
Abstract
In the crustacean Daphnia magna, studying homology-directed repair (HDR) is important to understand genome maintenance during parthenogenesis, effects of environmental toxicants on the genome, and improvement of HDR-mediated genome editing. Here we developed a transgenic D. magna that expresses green fluorescence protein (GFP) upon HDR occurrence. We utilized the previously established reporter plasmid named DR-GFP that has a mutated eGFP gene (SceGFP) and the tandemly located donor GFP gene fragment (iGFP). Upon double-strand break (DSB) introduction on SceGFP, the iGFP gene fragment acts as the HDR template and restores functional eGFP expression. We customized this reporter plasmid to allow bicistronic expression of the mCherry gene under the control of the D. magna EF1α-1 promoter/enhancer. By CRISPR/Cas-mediated knock-in of this plasmid via non-homologous joining, we generated the transgenic D. magna that expresses mCherry ubiquitously, suggesting that the DR-GFP reporter gene is expressed in most cells. Introducing DSB on the SceGFP resulted in eGFP expression and this HDR event could be detected by fluorescence, genomic PCR, and quantitative reverse-transcription PCR, suggesting this line could be used for evaluating HDR. The established reporter line might expand our understanding of the HDR mechanism and also improve the HDR-based gene-editing system in this species.
Similar content being viewed by others

Introduction
Genomes are threatened by endogenously generated chemicals like reactive oxygen species and exogenous compounds such as mutagenic agents and radiation1, which can lead to DNA double-strand breaks (DSBs). To ensure genetic stability and cellular viability, repairing the DSBs is essential. The DNA repair mainly occurs through non-homologous end joining (NHEJ) and homology-directed repair (HDR)2. The NHEJ leads to ligation of the two ends of the DNA strand during which insertion or deletion of nucleotides (indels) can often occur at the cleavage site. The HDR repairs the DSBs by using information copied from undamaged DNA that has an identical or homologous sequence (homology)3 This homology-directed repair system can be divided into four sub-pathways based on the mechanistic difference: double-strand break repair (DSBR), synthesis-dependent strand annealing (SDSA), break-induced replication (BIR), and single-strand annealing (SSA)2. First, in the DSBR, the formation of an intermediate structure with the Holliday junctions (HJs) leads to the generation of crossover and non-crossover products4. Second, the SDSA exclusively generates the non-crossover products due to the lack of formation of the HJ structure5. Third, when only one ended DSB site has a sequence similar to that of the template, BIR occurs for non-reciprocal translocation of genetic information from the template strand6. Fourth, when the DSB is induced between the tandem repeats of the highly homologous regions, the SSA repairs the DNA by pairing the homologous region followed by deletion of unpaired DNA and the intervening region7. In the mitotically proliferating cells, the SDSA is known to be the most common among the HDR sub-pathways8.
The HDR also plays an important role in the field of genome editing due to its nature of high fidelity and accuracy. Unlike NHEJ, HDR avoids multiple integrations of a donor DNA and indels between transgene and surrounding genomic region. Thus, following DSB using the programmable nuclease for instance TALEN or CRISPR-Cas, a precise genome modification such as the codon replacements or the seamless integration of the fluorescent reporter gene can be achieved by using a donor DNA flanked with right and left locus-specific homology arms as a template9,10.
The water flea Daphnia magna is a small freshwater crustacean found in broad continents such as Europe, the Middle East, Central Asia, Africa, and North America11. The genus Daphnia reproduces by parthenogenesis under favorable environmental conditions but switches it to sexual reproduction in response to environmental stimuli such as shortened photoperiod, a lack of food, and/or increased population density12. The sequenced genome of Daphnia reveals highly duplicated genes, resulting in tandem gene clusters13. These tandem clusters may serve as a template for HDR-based repair to attenuate the effect of deleterious mutations during the parthenogenetic cycle, which suggests that Daphnia may have a unique HDR mechanism.
Daphnia magna occupies an important position in the freshwater food chain and is highly sensitive to chemicals, which makes this species a model in environmental and toxicological studies. The effects of genotoxicants have been investigated at the phenotypic level14,15. To understand their actions at the molecular level, it is important to study the DNA repair mechanism in this species. In the field of genome editing, the HDR-based knock-in of the exogenous DNA fragments has been reported in D. magna16 as well as NHEJ-mediated knock-in17,18. The HDR-based knock-in efficiency was low probably due to competition with the NHEJ pathway. To test this hypothesis, disruption DNA ligase IV which is the conserved component of the NHEJ pathway has been attempted16 However, its effect was not fully evaluated due to the lack of a method for quantifying the HDR event in vivo. Therefore, a system to evaluate and quantify the HDR event is a necessity.
Fluorescence live imaging of the HDR event is essential not only for investigating how and where the genome integrity is maintained in living organisms but also for evaluating the HDR activity by manipulating the components for DNA repair. The direct repeat GFP (DR-GFP) reporter assay has been established for fluorescence-based visualization of the HDR activity19. The DR-GFP reporter system is composed of two mutated eGFP genes (Fig. 1, Genotype). The upstream eGFP gene named SceGFP contains a recognition site of the rare-cutting I-SceI restriction enzyme. This recognition site contains two in-frame stop codons to terminate the protein expression. At downstream of the SceGFP, there is another mutated eGFP fragment termed internal GFP or iGFP that is an 812-bp internal GFP fragment. The HDR event can be detected by introducing a double-strand break (DSB) with I-SceI in the inactive SceGFP gene. The cleavage site will be repaired by HDR using iGFP as the template. Among the HDR sub-pathways, this DR-GFP system can visualize the non-crossover events that are mediated by the DSBR and SDSA20, suggesting that this reporter can visualize the major HDR events spatiotemporally in vivo. This reporter has been applied to study the factors that contribute to HDR in mouse21 and to study the role of a transcriptional repressor protein in HDR using C. elegans models22.

The DR-GFP reporter assay and its potential application. The DR-GFP reporter consists of two tandem repeats of mutated GFP. The first repeat (SceGFP, solid green) contains an I-SceI recognition site with stop codons embedded (blue line). The second repeat (iGFP, striped-green) lacks 5′and 3′sequence. The introduction of DSB at the I-SceI site induces HDR-based DNA repair, utilizing homologous sequences from the iGFP, which in turn resulted in a functional eGFP gene (left box figure). The eGFP fluorescence signal will be observed in D. magna transgenic reporter as a result of the HDR event (middlebox figure). The possible application of the transgenic reporter is to elucidate the effect of various stimuli on the HDR level (right box).
Here we integrated the DR-GFP reporter system in the D. magna genome (Fig. 1, Genotype). We confirmed its functionality by introducing DSBs at the SceGFP region with the CRISPR/Cas9 system and detecting the eGFP signal spatiotemporally (Fig. 1, Phenotype). Furthermore, we could detect the repaired eGFP gene by genomic PCR and qPCR, which adds merit to this system to be utilized for the evaluation of the HDR event. By applying different stimuli (Fig. 1, Stimuli), the established transgenic Daphnia might contribute to various scientific fields such as ecotoxicology, genome editing, and evolutionary biology.
Materials and methods
Daphnia magna strain and culture condition
Wild type D. magna (NIES clone) was obtained from the National Institute of Environmental Studies (NIES, Tsukuba, Japan) and has been maintained in the laboratory for many generations. The D. magna was cultured under the following conditions: 80 juveniles (less than 24 h old) were collected and cultured in 5 L Artificial Daphnia Medium (ADaM)23 at temperature 22–24 °C, under 16 h/8 h of light/dark photoperiod. D. magna were fed daily with 8 × 109 cells of Chlorella vulgaris (Oita Medaka Biyori, Oita, Japan) and 3 mg of baker’s yeast (Marusan Pantry, Ehime, Japan) during the first week. Later, juveniles were removed daily and amounts of chlorella and yeast extract were doubled. The culture medium was changed once a week.
Customization of reporter plasmid
To visualize the HDR by fluorescence in D. magna, a reporter donor plasmid pEF1α-1::mCherry-2A-DR-GFP was constructed by customizing the previously established pDRGFP plasmid (Addgene No.26475)19 (Fig. 2A, B). To allow ubiquitous expression in D. magna, the original chicken β-actin promoter was replaced with a 2.3 kb of D. magna elongation factor 1 α-1 (EF1α-1) promoter/enhancer, including the transcription start site, the complete first intron, and part of the second exon with a start codon17,24. In addition, to recapitulate EF1α-1 endogenous expression, a full-length EF1α-1 3′ UTR was added downstream of the reporter. We retained the original two mutated eGFP fragments (SceGFP and iGFP) along with their nuclear localization sequence to distinguish the eGFP-expressing cells individually.

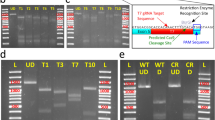
DR-GFP reporter system. (A) The donor plasmid design. The direct repeat of differentially mutated eGFP (DR-GFP) consists of mutated (SceGFP) and 5′ and 3′-lacking sequence of eGFP (iGFP), both are indicated in light green boxes. Three repeats (two complete and one partial) of the simian virus 40 large T-antigen nuclear localization signal (SV40 NLS) are included in the SceGFP sequence (Supplementary Figure S1). The reporter system is expressed ubiquitously under D. magna EF1α-1 promoter/enhancer (grey arrow). The red fluorescence protein mCherry-coding sequence is placed upstream of the DR-GFP system. mCherry and DR-GFP are bicistronically expressed using Thosea asigna virus 2A (T2A) peptide indicated in the yellow box. SceGFP contains a recognition site of the 18 bp I-SceI restriction enzyme and in-frame two stop codons indicated in the blue underline and red letter respectively. SceI gRNA was designed to correspond with the I-SceI recognition site (italic) upstream of the PAM sequence (small letter). The cleavage site of SceI gRNA was indicated by a black triangle. (B) The alignment between eGFP, iGFP, and SceGFP sequences. Blue areas indicate the I-SceI site. (C) The diagram of the DR-GFP system for reporting HDR events. Double-strand break (DSB) is introduced in the I-SceI site by the Cas9-gRNA complex. Following homology-directed repair (HDR) occurrence, iGFP will serve as a repair template, leading to SceGFP repair indicated by eGFP expression green box.
DR-GFP reporter will function when the DSB is introduced in the I-SceI site. By SDSA or non-crossover DSBR subpathway of HDR, SceGFP will use iGFP as a repair template resulting in the functional eGFP expression (Fig. 2C). A red fluorescent protein gene mCherry ORF was fused upstream of the SceGFP via a sequence encoding Thosea asigna virus 2A (T2A), which can lead to bicistronic expression of both mCherry and mutated/repaired eGFP25. Lastly, for the integration into D. magna genome, a 200 bp sequence of scarlet gene harboring a gRNA target sequence26,27 was added. The complete nucleotide sequence of the customized DR-GFP reporter and the deduced amino sequence is provided in Supplementary Figure S1. All assemblies were performed using GeneArt Cloning & Assembly (Invitrogen, Carlsbad, USA). The constructed donor plasmid was purified using FastGene Plasmid Mini Kit (Nippon Genetics, Tokyo, Japan) and sequenced. The donor plasmid used for microinjection was purified using PureYield Miniprep (Promega, Madison, USA) followed by phenol–chloroform purification, two times ethanol washing, and was re-suspended with ultrapure water (Invitrogen).
In vitro RNA synthesis
Guide RNAs (gRNAs) were synthesized using a cloning-free method from PCR-amplified template DNA as previously described27. The sense synthetic oligonucleotide containing a T7 promoter sequence, a gene-specific target sequence, and the first 20 nt of the Cas9 binding scaffold are shown in Table 1. gRNAs were synthesized using the MegaScript T7 Transcription Kit (Invitrogen), purified using Roche Mini Quick Spin RNA Column (Roche, Mannheim, Germany) followed by phenol/chloroform extraction, ethanol precipitation.
For Cas9 mRNA synthesis, a template DNA containing T7 promoter sequence was PCR amplified from pCS-Dmavas-Cas928. Capped mRNA synthesis and poly(A) tail addition were performed using mMessage mMachine T7 kit and Poly(A) Tailing Kit (Invitrogen) respectively. Synthesized mRNA was column purified, followed by phenol/chloroform extraction and ethanol precipitation. mRNA integrity and the addition of poly(A) tails were confirmed by denaturing formaldehyde gel electrophoresis.
Generation of HDR reporter transgenic Daphnia magna
For the generation of the DR-GFP line, the customized DR-GFP reporter plasmid was integrated into D. magna genome by utilizing the CRISPR/Cas-mediated knock-in via non-homologous end-joining17. Microinjection into Daphnia embryos was performed following an established protocol using the S. pyrogenes-originated Cas9 proteins17,29. The Cas9 proteins were expressed in E. coli strain BL21 (DE3) and purified following established protocol30. Fifty nanograms per microliter of purified donor plasmid was co-injected with 2 µM scarlet targeting gRNA, and 1 µM Cas9 protein. Shortly before the microinjection, Cas9 protein and gRNA were incubated at 37 °C for 5 min to form a ribonucleoprotein (RNP) complex. Microinjection was performed within two hours after the preparation of the solution. After injection, the intact eggs were transferred, cultured individually in a sterile 96 well plate, and put in an incubator at 22 °C with 16 h/8 h of light/dark photoperiod for 3 days. Each well of the 96 well-plate was filled with 100 μL of M4-sucrose. Transgenic candidates were screened based on the mCherry expression in the ovary of injected embryos (G0). mCherry expressing offspring were cultured and genotyping was performed using the second generation offsprings (G2).
Genotyping
Daphniids were collected and homogenized in 500 µL lysis buffer (50 mM Tris-HCl pH 7.5, 20 mM EDTA pH 8.0, 100 mM NaCl, 1% SDS) using MicroSmash homogenizer (TOMY, Tokyo, Japan) at 3000 rpm for 1.5 min with the presence of 0.15 mg/mL Proteinase K (Nacalai Tesque, Kyoto, Japan). The homogenized daphniids (lysate) were shaken overnight at 55 °C, 450 rpm using an incubator shaker (Bioshaker M-BR-022UP, TAITEC, Tokyo, Japan). To obtain genomic DNA (gDNA), the lysate was purified using phenol/chloroform extraction, precipitated with isopropanol, rinsed twice with 70% ethanol, and dissolved in 50 µL TE buffer before being used as a template for genomic PCR. The PCR was performed by using an Ex Taq Hot-Start DNA polymerase (TaKaRa) with primer sets amplifying the target region as described in Table 2.
Functional analysis of DR-GFP reporter transgenic D. magna
To demonstrate the functionality of the DSB reporter in Daphnia, we designed a gRNA named SceI gRNA that introduced DSB at 2 bp upstream of the I-SceI digestion site that was previously used for I-SceI-dependent DSBs19.
To confirm whether Cas9 was active during microinjection, we also co-injected the SceI gRNA with another gRNA targeting Distal-less (Dll) gene. Previously RNAi-mediated knockdown of Dll in embryos of D. magna led to a distinct phenotype “truncation of second antennae” and the level of this truncation corresponded to the degree of impairment of this gene29. The Dll gRNA was designed to target the upstream of the homeodomain region in exon 2 (Supplementary Figure S2), as this region is highly conserved among arthropod29 and considered important for Dll function31,32.
The phenotypes of the second antennae of the injected embryos were observed 48 h post-injection (hpi) and categorized as normal, mild, medium, or strong truncation following the previous study. In normal phenotype, second antennae consist of a protopodite (P), carrying a dorsal and ventral ramus. Each ramus has three segments, Terminal (T), Middle (M), and Basal (B). There is an additional small wedge-shaped segment (w) between B and P. Mild truncation exhibit, a part of M and full B (ventral ramus), full M, B, and w (dorsal ramus). Medium truncation: a trace of B (ventral), B, and w (dorsal). Strong truncation: only a trace of B and w (dorsal)29.
Fluorescence photography
Fluorescence images were photographed using the Leica DC500 CCD Digital Camera mounted on the Leica M165FC fluorescence microscope (Leica Microsystem, Wetzlar, Germany). The mCherry filter (Leica Microsystem) was used to screen the DR-GFP line. The GFP3 filter (Leica Microsystem) was used to observe repaired SceGFP after the HDR.
Total RNA extraction
Injected and uninjected embryos were collected at 48 h post-injection. Injected embryos were sorted based on the presence of eGFP expression and its total RNA was extracted in the presence of Sepasol-RNA I solution (Nacalai Tesque) according to the manufacturer’s protocol. The total RNA was purified using phenol/chloroform extraction, precipitated with 70% ethanol, dissolved in UltraPure water (Invitrogen), and directly subjected to reverse transcriptase reaction.
Quantitative reverse-transcription PCR (qPCR)
cDNAs were synthesized from 1 µg of purified total RNA using random primers and PrimeScript II 1st strand cDNA Synthesis Kit (TaKaRa). The β-actin gene was amplified by PCR to confirm the absence of genomic DNA contamination as previously described33. To quantify repaired SceGFP mRNA level, qPCR was performed in StepOnePlus (Applied Biosystem) using KOD SYBR® qPCR Mix (Toyobo, Osaka, Japan) with primers as listed in Table 2. The cycling condition is as follows: 2 min at 98 °C, followed by a total of 40 cycles of 98 °C for 10 s, 60 °C for 10 s, and 68 °C for 30 s. Primers’ specificity was confirmed by melting curve analysis and agarose gel electrophoresis. The expression level of eGFP was normalized using that of mCherry that is controlled under the same promoter.
Results
The genotype of HDR reporter transgenic Daphnia
To establish the HDR reporter transgenic Daphnia (DR-GFP line), we co-injected reporter plasmid and the RNP complex into 29 eggs. The 10 injected embryos survived until adult, from which 9 produced offspring with a white eye that is the typical phenotype of the scarlet mutant, indicating that the Cas9 RNP induced DSBs at the targeted site. Of the 9, one adult produced offspring with ubiquitous mCherry fluorescence, suggesting germline transmission of reporter plasmid (Fig. 3A). This fluorescence pattern also indicates that this reporter system enables us to detect the HDR event in most types of cells. We cultured this potentially transgenic line for genotyping.

The phenotype and genotype of the DR-GFP line (A) Comparing phenotypes of the DR-GFP line with the wild type Daphnia. The top and lower rows show D. magna obtained from wild type and DR-GFP lines respectively. The image in each column was photographed using either of bright field, mCherry, or GFP3 filter. The region inside the white-dashed line is the gut, in which ingested chlorella emits slight red autofluorescence both in wild type and DR-GFP. Widespread mCherry fluorescence was observed only in the transgenic line, while eGFP fluorescence was not observed. Black arrow indicates loss of black eye pigment due to disrupted scarlet allele. (B) Schematic representation of the integration site of the DR-GFP donor plasmid. A part of the RHOGAP (Rho-GTPase activating protein) gene is shown upstream of scarlet. The DR-GFP donor plasmid was integrated into exon 3 of the scarlet gene. A and C indicate the 5′ and 3′ junction regions of donor plasmid and genome, B indicated the internal region of donor plasmid (mCherry gene). (C) PCR result was visualized by gel electrophoresis. The first lane is the marker, followed by fragments A, B, C, and β-actin (beta-actin) for both wild type and DR-GFP line Daphnia. In the sample using DR-GFP line Daphnia, all fragments except the 5′ junctions were amplified. The full length of the gel is presented in Supplementary Figure S4 (D) 20 bp deletion and 8 bp insertion were detected in the 3′ junction of plasmid integration and another allele of scarlet respectively. The red-colored nucleotides and black triangle indicated the St gRNA target and DSB site respectively.
To investigate whether NHEJ-mediated knock-in occurred, genotyping was performed using the genome of the potentially transgenic line. We amplified the mCherry fragment, 5′ and 3′ junctions between the transgene and its surrounding region by PCR (Fig. 3B). The expected size of the mCherry fragments was obtained only in the potential transgenic line (Fig. 3C, fragment B, DR-GFP line). The 3′ junction region was also amplified in this line by using forward primer targeted at the downstream of EF1α-1 3′ UTR of the donor plasmid and reverse primer targeted exon 8 of scarlet gene locus (Fig. 3B, C, fragment C, DR-GFP line). Sequencing of this PCR product confirmed the integration of the reporter plasmid at the scarlet locus and revealed 20 bp deletion and 8 bp insertion at the 3′ side of the integrated cassette (Fig. 3D, 3′ junction). Consistent with the white-eyed phenotype, another allele contained indel mutation at the DSB site (Fig. 3D, 2nd allele). We were unable to amplify the 5′ junction region even if the forward primer was designed at 3157 bp upstream and 2610 bp downstream of the DSB site (Fig. 3B, C, fragment A, DR-GFP line). This suggests that large deletion occurred at the 5′ side of the integration site. Nevertheless, amplification and sequencing of the full-length of the DR-GFP gene cassette demonstrated the integration of the intact DR-GFP reporter (Supplementary Figure S1).
The DSB near the I-SceI site leads to the generation of eGFP-positive cells in embryos of the DR-GFP line
To examine whether the DR-GFP reporter gene is functional in the established line, we attempted to introduce the DSB near the I-SceI site. Seventy-four eggs were co-injected with 1 µM Cas9 protein and gRNA mixtures (SceI gRNA and Dll gRNA, 2 µM each). Dll gRNA was used as a marker for Cas9 activity during microinjection as described in Materials and methods and Supplementary Table S1. Forty-three embryos survived until the 48 hpi stage and 41 (95%) showed truncation of the second antennae (Table 3) from which, 22 embryos (54%) showed the strong phenotype29, indicating that Cas9 was active during injection and could introduce DSBs on the genome. Of the 41, 33 (80%) showed strong nuclear-localized eGFP fluorescence in the tissues such as the head and thoracic appendages (Fig. 4). In contrast, embryos injected with Cas9 RNP including the unrelated St gRNA (Fig. 4) and Dll gRNA did not show intense and nuclear-localized GFP signal, indicating that the recovery of the eGFP fluorescence occurred by injection of Cas9 protein and SceI gRNA.

Detection of eGFP-positive cells of DR-GFP transgenic D. magna following the DSB of the SceGFP. DR-GFP line was co-injected with Cas9 protein, SceI gRNA, and Dll gRNA. The injection of Cas9 protein with St gRNA and Dll gRNA was performed as a control. The first row shows uninjected control, while the second, third, and fourth rows show injected individually Daphnia. Images were taken using the bright field and the GFP3 filter. All daphniids were photographed at 48 h post-injection. The red dashed line shows the second antennae region, which was truncated because of Dll gRNA injection. The weak background green fluorescence observed throughout the body of all samples was coming from autofluorescence. The repaired SceGFP was controlled under EF1α-1 promoter/enhancer and contains NLS, resulting in abundantly expressed and nuclear-localized eGFP expression (yellow triangles).
The embryos showing the nuclear-localized fluorescence signals have a functional eGFP gene repaired by HDR
To confirm whether HDR occurred at the genomic level, we extracted genome DNA from uninjected embryos and injected embryos that showed nuclear-localized eGFP fluorescence. PCR was then performed with a forward primer in the mCherry region and a reverse primer that recognizes specifically the sequence of the repaired SceGFP (Fig. 5A, B). Because the reverse primer also can bind to the iGFP sequence that is a template for HDR (Fig. 5A), we expected two bands would appear upon genomic PCR. A higher size band (2,843 bp) was present in all samples, indicating amplification from iGFP sequence (Fig. 5C, ii), while a lower size band (1048 bp) indicating amplification from repaired SceGFP sequence was obtained only from embryos injected with Cas9 and SceI gRNA (Fig. 5C, i). These results also suggest the repair of SceGFP by Cas9 and SceI gRNA.

The genotype of DR-GFP transgenic after DSB introduction. (A) PCR was performed on uninjected and injected DR-GFP genome using primer pairs indicated by arrows. The forward primer is attached in the mCherry region (pink area) while the reverse primers are attached in two locations, the repaired SceGFP (yellow area) and iGFP (light green). (B) Alignment between SceGFP and repaired SceGFP or eGFP. The reverse primer was designed to bind specifically to the repaired SceGFP (underline). (C) Gel electrophoresis result. The most left lane indicated the DNA marker (m) followed by amplified genome fragments (lanes 1–9) from Cas9 Protein and SceI gRNA injected embryos. Uninjected DR-GFP (lanes 10–12) was used as the negative control. The primer set amplified the repaired SceGFP region with the length 1048 bp (i). The reverse primer was also attached to the iGFP region, which resulted in a 2843 bp length PCR product (ii). The full length of the gel is presented in Supplementary Figure S4.
We also attempted to develop a qPCR-based method that can detect the repaired SceGFP expression. We designed a forward primer that binds to the T2A-coding sequence of DR-GFP reporter locus, and a reverse primer that specifically binds to repaired SceGFP sequence (Fig. 6A). As a model to test this system, we used Cas9-mRNA and Cas9 protein for introducing the DSB on the SceGFP because mutagenesis efficiency with Cas9 mRNA was lower than that with Cas9 protein17, which suggested Cas9 mRNA induces DSB occurrence to a lesser extent. We introduced the DSB at the SceGFP following either optimum condition of Cas9 mRNA (500 ng/µL Cas9 mRNA and 50 ng/μL gRNA) or Cas9 protein injection (1 µM Cas9 protein and 2 μM gRNA)17,28. The Dll gRNA was co-injected to evaluate the Cas9 activity in each injection. We confirmed 54% of Cas9 protein injected embryos showed a strong phenotype of second antennae truncation while Cas9 mRNA could only introduce a mild phenotype (Tables 3 and 4). This result implied that Cas9 protein had stronger activity to introduce DSB. Subsequently, the level of repaired SceGFP was analyzed using qPCR. By Cas9 protein injection, we observed significantly higher expression of repaired SceGFP (~ fivefold) relative to Cas9 mRNA injection. Moreover, neither repaired SceGFP signal nor amplification was detected in uninjected embryos as well as scarlet gRNA injected embryos (Fig. 6B, Supplementary Figure S3). Our result shows that qPCR can be used to detect the functional eGFP repaired by HDR.

Detection of the functional eGFP transcript by qPCR. (A) The position of primers and the region used for quantifying the repaired SceGFP level (above) were shown in the black line. The alignment showed that the reverse primer was designed to specifically bind to repaired SceGFP fragment (underline). (B) Level of repaired SceGFP between injected and uninjected samples after the introduction of DSB. The value was quantified by qPCR. There was a significant difference between Cas9 protein and mRNA injection. The values are means and error bars represent standard error (N = 3). *p < 0.05 (Student’s t-test). In uninjected embryos and ones injected with Cas9 protein and scarlet gRNA, the repaired SceGFP mRNA was not detected (ND).
Discussion
Here, we successfully integrated the DR-GFP system into D. magna genome and visualized HDR occurrence in vivo. We evaluated the functionality of this reporter system by introducing targeted DSB in the reporter site. We observed the eGFP signal and detected PCR products from the repaired eGFP gene in the injected daphniids, demonstrating evidence of detection of HDR in situ based on the eGFP fluorescence. We could also detect the repaired eGFP by qPCR that is potentially used for quantitative measurement of the HDR level following DNA DSB occurrences in the future. Furthermore, ubiquitous expression of mCherry that is bicistronically expressed with the DR-GFP suggests that this reporter system enables us to detect the HDR event in most types of cells.
In this study, we used the DR-GFP line to compare Cas9 mRNA and Cas9 protein ability to introduce DSB and induce HDR in D. magna. By using qPCR, we detected significantly higher repaired SceGFP expression in the Cas9 protein injected sample (Fig. 6B). This result was in line with a difference in DSB-inducing activity between Cas9 protein and mRNA in D. magna17. Recently, genome editing techniques using the CRISPR/Cas system are rapidly developing with the emergence of novel and smaller Cas9 proteins, such as Staphylococcus aureus derived Cas9 (SaCas9)35 and synthetic RNA-guided nuclease (sRGN, surgeons)36. As the DR-GFP system was also used to measure HDR DSB and single-strand break (SSB)-inducing activity using Cas9 and Cas9 nickase respectively34 in HEK293T cells, we anticipate our DR-GFP Daphnia could be a promising tool for the evaluation of new genome editing tools (Fig. 1, genome editors).
The DR-GFP system also has been applied for the screening of genotoxicants such as heavy metals37, FDA-approved drugs for cancer therapy38 in addition to evaluation of the sensitivity of the cancer cell to gamma-ray irradiation39. The aquatic ecosystem is constantly exposed to genotoxicants, and D. magna has been long used as a workhorse for ecotoxicology analysis as a biosensor. As the test guideline for acute or chronic toxicity test for Daphnia is well established by following the OECD Guideline40,41 we believe our DR-GFP Daphnia may be suitable for screening genotoxicants in vivo (Fig. 1, genotoxicants).
In recent years, several transgenic animals containing the DR-GFP system have already been established for analyzing the molecular mechanisms of HDR. For instance the generation of DR-GFP reporter mouse for analyzing the HDR frequencies in primary cell types derived from diverse lineages21. In C. elegans22, the DR-GFP system was integrated into the genome to identify a novel role of protein for promoting HDR. Daphnia might have a unique DNA repair mechanism to neutralize the genetic drawbacks because its asexual ability might lead to the accumulation of deleterious mutations due to the absence of recombination events via mating. We anticipate the prospect of utilizing this transgenic Daphnia for studying the function and roles of HDR in asexual reproduction by manipulating the components of HDR machinery. The result would contribute to a further understanding of evolutionary genomics (Fig. 1, manipulated components).
The HDR efficiency reported in mammals and plants is lower compared to NHEJ42,43,44 because it takes a longer time to complete than NHEJ42 and functions only during S and G2 phases when the sister chromatid, the main template to repair DSB, is present45. Thus, several approaches have been developed to enhance genome editing by HDR such as inhibiting46 or knocking out the key factor of NHEJ16, synchronizing and capturing cells at the certain phases47, and modifying the Cas9 by fusing it with a key protein necessary in the HDR steps48. To evaluate the effects of these approaches on the HDR activity, the reporter system for visualizing the HDR event has been used in mammalian cells49. We suggest the potential use of DR-GFP Daphnia for optimizing HDR efficiency for instance by impairing the NHEJ repair genes (Ku70 or Lig4) (Fig. 1. manipulated components).
We also acknowledge the limitations of this reporter system. First, in live imaging, it may be difficult to detect the eGFP signals from mutated cells that are located deep inside the tissues, which may lower sensitivity for detecting eGFP positive cells and their quantification. This limitation could be addressed by sorting and counting the eGFP-positive cells using fluorescence-activated cell sorting (FACS). Second, the DR-GFP reporter system can only visualize the presence of HDR events at the reporter locus. This situation may affect the sensitivity for detection of the HDR triggered by environmental genotoxicants or mutagenic agents that may introduce random DSB throughout the genome. Therefore, other approaches to globally visualize HDR events in Daphnia may be developed. For instance, fusing the Förster resonance energy transfer (FRET) system in HDR key proteins to provide spatiotemporal visualization of their function50. Third, for a comprehensive understanding of the DNA repair mechanism in this species, reporters for detection of the other DNA repair pathways such as NHEJ and SSA need to be developed. This limitation can be addressed by utilizing other reporter systems, such as “traffic light”, a dual fluorescence-based reporter which can visualize HDR and NHEJ repair pathways51. However, despite these possible limitations, DR-GFP reporter Daphnia would offer a valuable tool for the evaluation of HDR in this ecologically important species.
References
Aguilera, A. & Gómez-González, B. Genome instability: A mechanistic view of its causes and consequences. Nat. Rev. Genet. 9, 204–217 (2008).
Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 17, 11–16 (2010).
Li, X. & Heyer, W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 18, 99–113 (2008).
Szostak, J. W., Orr-Weaver, T. L., Rothstein, R. J. & Stahl, F. W. The double-strand-break repair model for recombination. Cell 33, 25–35 (1983).
Allers, T. & Lichten, M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 106, 47–57 (2001).
Morrow, D. M., Connelly, C. & Hieter, P. ‘Break copy’ duplication: A model for chromosome fragment formation in saccharomyces cerevisiae. Genetics 147, 371–382 (1997).
Lin, F. L., Sperle, K. & Sternberg, N. Model for homologous recombination during transfer of DNA into mouse L cells: Role for DNA ends in the recombination process. Mol. Cell. Biol. 4, 1020–1034 (1984).
Andersen, S. L. & Sekelsky, J. Meiotic versus mitotic recombination: Two different routes for double-strand break repair. BioEssays 32, 1058–1066 (2010).
Yeh, C. D., Richardson, C. D. & Corn, J. E. Advances in genome editing through control of DNA repair pathways. Nat. Cell Biol. 21, 1468–1478 (2019).
Chu, C. et al. Homologous recombination-mediated targeted integration in monkey embryos using TALE nucleases. BMC Biotechnol. 19, 1–10 (2019).
Bekker, E. I. et al. Phylogeography of Daphnia magna Straus (Crustacea: Cladocera) in Northern Eurasia: Evidence for a deep longitudinal split between mitochondrial lineages. PLoS ONE 13, 1–20 (2018).
Herbert, P. D. N. The population biology of Daphnia (Crustacea, Daphnidae). Biol. Rev. 53, 387–426 (1978).
Colbourne, J. K. et al. The ecoresponsive genome of Daphnia pulex. Science (80-. ). 331, 555–561 (2011).
Arao, T. et al. Production of genome-edited Daphnia for heavy metal detection by fluorescence. Sci. Rep. 10, 1–10 (2020).
Törner, K., Nakanishi, T., Matsuura, T., Kato, Y. & Watanabe, H. Genomic integration and ligand-dependent activation of the human estrogen receptor α in the crustacean Daphnia magna. PLoS ONE 13, 1–11 (2018).
Nakanishi, T., Kato, Y., Matsuura, T. & Watanabe, H. TALEN-mediated homologous recombination in Daphnia magna. Sci. Rep. 5, 1–10 (2015).
Kumagai, H., Nakanishi, T., Matsuura, T., Kato, Y. & Watanabe, H. CRISPR/Cas-mediated knock-in via nonhomologous end-joining in the crustacean Daphnia magna. PLoS ONE 12, 1–12 (2017).
Nakanishi, T., Kato, Y., Matsuura, T. & Watanabe, H. TALEN-mediated knock-in via non-homologous end joining in the crustacean Daphnia magna. Sci. Rep. 6, 1–7 (2016).
Pierce, A. J., Johnson, R. D., Thompson, L. H. & Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells service. Genes Dev. 13, 2633–2638 (1999).
Paliwal, S., Kanagaraj, R., Sturzenegger, A., Burdova, K. & Janscak, P. Human RECQ5 helicase promotes repair of DNA double-strand breaks by synthesis-dependent strand annealing. Nucl. Acids Res. 42, 2380–2390 (2014).
Kass, E. M., Helgadottir, H. R., Chen, C., Barbera, M. & Wang, R. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. 110, 5564–5569 (2013).
Johnson, N. M., Lemmens, B. B. L. G. & Tijsterman, M. A Role for the malignant brain tumour (MBT) domain protein LIN-61 in DNA double-strand break repair by homologous recombination. PLoS Genet. 9, e1003339 (2013).
Klüttgen, B., Dülmer, U., Engels, M. & Ratte, H. T. ADaM, an artificial freshwater for the culture of zooplankton. Water Res. 28, 743–746 (1994).
Kato, Y., Matsuura, T. & Watanabe, H. Genomic integration and germline transmission of plasmid injected into crustacean Daphnia magna Eggs. PLoS ONE 7, 1–7 (2012).
Kumagai, H., Matsuura, T., Kato, Y. & Watanabe, H. Development of a bicistronic expression system in the branchiopod crustacean Daphnia magna. Genesis 55, 1–6 (2017).
Izzatur, N., Ismail, B., Kato, Y., Matsuura, T. & Id, H. W. Generation of white-eyed Daphnia magna mutants lacking scarlet function. PLoS ONE 11, 1–11 (2018).
Adhitama, N., Matsuura, T., Kato, Y. & Watanabe, H. Monitoring ecdysteroid activities using genetically encoded reporter gene in Daphnia magna. Mar. Environ. Res. 140, 375–381 (2018).
Nakanishi, T., Kato, Y., Matsuura, T. & Watanabe, H. CRISPR/Cas-mediated targeted mutagenesis in Daphnia magna. PLoS ONE 9, 1–7 (2014).
Kato, Y. et al. Development of an RNA interference method in the cladoceran crustacean Daphnia magna. Dev. Genes Evol. 220, 337–345 (2011).
Honda, Y., Tsuchiya, K., Sumiyoshi, E., Haruta, N. & Sugimoto, A. Tubulin isotype substitution revealed that isotype combination modulates microtubule dynamics in C. elegans embryos. J. Cell Sci. 130, 1652–1661 (2017).
Panganiban, G. & Rubenstein, J. L. R. Developmental functions of the Distal-less/Dlx homeobox genes. Development 129, 4371–4386 (2002).
McGinnis, W. & Krumlauf, R. Homeobox genes and axial patterning. Cell 68, 283–302 (1992).
Kato, Y. et al. A 5′ UTR-overlapping LncRNA activates the male-determining gene doublesex1 in the crustacean Daphnia magna. Curr. Biol. 28, 1811-1817.e4 (2018).
Vriend, L. E. M., Jasin, M. & Krawczyk, P. M. Assaying break and nick-induced homologous recombination in mammalian cells using the DR-GFP reporter and cas9 nucleases. Methods Enzymol. 546, 175–191 (2014).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Schmidt, M. J. et al. Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases. Nat. Commun. 12 (2021).
Deininger, P. L. et al. Heavy metal exposure influences double strand break DNA repair outcomes. PLoS ONE 11, e0151367 (2016).
Shahar, O. D. et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucl. Acids Res. 42, 5689–5701 (2014).
Osaki, J. H., Espinha, G., Magalhaes, Y. T. & Forti, F. L. Modulation of RhoA GTPase activity sensitizes human cervix carcinoma cells to γ-radiation by attenuating DNA repair pathways. Oxid. Med. Cell. Longev. 2016 (2016).
OECD. Test guideline No. 202: Daphnia sp. acute immobilisation test. OECD Guidel. Test. Chem. (2004).
OECD. Test guideline No. 211: Daphnia magna reproduction test. OECD Guidel. Test. Chem. (2012).
Mao, Z., Bozzella, M., Seluanov, A. & Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. Comp. nonhomologous end Join. Homol. Recomb. Hum. cells 7, 1765–1771 (2008).
Mao, Zhiyong, Michael Bozzella, Andrei Seluanov, and V. G. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Int. J. Biochem. Mol. Biol. 1, 1–11 (2010).
Puchta, H. The repair of double-strand breaks in plants: Mechanisms and consequences for genome evolution. J. Exp. Bot. 56, 1–14 (2005).
Pardo, B., Gómez-González, B. & Aguilera, A. DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 66, 1039–1056 (2009).
Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015).
Lin, S., Staahl, B. T., Alla, R. K. & Doudna, J. A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, 1–13 (2014).
Charpentier, M. et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat. Commun. 9, 1–11 (2018).
Słabicki, M. et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 8, e1000408 (2010).
Gibbs, D. R. & Dhakal, S. Homologous recombination under the single-molecule fluorescence microscope. Int. J. Mol. Sci. 20, 1–16 (2019).
Certo, Michael T.Byoung Y.Ryu , James E. Annis, Mikhail Garibov, Jordan V. Jarjour, David J. Rawlings, A. M. S. Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods 8, 671–676 (2018).
Acknowledgements
pDRGFP plasmid was a gift from Maria Jasin (Addgene plasmid #26475). This work was supported by funding from the Japan Society for Promotion of Science (Grant Nos. 20H04923, 19H05423, 18H04884, 17H05602, 17K19236, and 17H01880). One of the authors (R.M.F) would like to acknowledge the Ministry of Education, Culture, Sports, Science and Technology (MEXT) for providing financial support under the Monbukagakusho/MEXT scholarship.
Author information
Authors and Affiliations
Contributions
R.M.F., N.A., Y.K., and H.W. conceived and designed the study. R.M.F. made the transgenic animal, performed the main experiments, and wrote the original draft. R.M.F., N.A., and Y.K edited the manuscript. H.W. supervised the experiments and finalized the manuscript. All authors discussed the results, commented, and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fatimah, R.M., Adhitama, N., Kato, Y. et al. Development of transgenic Daphnia magna for visualizing homology-directed repair of DNA. Sci Rep 12, 2497 (2022). https://doi.org/10.1038/s41598-022-06526-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06526-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
