stress (TDH2, VTC4, PRB1, HSP31, HSP12, TOH1, DUR1,2 & RGI1)25. Yeast cells under DNA replication stress show sensitivity to hydroxyurea (HU). To check if the eIF5G31R mutation is sensitive to DNA replication stress, we checked the growth on 25 mM HU. The eIF5G31R mutation was sensitive to HU and the sensitivity was partially rescued by a high copy overexpression of URA6 gene (Fig. 2c). The DNA replication stress also induces DNA damage that can be tested by a TUNEL assay. Consistently, the eIF5G31R mutant cells showed a twofold increase in the TUNEL positive cells (Fig. 2d).
eIF5G31R mutation causes oxidative stress
The catalase-T enzyme (CTT1) showed higher abundance in the eIF5G31R mutant as per our iTRAQ analysis. The higher abundance of the catalase-T enzyme indicates oxidative stress in the eIF5G31R mutant cells. To test this, we first checked the levels of catalase-T by adding a C-terminal 3xHA tag and analyzed the plasmid-borne expression of this modified gene in the WT and eIF5G31R mutant yeast strain by Western blot using anti-HA tag antibody. Consistent with the iTRAQ data analysis, the Western blot showed 1.3-fold upregulation of catalase-T protein (Fig. 3a, left). We also checked the transcription level of CTT1 mRNA by RT-qPCR using CTT1 ORF specific oligonucleotides. There was ~ threefold higher expression of CTT1 mRNA in the eIF5G31R mutant cells (Fig. 3a, right). The discrepancy between the CTT1 mRNA expression and protein expression could be because of the presence of the four upUUG codons (− 105, − 76, − 66 and − 40) in the 112 nucleotides long 5′ UTR of CTT1 mRNA. The upUUG codons at the − 105 and − 66 position are in-frame with the main AUG start codon, whereas the upUUG codon − 76 and − 40 are out-of-frame with the main AUG start codon. Since the upUUG codon at the − 105 position is very close to the 5′ cap of the mRNA and could be skipped by the eIF5G31R mutant containing 48S scanning ribosome. However, the eIF5G31R mutant could initiate translation from the out-of-frame − 76 or − 40 upUUG codon causing premature termination, whereas initiation from the in-frame upUUG codon at − 66 position could result in translation of the catalase-T protein. Taken together, this results in ~ 1.3-fold increase in the catalase-T protein expression as the CTT1 mRNA levels are high. These results suggest eIF5G31R mutant cells must be under oxidative stress and in response to this stress, the expression of the CTT1 transcript could have been upregulated. The yeast cells under oxidation stress should show sensitivity to H2O2. Consistently, the eIF5G31R mutant showed strong sensitivity to H2O2 treatment (Fig. 3b). The sensitivity to oxidative stress was partially rescued in the eIF5G31R mutant cells when the CTT1 or SOD1/SOD2 genes were overexpressed from a high copy plasmid (Fig. 3c). Moreover, the oxidative stress was exacerbated in the eIF5G31R mutant cells when the CTT1 gene was deleted (Fig. 3d). The oxidative stress in the eIF5G31R mutant may have been triggered by the accumulation of reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), the hydroxyl radicals (OH·), and the superoxide anion (O2·−). To check if the eIF5G31R mutant has higher ROS levels, we treated the eIF5G31R mutant cells with H2DCFDA and MitoSOX red dye to check cytosolic and mitochondrial ROS levels, respectively. The 1.48-fold high fluorescence for H2DCFDA and 2.06-fold high fluorescence for MitoSOX red suggests high ROS levels in the eIF5G31R mutant cells (Fig. 3e,f).

eIF5G31R mutant causes oxidative stress. (a) Quantitation of CTT1 expression. Yeast strain YP823 transformed with pYcplac33-CTT1-3xHA (A1144) construct along with an empty vector pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were processed as Fig. 2a. CTT1p was identified by Western blot using an anti-HA antibody and normalized with blot stained by CBB (left). The CTT1 mRNA was quantitated by RT-qPCR using CTT1 ORF specific oligonucleotides oPA1113 and oPA1114 (right) as described in Fig. 2a (right). The error represents an average deviation. (b–d) Growth analysis in the presence of H2O2. Yeast strain as in Fig. 2c was serially diluted and spotted on SD, leucine, uracil or SD, leucine, uracil plus 3 mM H2O2 plates and incubated at 30 °C for 2 days (b). Yeast strain YP823 carrying EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were transformed with either high copy EV pRS425 (B1378), pRS425-CTT1 (A1149), pRS425SOD1-SOD2 (A1170) constructs were grown overnight, serially diluted, and spotted on SD, uracil or SD, uracil plus 3 mM H2O2 plates (c). Yeast strain YP939 (ctt1∆) and its isogenic WT strain (YP823) were transformed with EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown overnight, serially diluted, and spotted on SD, leucine or SD, leucine plus 2 mM H2O2 plates (d). (e,f) Analysis of oxidative stress. Yeast strain YP823 was transformed with either EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown in SD plus leucine, uracil medium till OD600 ~ 0.8 at 30 °C. 200 μl cells suspension were stained with either 100 μM of H2DCFDA to check cytosolic ROS or 5 μM of MitoSOX Red to check mitochondrial ROS. Fluorescence microscopic imaging was performed using FITC (Em λ529) filter to measure H2DCFDA fluorescence and TRITC (Em λ580) filters to measure MitoSOX Red fluorescence. The values are average (n = 300) along with the standard error of the mean (SEM). Statistical differences were determined by the two-tailed Student’s t-test.
Lower levels of glutathione and YAP1 gene expression exacerbate the oxidative stress in the eIF5G31R mutant
Our data showed higher levels of ROS in the eIF5G31R mutant cells. Catalase, superoxide dismutase, glutathione peroxidase, and thioredoxin system neutralize the intracellular ROS47,48,49. Our iTRAQ data analysis showed 20% downregulation of GSH1 protein levels in the eIF5G31R mutant (Supplementary Sheet 2), suggesting that there may be inadequate levels of glutathione to detoxify the cellular ROS. We check the intracellular glutathione levels by monochlorobimane dye, which reacts with glutathione in the presence of glutathione-S-transferase to form an adduct that shows fluorescence at 380 nm50,51. Microscopic image analysis showed a 30% reduction of glutathione level in the eIF5G31R mutant cells (Fig. 4a). The growth defect associated with the eIF5G31R mutant was partially rescued in the presence of the 5 mM glutathione (Fig. 4b). However, the high copy overexpression of GSH1 and GSH2 genes did not rescue the slow growth of the eIF5G31R mutant (data not shown). It could be possible that the other components of the redox system critical for ROS detoxification are not fully expressed in the eIF5G31R mutant cells.

Lower level of glutathione and YAP1 gene expression exacerbates oxidative stress in eIF5G31R mutant cells. (a) Glutathione quantification. Yeast strain YP823 carrying EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were grown in SD plus leucine, uracil medium till OD600 ~ 0.8 at 30 °C. 200 μl cells suspension were stained with 30 μM of monochlorobimane (mCBM) followed by fluorescence microscopic imaging at Em λ460. The values are average (n = 300) along with the standard error of the mean (SEM). (b) Growth analysis in the presence of glutathione. Yeast strains as per (a) were spotted on either SD plus leucine, uracil plus 2 mM H2O2 or supplemented with 5 mM glutathione plates and incubated at 30 °C for 3 days. (c) Quantification of YAP1-LacZ expression. Yeast strain YP823 carrying EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were transformed with pYCplac111-YAP1_LacZ (A1311) or pYCplac111-YAP1-upTTGless_LacZ (A1312) construct, were grown till OD600 ~ 0.8 at 30 °C and the β-galactosidase activity was measured as per Fig. 2b. The error represents an average deviation. (d) Growth analysis in the presence of H2O2. Yeast strain YP823 carrying EV pYcplac22 (A823) or pYcplac22-eIF5G31R (A838) constructs were transformed with either high copy EV pRS425 (B1378) or (h.c) pRS425-YAP1 construct (A1346), grown overnight, serially diluted, and spotted on SD, uracil plus 2 mM H2O2 as per Fig. 3c. Statistical differences were determined by the two-tailed Student’s t-test. ns non-significant.
YAP1 is a AP-1 family transcription factor that regulates ~ 28 genes associated with ROS detoxification, including GSH1, GSH2, GLR1, SOD1, SOD2, GPX2, TSA1, TRR1, AHP1, and TRX2. These genes are involved in glutathione biosynthesis, superoxide dismutase activity, glutathione peroxidase function, thioredoxin, and thioredoxin peroxidases action52. The YAP1 protein was not identified in our iTRAQ or 2D-PAGE data analysis. However, analysis of the YAP1 gene transcript suggests the presence of six out-of-frame upUUG codons at the 5′ UTR that may affect its translation. We made YAP1-LacZ reporter constructs with native 5′ UTR region containing the six upUUG codons (A1311) and the modified construct devoid of the six upUUG codons (A1312) and these constructs were transformed in the WT or eIF5G31R mutant cells. The eIF5G31R mutant showed 2.4-fold less YAP1 reporter expression from the native constructs. However, upon removing six upUUG codons, the reporter expression in the eIF5G31R mutant was similar to the WT (Fig. 4c). This result suggests that the YAP1 protein expression was downregulated in the eIF5G31R mutant, which may have affected the transcription of the target genes involved in the redox pathway. Consistently, we observed down-regulation of the AHP1 mRNA (Supplementary Fig. S6) and AHP1 protein levels that protect against oxidative damage53 (Table 2). However, overexpression of the YAP1 gene from a high copy plasmid should increase the YAP1 protein levels and show a detoxifying effect on ROS. Consistently, the YAP1 overexpression showed partial growth rescue of the eIF5G31R mutant in the presence of H2O2 (Fig. 4d).
Discussion
Previously, we reported that the eIF5G31R mutation recognizes UUG codon but does not initiate translation from CUG or GUG codon18. In this study, we decipher the underpinning effect of the eIF5G31R mutation on UUG codon recognition that alters cellular proteome in dominant conditions. Our iTRAQ data suggests that seven proteins abundance was downregulated, while nineteen proteins abundance was upregulated. The most striking of them was the 0.55-fold downregulation of the uridylate kinase protein (URA6), which takes part in the de-novo pyrimidine biosynthesis pathway. This downregulation of uridylate kinase was not due to the lower levels of URA6 transcript; rather, our analysis suggests that the utilization of the upstream UUG codons could generate out-of-frame upORFs that impedes translation initiation from the URA6 main ORF. Removal of all the four upUUG codons from the URA6 transcript caused an overall reduction in the uridylate kinase translation in the WT cells. We suspect mutating the upUUG codons may have changed the secondary structure of the mRNA resulting in the lower translation of the URA6 transcript. However, the relative expression of uridylate kinase increased in the eIF5G31R mutant cells after removing the four upUUG codons (Fig. 2a). The eIF5G31R mutant cells could have unsuccessfully tried to mitigate the low abundance of uridylate kinase by upregulating the URA6 mRNA expression to relieve the lower nucleotide availability stress. The upregulation of DNA replication stress markers (TDH2, VTC4, PRB1, HSP31, HSP12, TOH1, DUR1,2 & RGI1)25 observed in our iTRAQ data is consistent with the sensitivity of the eIF5G31R mutant to hydroxyurea because of the low uridylate kinase expression and the attendant DNA damage (Fig. 2c,d).
The upregulations of catalase-T in the eIF5G31R mutant proteome suggest oxidative stress. Consistently, the eIF5G31R mutant showed sensitivity to H2O2, which was partially rescued by the high copy overexpression of either CTT1 or SOD1/SOD2 genes (Fig. 3b,c). The sensitivity to H2O2 is consistent with the high cytosolic and mitochondrial ROS levels in the eIF5G31R mutant (Fig. 3e,f). Compounding this oxidative stress, glutathione levels were 30% lower in the eIF5G31R mutant cells (Fig. 4a), and supplementing 5 mM glutathione could partially rescue its growth defect (Fig. 4b). However, we could not identify a major source of ROS generation in the eIF5G31R mutant through our proteome analysis. Previous report from Salmon and co-workers, suggest that the DNA damage alone can elicit an increase in the intracellular ROS levels54. Thus the DNA damage observed in the eIF5G31R mutant (Fig. 2d) could trigger an intracellular ROS generation, and the components involved in ROS detoxification were not optimum due to the downregulation of regulatory genes, thus tipping the balance towards oxidative stress. YAP1 is an AP-1 family transcription factor that regulates ~ 28 genes associated with ROS detoxification52 and the YAP1-LacZ reporter expression was observed to be downregulated in the eIF5G31R mutant because of the upUUG codon utilization. Consistently, the overexpression of the YAP1 gene partially rescued the sensitivity to oxidative stress (Fig. 4c,d).
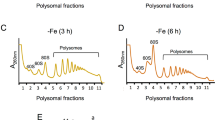
Our results indicate that the defect in translation initiation fidelity in the eIF5G31R mutant re-programs the cellular pathways to cause DNA damage and oxidative stress. Moreover, the upregulation of proteins involved in the protein degradation and micro-autophagy pathways suggests an enhanced turnover of proteins in the eIF5G31R mutant cells. Consistently, we observed enhanced degraded products of FBP1, PDC1 and ENO2 proteins in 2D-PAGE MALDI-TOF/MS analysis (Table 2). We did not observe significant changes in the 40S, 60S, and 80S ribosomal (monosome) peaks in the eIF5G31R mutant (Supplementary Fig. S2), suggesting that the mutation may not have affected the general assembly of PIC during the translation initiation process and only affected the 5′ to 3′ mRNA scanning where the eIF5G31R mutation preferentially selected UUG as initiation codon. In support of our finding, the cryo-EM data suggest that the N-terminal domain of eIF5 displaces eIF1 from the P-site of the 48S complex during AUG codon recognition. It is suggested that the eIF5G31R mutation may prematurely displace eIF1 while scanning the mRNA from 5′ to 3′ direction and stabilizing the base pairing of Met-tRNAi anticodon with the UUG codon4. Furthermore, the 35S-Methionine labelling experiment suggested a less than 10% reduction in the protein biosynthesis in the eIF5G31R mutant cells (data not shown). This subtle decrease in the protein translation may be an effect of oxidative stress in the eIF5G31R mutant, as oxidative stress increases ribosomal transition time on mRNA and shows an inhibitory effect on translation elongation55. Consistently, we observed lower polysomes in the eIF5G31R mutant (Supplementary Fig. S2).
Earlier in-vitro biochemical data suggest that the eIF5G31R mutation disfavors the AUG base pairing while favors UUG base pairing in the ‘Closed/PIN’ conformation with the Met-tRNAi56,57, suggesting a possible mechanism of UUG codon recognition. However, it is not clear whether the eIF5G31R mutant also disfavors AUG codon recognition in-vivo. Our data can resolve this issue; as upon removal of the upUUG codons from the URA6-LacZ or YAP1-LacZ reporter constructs, the levels of reporter expression initiated from the AUG codon in the eIF5G31R mutant and WT cells were essentially identical (Figs. 2b, 4c), suggesting that the eIF5G31R mutant can recognize AUG start codon in-vivo. Thus the dominant slow-growth phenotype and the recessive lethality could be because of the additional UUG codon recognition and the attendant proteome change in the eIF5G31R mutant. The eIF2γN135D mutation confers a Sui− phenotype, a slow-growth phenotype, and Met-tRNAi binding defects that are consistent with its Gcd− phenotype (de-repression of GCN4 expression). However, the slow-growth phenotype of the eIF2γN135D mutations is suppressed by the overexpression of Met-tRNAi but not by the eIF1 overexpression, which only suppresses the Sui− phenotype17. Taken together, it appears that the slow growth phenotype of the eIF2γN135D mutation is because of the TC formation defects. However, the dominant slow growth phenotype in the eIF5G31R mutant is primarily because of the UUG codon recognition (Sui− phenotype) and the attendant proteome change that causes oxidative stress and DNA damage, as the eIF5G31R mutation does not have TC formation defect and show Gcn− phenotype19. However, we do not imply that oxidative stress and DNA damage are the only defects caused by the upUUG codon recognition in the eIF5G31R mutant cells. It could be possible that the mRNA transcript having multiple out of frame upUUG codons (examples, URA6 or YAP1) may be disproportionately affected compared to the mRNA transcript having a single out of frame upUUG codon. Moreover, not all upUUG codons can be utilized efficiently to initiate translation by the Sui− mutants as the sequence context surrounding the upUUG codon may also play an important role58. Consistently, the eIF5G31R mutation showed ~ 2.6-fold decrease in UUG codon recognition efficacy when the UUG codon is in a poor sequence context (Supplementary Fig. S7). Probably genome-wide ribosomal profiling of the Sui− mutants will give more clarity on global upUUG codon utilization in different sequence contexts and identification of affected proteins involved in various cellular pathways.
The significance of studying defects in the translation initiation process is that it uncovers the fundamentals of the information decoding process and sheds light on the diseases associated with this process. Diseases such as Alzheimer’s, amyotrophic lateral sclerosis, Vanishing White Matter (VWM) are linked with the defects associated with integrated stress response (ISR) that lower TC levels59. Mutations isolated in the human eIF2γ subunit are associated with the X-linked intellectual disability characterized by mental deficiency, epilepsy, hypogenitalism, microcephaly, and obesity (MEHMO syndrome)60,61,62,63. Interestingly, the yeast model of MEHMO syndrome mutations shows reduced TC levels and increased translation initiation at non-AUG codon. It is suggested that the difference in VWM disease and MEHMO syndrome may be due to the altered cellular proteome caused by the non-AUG codon initiation in the MEHMO syndrome patients60. Thus our study on translation initiation fidelity defective eIF5G31R mutant that uncovered DNA damage and oxidative stress due to altered proteome could serve as an impetus to investigate further the proteome change and the attendant cellular defects caused by the eIF2γ mutants in the MEHMO syndrome patients.
References
Hinnebusch, A. G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 75, 434–467 (2011).
Hinnebusch, A. G. The scanning mechanism of eukaryotic translation Initiation. Annu. Rev. Biochem. 83, 779–812 (2014).
Llácer, J. L. et al. Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol. Cell 59, 399–412 (2015).
Llácer, J. L. et al. Translational initiation factor eIF5 replaces eIF1 on the 40S ribosomal subunit to promote start-codon recognition. Elife 7, e39273 (2018).
Fekete, C. A. et al. The eIF1A C-terminal domain promotes initiation complex assembly, scanning and AUG selection in vivo. EMBO J. 24, 3588–3601 (2005).
Alone, P. V. & Dever, T. E. Direct binding of translation initiation factor eIF2gamma-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2B epsilon. J. Biol. Chem. 281, 12636–12644 (2006).
Maag, D., Fekete, C. A., Gryczynski, Z. & Lorsch, J. R. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell 17, 265–275 (2005).
Chang, K.-J. & Wang, C.-C. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J. Biol. Chem. 279, 13778–13785 (2004).
Chen, S.-J.J., Lin, G., Chang, K.-J.J., Yeh, L.-S.S. & Wang, C.-C.C. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J. Biol. Chem. 283, 3173–3180 (2008).
Iwasaki, S., Floor, S. N. & Ingolia, N. T. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature 534, 558–561 (2016).
Guenther, U. P. et al. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature 559, 130–134 (2018).
Costello, J. et al. Global mRNA selection mechanisms for translation initiation. Genome Biol. 16, 1–21 (2015).
Mitchell, S. F. et al. The 5’-7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol. Cell 39, 950–962 (2010).
Gupta, N., Lorsch, J. R. & Hinnebusch, A. G. Yeast Ded1 promotes 48S translation preinitiation complex assembly in an mRNA-specific and eIF4F-dependent manner. Elife 7, 1–25 (2018).
Castilho-Valavicius, B., Yoon, H. & Donahue, T. F. Genetic characterization of the Saccharomyces cerevisiae translational initiation suppressors sui1, sui2 and SUI3 and their effects on HIS4 expression. Genetics 124, 483–495 (1990).
Huang, H. K., Yoon, H., Hannig, E. M. & Donahue, T. F. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 11, 2396–2413 (1997).
Alone, P. V., Cao, C. & Dever, T. E. Translation initiation factor 2gamma mutant alters start codon selection independent of Met-tRNA binding. Mol. Cell. Biol. 28, 6877–6888 (2008).
Antony, A. C., Ram, A. K., Dutta, K. & Alone, P. V. Ribosomal mutation in helix 32 of 18S rRNA alters fidelity of eukaryotic translation start site selection. FEBS Lett. 593, 852–867 (2019).
Antony, A. C. & Alone, P. V. Defect in the GTPase activating protein (GAP) function of eIF5 causes repression of GCN4 translation. Biochem. Biophys. Res. Commun. 486, 1110–1115 (2017).
Antony, A. C. & Alone, P. V. Fidelity of HIS4 start codon selection influences 3-amino-1,2,4-triazole sensitivity in GTPase activating protein function defective eIF5. J. Genet. 97, 953–964 (2018).
Gietz, R. D., Sugino, A. & Akio, S. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 (1988).
Gueldener, U., Heinisch, J., Koehler, G. J., Voss, D. & Hegemann, J. H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 30, e23 (2002).
Christianson, T. W., Sikorski, R. S., Dante, M., Shero, J. H. & Hieter, P. Multifunctional yeast high-copy-number shuttle vectors. Gene 110, 119–122 (1992).
Perez-Riverol, Y. et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 47, D442–D450 (2019).
Tkach, J. M. et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 14, 966–976 (2012).
Uttenweiler, A., Schwarz, H., Neumann, H. & Mayer, A. The vacuolar transporter chaperone (VTC) complex is required for microautophagy. Mol. Biol. Cell 18, 166–175 (2007).
Jager, S., Strayle, J., Heinemeyer, W. & Wolf, D. H. Cic1, an adaptor protein specifically linking the 26S proteasome to its substrate, the SCF component Cdc4. EMBO J. 20, 4423–4431 (2001).
Kumánovics, A. et al. Identification of FRA1 and FRA2 as genes involved in regulating the yeast iron regulon in response to decreased mitochondrial iron-sulfur cluster synthesis. J. Biol. Chem. 283, 10276–10286 (2008).
Goder, V. & Melero, A. Protein O-mannosyltransferases participate in ER protein quality control. J. Cell Sci. 124, 144–153 (2011).
Altmann, M., Schmitz, N., Berset, C. & Trachsel, H. A novel inhibitor of cap-dependent translation initiation in yeast: p20 competes with eIF4G for binding to eIF4E. EMBO J. 16, 1114–1121 (1997).
Sertil, O., Kapoor, R., Cohen, B. D., Abramova, N. & Lowry, C. V. Synergistic repression of anaerobic genes by Mot3 and Rox1 in Saccharomyces cerevisiae. Nucleic Acids Res. 31, 5831–5837 (2003).
Vergauwen, B., Pauwels, F. & Van Beeumen, J. J. Glutathione and catalase provide overlapping defenses for protection against respiration-generated hydrogen peroxide in Haemophilus influenzae. J. Bacteriol. 185, 5555–5562 (2003).
Malys, N. & McCarthy, J. E. G. Dcs2, a novel stress-induced modulator of m7G pppX pyrophosphatase activity that locates to P bodies. J. Mol. Biol. 363, 370–382 (2006).
Skoneczna, A., Miciałkiewicz, A. & Skoneczny, M. Saccharomyces cerevisiae Hsp31p, a stress response protein conferring protection against reactive oxygen species. Free Radic. Biol. Med. 42, 1409–1420 (2007).
Fakas, S. Lipid biosynthesis in yeasts: A comparison of the lipid biosynthetic pathway between the model nonoleaginous yeast Saccharomyces cerevisiae and the model oleaginous yeast Yarrowia lipolytica. Eng. Life Sci. 17, 292–302 (2017).
Pallotta, M. L., Valenti, D., Iacovino, M. & Passarella, S. Two separate pathways for d-lactate oxidation by Saccharomyces cerevisiae mitochondria which differ in energy production and carrier involvement. Biochim. Biophys. Acta BBA Bioenergy 1608, 104–113 (2004).
Lagoskyl, P. A., Taylor, G. R. & Haynes, R. H. Molecular characterization of the Saccharomyces cerevisiae dihydrofolate reductase gene (DFRI). Nucleic Acids Res. 15, 10355 (1987).
Huang, Z. et al. OLE1 reduces cadmium-induced oxidative damage in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 365, 193 (2018).
Grishin, A. V., Rothenberg, M., Downs, M. A. & Blumer, K. J. Mot3, a Zn finger transcription factor that modulates gene expression and attenuates mating pheromone signaling in Saccharomyces cerevisiae. Genetics 149, 879–892 (1998).
Zhang, S., Sanyal, I., Bulboaca, G. H., Rich, A. & Flint, D. H. The gene for biotin synthase from Saccharomyces cerevisiae: Cloning, sequencing, and complementation of Escherichia coli strains lacking biotin synthase. Arch. Biochem. Biophys. 309, 29–35 (1994).
García-Ramírez, J. J., Santos, M. A. & Revuelta, J. L. The Saccharomyces cerevisiae RIB4 gene codes for 6,7-dimethyl-8-ribityllumazine synthase involved in riboflavin biosynthesis. J. Biol. Chem. 270, 23801–23807 (1995).
Fatica, A., Oeffinger, M., Tollervey, D. & Bozzoni, I. Cic1p/Nsa3p is required for synthesis and nuclear export of 60S ribosomal subunits. RNA 9, 1431–1436 (2003).
Ogg, S. C., Poritz, M. A. & Walter, P. Signal recognition particle receptor is important for cell growth and protein secretion in Saccharomyces cerevisiae. Mol. Biol. Cell 3, 895–911 (1992).
McNew, J. A. et al. Gos1p, a Saccharomyces cerevisiae SNARE protein involved in Golgi transport. FEBS Lett. 435, 89–95 (1998).
Jiang, Z. R., Abaigar, L. T., Huang, S. H., Cai, B. & Jong, A. Y. Molecular characterization of Saccharomyces cerevisiae URA6 gene. DNA sequence, mutagenesis analysis, and cell cycle regulation relevant to its suppression mechanism to cdc8 mutation. J. Biol. Chem. 266, 18287–18293 (1991).
Patricia, L. & Francois, L. Genetic characterization and isolation of the Saccharomyces cerevisiae gene coding for uridine monophosphokinase. Mol. Gen. Genet. 205, 74–81 (1986).
Jamieson, D. J. Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast Chichester Engl. 14, 1511–1527 (1998).
Kurutas, E. B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 15, 71 (2016).
He, L. et al. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 44, 532–553 (2017).
Machado, M. D. & Soares, E. V. Assessment of cellular reduced glutathione content in Pseudokirchneriella subcapitata using monochlorobimane. J. Appl. Phycol. 24, 1509–1516 (2012).
Bankapalli, K. et al. Robust glyoxalase activity of Hsp31, a ThiJ/DJ-1/PfpI family member protein, is critical for oxidative stress resistance in Saccharomyces cerevisiae. J. Biol. Chem. 290, 26491–26507 (2015).
Perrone, G. G., Tan, S. X. & Dawes, I. W. Reactive oxygen species and yeast apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 1783, 1354–1368 (2008).
Lee, J., Spector, D., Godon, C., Labarre, J. & Toledano, M. B. A new antioxidant with alkyl hydroperoxide defense properties in yeast. J. Biol. Chem. 274, 4537–4544 (1999).
Salmon, T. B., Evert, B. A., Song, B. & Doetsch, P. W. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 32, 3712–3723 (2004).
Shenton, D. et al. Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J. Biol. Chem. 281, 29011–29021 (2006).
Saini, A. K. et al. Eukaryotic translation initiation factor eIF5 promotes the accuracy of start codon recognition by regulating Pi release and conformational transitions of the preinitiation complex. Nucleic Acids Res. 42, 9623–9640 (2014).
Martin-Marcos, P. et al. Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy. RNA 20, 150–167 (2014).
Martin-Marcos, P., Cheung, Y.-N. & Hinnebusch, A. G. Functional elements in initiation factors 1, 1A, and 2β discriminate against poor AUG context and non-AUG start codons. Mol. Cell. Biol. 31, 4814–4831 (2011).
Costa-Mattioli, M. & Walter, P. The integrated stress response: From mechanism to disease. Science 368, 5314 (2020).
Young-Baird, S. K. et al. Suppression of MEHMO syndrome mutation in eIF2 by small molecule ISRIB. Mol. Cell 77, 875–886 (2020).
Young-Baird, S. K., Shin, B.-S. & Dever, T. E. MEHMO syndrome mutation EIF2S3-I259M impairs initiator Met-tRNAiMet binding to eukaryotic translation initiation factor eIF2. Nucleic Acids Res. 47, 855–867 (2019).
Skopkova, M. et al. EIF2S3 mutations associated with severe X-linked intellectual disability syndrome MEHMO. Hum. Mutat. 38, 409–425 (2017).
Borck, G. et al. eIF2γ mutation that disrupts eIF2 complex integrity links intellectual disability to impaired translation initiation. Mol. Cell 48, 641–646 (2012).
Krzywinski, M. et al. Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Acknowledgements
This work was supported by the intramural grant from the Department of Atomic Energy, Government of India to P.V.A. and the intramural grant from Department of Biotechnology to ARS. We acknowledge the support of the ILS proteomics facility to carry out proteomics work. We thank Narottam Acharya for reagents and advice.
Author information
Authors and Affiliations
Contributions
P.V.A. conceived the study and designed the experiments; A.K.R., M.M. and R.R.R. performed the experiments; P.V.A., A.K.R., and A.R.S. analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ram, A.K., Mallik, M., Reddy, R.R. et al. Altered proteome in translation initiation fidelity defective eIF5G31R mutant causes oxidative stress and DNA damage. Sci Rep 12, 5033 (2022). https://doi.org/10.1038/s41598-022-08857-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08857-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
