
Abstract
Mpro, the main protease of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is essential for the viral life cycle. Accordingly, several groups have performed in silico screens to identify Mpro inhibitors that might be used to treat SARS-CoV-2 infections. We selected more than five hundred compounds from the top-ranking hits of two very large in silico screens for on-demand synthesis. We then examined whether these compounds could bind to Mpro and inhibit its protease activity. Two interesting chemotypes were identified, which were further evaluated by characterizing an additional five hundred synthesis on-demand analogues. The compounds of the first chemotype denatured Mpro and were considered not useful for further development. The compounds of the second chemotype bound to and enhanced the melting temperature of Mpro. The most active compound from this chemotype inhibited Mpro in vitro with an IC50 value of 1 μM and suppressed replication of the SARS-CoV-2 virus in tissue culture cells. Its mode of binding to Mpro was determined by X-ray crystallography, revealing that it is a non-covalent inhibitor. We propose that the inhibitors described here could form the basis for medicinal chemistry efforts that could lead to the development of clinically relevant inhibitors.
Similar content being viewed by others
Introduction
In December 2019, the Chinese health authorities reported the emergence of a disease, now referred to as COVID-19, that was declared a pandemic by the World Health Organization just a few months later1. The causative agent of COVID-19 is a novel coronavirus named SARS-CoV-22.
Similar to other coronaviruses, SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA virus. Its genome contains at least nine open reading frames (ORFs), of which ORF1a and ORF1b give rise to polyproteins pp1a and pp1ab, respectively3. A main protease (Mpro, also called 3C-like protease) and a papain-like protease (PLpro) cleave these polyproteins into characteristic nonstructural proteins4. Mpro is essential for the viral life cycle, as it is needed to generate the mature forms of most of the nonstructural proteins of the virus; it contains a cysteine-histidine dyad at its catalytic center and cleaves its substrates at sites comprising a glutamine followed by a residue with a small side chain. Mpro is highly conserved within the coronavirus family and is considered a good target for the development of drugs that could treat COVID-19 patients5,6,7.
Most efforts to identify Mpro inhibitors have focused on repurposing or modifying inhibitors of Mpro-related proteases8,9,10,11,12,13,14,15,16,17,18,19. Among the most potent of these inhibitors are GC376, boceprevir/telaprevir analogues and the PF-00835231 and PF-07321332 compounds; they are all irreversible inhibitors that form a covalent bond with the thiol group of the catalytically important residue Cys145. GC376, originally developed as a Norwalk virus 3CLpro inhibitor20,21, inhibits SARS-CoV-2 Mpro in vitro with a half maximal inhibitory concentration (IC50) of 30 ± 8 nM12,13. Analogues of boceprevir and telaprevir, two inhibitors approved for the treatment of hepatitis C, inhibit Mpro with IC50 values as low as 7.6 ± 0.1 nM in vitro and show activity in a mouse model of SARS-CoV-2 infection22. Finally, PF-00835231 and PF-07321332 are related compounds derived from an inhibitor of the Mpro of SARS-CoV-114,15. PF-00835231 requires continuous intravenous infusion as a prodrug to achieve effective doses in the plasma of human patients23, while PF-07321332 can be administered orally and promises to have a significant impact on the course of the COVID-19 pandemic15.
De novo Mpro inhibitors have been identified by either in silico or physical screens. Three in silico screening studies are particularly relevant here: two studies that ranked more than one billion compounds each, but did not validate the identified hits24,25, and a third study that ranked 6.5 million compounds and validated seven top compounds, of which the most potent exhibited an IC50 of 4.2 μM in vitro26. Among the studies that employed physical screens to identify Mpro inhibitors, one study screened a small library of chemical fragments for binding to Mpro by X-ray crystallography and identified several hits, which, however, were not developed further27. A second study screened a DNA-encoded library and identified compounds that inhibited Mpro with inhibition constants in the 35–45 nM range28. These inhibitors bound to Mpro covalently, as seen with the repurposed inhibitors, presumably because reactive, electrophilic compounds were included in the DNA-encoded library.
Covalent inhibitors often require extensive development to limit off-target effects and associated toxicity. Therefore, the development of non-covalent Mpro inhibitors, as an alternate strategy, is also worth pursuing. Here we characterized selected hits from the two very large in silico screens cited above24,25. The vast majority of the examined hits did not bind to or inhibit Mpro in vitro. However, a handful of hits were validated and these served as starting points for the development of a non-covalent Mpro inhibitor that was capable of inhibiting SARS-CoV-2 replication in tissue culture cells.
Results
Validation of putative SARS-CoV-2 Mpro inhibitors identified by in silico screens
To validate candidate inhibitors of SARS-CoV-2 Mpro identified by in silico screens, we utilized an in vitro protease activity assay. An expression plasmid encoding the Mpro protease with a C-terminal His-tag8 was introduced in the E. coli strain BL21-Gold (DE3) and recombinant Mpro was expressed and purified by affinity and size exclusion chromatography (Supplementary Fig. S1A). Protease activity was determined using a fluorescence resonance energy transfer (FRET) assay. The substrate peptide contained at its N-terminus a fluorescent dye (HiLyte-Fluor488) and at its C-terminus a quencher (QXL520); cleavage of the peptide by Mpro led to an increase in fluorescence intensity. The protease and fluorogenic substrate were assayed at 22 °C at final concentrations of 100 nM and 500 nM, respectively. Fluorescence intensity was measured every 10 min; in the absence of an Mpro inhibitor, fluorescence intensity increased linearly during the first 60 min of the reaction, whereas in the presence of the potent GC376 inhibitor12,13 no increase in fluorescence intensity was observed (Supplementary Fig. S1B).
We examined hits from two in silico screening studies24,25 that ranked the docking of molecules present in the REAL Space or ZINC chemical compound libraries29,30. In the first study, two in silico screens were performed, using two different three-dimensional structures to encompass the conformational flexibility of the active site of Mpro24. The first screen, referred to as screen 1A, used the structure of Mpro described by Jin et al.10 (pdb id: 6lu7) as target, while the second screen, referred to as screen 1B, used the structure of Dai et al.9 (pdb id: 6m0k) with minor changes in the side-chain rotamers of residues Ser46, Met49 and Cys145 to capture a more open conformation of the active site. The two screens docked the same library of compounds; nevertheless, comparison of the top 1,000 hits of each screen, revealed an overlap of only 12 compounds. The second study performed one in silico screen, hereafter referred to as screen 2, of 1.3 billion compounds25, using the structure of Mpro described by Jin et al.10 (pdb id: 6lu7) as target.
From the 3,808 top-ranking compounds of screen 1A, 195 compounds were manually selected for on-demand synthesis aiming for chemical diversity and drug-like features (Supplementary Table 1); whereas, from the 3,851 top-ranking compounds of screen 1B, 226 compounds were selected for on-demand synthesis (Supplementary Table 2). In addition, guided by the results of a crystallographic fragment screen27 that showed a fragment containing a nitrile group deep in the active site of Mpro (pdb id: 5r82), we identified all the nitrile-containing compounds among the top 20,000 hits of screens 1A and 1B. This list included 253 compounds, 45 of which were selected for on-demand synthesis (Supplementary Table 3). Twelve of the 15 top-ranking compounds from screen 1A were also selected for on-demand synthesis (Supplementary Table 4). Finally, from screen 2, eight of the 15 top-ranking hits were selected for on-demand synthesis (Supplementary Table 4).
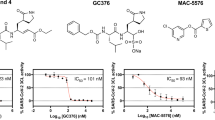
In total, we had 486 compounds synthesized and all these compounds were assayed at a final concentration of 40 μM for their ability to inhibit the protease activity of Mpro. Remarkably, only five compounds inhibited Mpro more than the pre-defined threshold level of 25% inhibition (Fig. 1A). The active compounds were: the diamino-quinazoline Z1037455358 (Fig. 1B), which is one of the 226 compounds selected from the 3,851 top-ranking compounds of screen 1B (Supplementary Table 2); the structurally-related nitriles Z637352244 and Z637352642 (Fig. 1B), which are two of the 45 nitrile-containing compounds selected from screens 1A and 1B (Supplementary Table 3); and the structurally-related dihydro-quinolinones ZINC000636416501 and ZINC000373659060 (Fig. 1B), which are two of the eight selected top-ranking compounds from screen 2 (Supplementary Table 4). All these five compounds were characterized further, as described below.

Development of Mpro inhibitors from in silico screening hits. (A) Graph showing the inhibitory activity of the five validated in silico screening hits tested at a final concentration of 40 μM in an in vitro protease-activity assay. After addition of the FRET substrate, fluorescence was acquired at 10 min intervals over 60 min. For each compound, the increase in fluorescence intensity was normalized to the DMSO control. GC376, a previously described Mpro inhibitor, served as a positive control. Blank, reaction omitting Mpro; RFU, relative fluorescence units. (B) Chemical structure of the five validated compounds. (C) Chemical structure of Z222979552, the most active dihydro-quinolinone compound obtained after two rounds of chemical structure similarity searches of the REAL space library of molecules. (D) Dose–response curves for compound Z222979552 examined at 0.4, 1, 3, 5, 10 and 20 μM final compound concentrations, in absence (solid lines) or presence (dashed lines) of 0.1 μg protein lysate. DMSO, GC376 and blank controls are as described above. (E) Thermal shift assay performed in the presence of DMSO or 20 μM of compounds Z222979552 or GC376. The graphs show the derivatives of the melting curves used to calculate the melting temperature of Mpro.
Characterization of the diamino-quinazoline and nitrile compounds
We first focused our efforts on compound Z1037455358, which contains a diamino-quinazoline core (Fig. 1B). The IC50 of this compound in the protease assay was 26 μM (Supplementary Fig. S2A). Analogues of Z1037455358 were identified using similarity and substructure searches of the REAL Space library of chemical compounds29; 108 of these analogues were selected for on-demand synthesis (Supplementary Table 5); however, none of the analogues were more potent than the parent compound in the biochemical assay. Therefore, we retained only the parent compound for further analysis.
Next, we examined the two structurally-related compounds Z637352244 and Z637352642, both of which contain a nitrile group (Fig. 1B). A nitrile group is also present in a fragment that was found to bind Mpro by crystallographic screening27. To identify more potent compounds, we selected 301 analogues for on-demand synthesis (Supplementary Table 6) and examined them at a final concentration of 40 μM for their ability to inhibit Mpro in the protease assay. Five analogues were more active than the original compounds (Supplementary Fig. S2B). We determined the IC50 concentrations of the original compounds and of these five analogues. The parent compounds Z637352244 and Z637352642 exhibited IC50 values of 22 and 223 μM, respectively (Supplementary Fig. S2C). Three analogues, Z56785964, Z637450230 and Z56786187, had IC50 values between 13–24 μM, whereas two analogues, Z2239054061 and Z637352638, had IC50 values of 6.7 and 7.5 μM, respectively (Supplementary Fig. S2C), representing a significant improvement over the parent compounds.
To further characterize the diamino-quinazoline and nitrile compounds, we examined their effect on the melting temperature of Mpro using a thermal shift assay (TSA). Briefly, Mpro, at a final concentration of 1 μM, was incubated for 20 min with the inhibitors, at a final concentration of 20 μM, and the melting temperature of Mpro was determined. Compounds that bind to Mpro should enhance its melting temperature31. Indeed, GC376 increased the melting temperature of Mpro by 19 °C (Supplementary Fig. S3A). Surprisingly, the parent nitrile-containing compounds decreased the melting temperature of Mpro, as did all their analogues, except for analogue Z637450230, which did not affect the melting temperature of Mpro (Supplementary Fig. S3A). The diamino-quinazoline compound Z1037455358 also decreased the melting temperature of Mpro (Supplementary Fig. S3B).
The decrease in the melting temperature of Mpro by the above compounds was of concern, as it could indicate that these compounds were destabilizing Mpro. Compounds that denature proteins non-specifically should lose inhibitory activity in the presence of excess carrier protein, since the latter can serve as a sink for compound sequestration. Indeed, the parent nitrile-containing compound Z637352244, its analogues Z56786187 and Z637450230, and the diamino-quinazoline compound Z1037455358, all lost activity, when the protease assay was performed in the presence of 1 μg cell lysate (Supplementary Fig. S2D). In contrast, GC376 maintained inhibitory activity in the presence of lysate and, interestingly, so did the dihydro-quinolinones compounds ZINC000636416501 and ZINC000373659060 (Supplementary Fig. S2D).
Characterization of the dihydro-quinolinone compounds
Compounds ZINC000636416501 and ZINC000373659060 are related to each other and contain a dihydro-quinolinone core (Fig. 1B). Encouraged by the fact that the activity of these two compounds was not affected by the presence of cell lysate, we obtained 157 analogues (Supplementary Table 7) and examined their ability to inhibit Mpro. Three analogues were significantly more potent than the parent compounds (Supplementary Fig. S4A). Specifically, compounds Z228770960, Z393665558 and Z225602086 had IC50 values of 4, 6 and 7.4 μM, respectively, whereas the parent compounds ZINC000373659060 and ZINC000636416501 had IC50 values of 58 and 93 μM, respectively (Supplementary Fig. S4B). Importantly, all three analogues retained their inhibitory activity against Mpro in the presence of cell lysate (Supplementary Fig. S4C).
The parent ZINC000636416501 and ZINC000373659060 compounds and their three active analogues were then examined for their ability to modulate the melting temperature of Mpro using the thermal shift assay. The parent compounds did not affect the melting temperature of Mpro (Supplementary Fig. S4D). However, the analogues increased the melting temperature of Mpro with the most active analogue, Z228770960, inducing an increase of 1.2 °C (Supplementary Fig. S4E). These results are consistent with the analogues forming stable complexes with Mpro.
To identify even more potent compounds, we performed a second round of analogue synthesis, using the first-round analogues Z228770960, Z393665558 and Z225602086 as starting points for structure similarity searches. A total of 113 second-round analogues were selected for on-demand synthesis (Supplementary Table 8), of which four were as potent or more potent than the analogues from the first round (Fig. 1C and Supplementary Fig. S5A). Specifically, compounds Z222979552, Z228166018, Z222977344 and Z222978028 had IC50 values of 1.0, 1.6, 2.0 and 5.8 μM, respectively, which inhibitory activity they retained in the presence of cell lysate (Fig. 1D and Supplementary Fig. S5B). The above four analogues also increased the melting temperature of Mpro by up to 1.2 °C (Fig. 1E and Supplementary Fig. S5C).
The first and second rounds of analogue synthesis resulted in about 15-fold and four-fold improvements in the IC50 values, respectively. However, additional chemical diversity of structures that we could gain from further rounds of compound similarity searches was limited. We therefore decided to characterize the most potent dihydro-quinolinone Mpro inhibitor, Z222979552 (IC50 = 1.0 μM), by determining its crystal structure in complex with Mpro and its antiviral activity in cell-based assays.
Structure of a dihydro-quinolinone inhibitor in complex with Mpro
The crystal structure of Mpro in complex with compound Z222979552 was solved at a resolution of 2.5 Å and shows the compound in the active site of Mpro (Fig. 2A; Table 1). Unlike the previously described high-affinity Mpro inhibitors, compound Z222979552 did not form a covalent bond with Mpro. Rather, binding was mediated by hydrogen bonds, pi-stacking and hydrophobic interactions. Hydrogen bonds were observed between the dihydro-quinolinone group and the side chains of Glu166, His163 and His172, as well as between the carbonyl group of the compound and the thiol group of Cys145 and the main chain of Glu166 (Fig. 2B). A T-type pi-stacking interaction was observed between the benzene ring and His41, while the dihydro-quinolinone group, the benzene group and the iodine atom participated in hydrophobic interactions with Asn142, Met49 and Met165, respectively (Fig. 2B).

Crystal structure of Mpro in complex with the dihydro-quinolinone compound Z222979552. (A) Surface representation of SARS-CoV-2 Mpro with secondary structure elements colored green and compound Z222979552 shown as a stick model. The omit electron density map corresponding to the ligand compound is shown (σ = 1.0). (B) Stick model of compound Z222979552 and of select residues at the active site of Mpro showing the key interactions between the protease and its inhibitor.
Inhibition of viral replication by a dihydro-quinolinone Mpro inhibitor
To further characterize compound Z222979552, we examined whether it could inhibit SARS-CoV-2 replication in Vero E6 cells. Consistent with its ability to inhibit Mpro in vitro, Z222979552 suppressed SARS-CoV-2 replication in Vero E6 cells, resulting in a more than a 100-fold decrease of SARS-CoV-2 titers at the highest concentration tested (Fig. 3A). In a second assay we monitored by immunofluorescence the presence of viral double-stranded RNA (dsRNA) in Vero E6 cells infected with SARS-CoV-2. Treatment of the cells with Z222979552 prevented the formation of dsRNA intermediates of SARS-CoV-2 RNA synthesis (Fig. 3B). We also performed a cytotoxicity assay with non-infected Vero E6 cells and observed that Z222979552 is not cytotoxic (Fig. 3C). Finally, we validated the cell-based assays using remdesivir as a positive control (Supplementary Fig. S6). Taken together, the above findings indicate that compound Z222979552 has antiviral activity in cells.

Compound Z222979552 reduces SARS-CoV-2 replication in Vero E6 cells in a dose-dependent manner. (A) SARS-CoV-2 titers upon treatment with compound Z222979552 or DMSO control. Viral titers are displayed as fifty-percent tissue culture infective dose (TCID50)/ml 24 h post-infection. TCID50 values correspond to the viral titers (log10 scale) required to kill fifty-percent of infected host cells. The experiment was performed in triplicate and the results are shown as means and standard deviations. (B) Immunofluorescence staining of infected Vero E6 cells for double-stranded RNA (dsRNA). The cells were treated with compound Z222979552 or DMSO and were stained 24 h post-infection. Green, dsRNA; blue, DAPI. One representative image out of three biological replicates is shown. (C) Z222979552-mediated cytotoxicity, determined using Vero E6 cells treated with the compound or DMSO for 24 h. The experiment was performed in triplicate and the results are shown as means and standard deviations.
Discussion
COVID-19 has had a significant impact on our society. The vaccines have significantly curtailed the spread of the pandemic32, but challenges still remain regarding vaccine acceptance by the public and durability of vaccine efficacy over time33,34. Thus, there continues to be an urgent need for novel medicines to treat COVID-19 and Mpro is considered a valuable target for the development of SARS-CoV-2 antivirals, because it is required for viral replication5,6,7. Equally importantly, Mpro is highly conserved in evolution5, which means that inhibitors developed against Mpro of one coronavirus may inhibit replication of multiple members of the coronavirus family. Indeed, SARS-CoV-2 Mpro inhibitors entering the clinic were derived from compounds that were originally developed as inhibitors of SARS-CoV-1 Mpro, but whose development was, unfortunately, halted, when the SARS-CoV-1 epidemic waned14,15,23.
All the inhibitors described here were obtained from the REAL Space or the ZINC chemical compound libraries, which together encompass more than 20 billion make-on-demand compounds29,30. Despite their large size, these two libraries represent a very small part of the universe of chemical space. Therefore, we anticipate that the compounds that we have identified can serve as starting points for medicinal chemistry efforts, particularly because these compounds have drug-like features and activity in cell-based assays. Moreover, their mechanism of action is understood from the crystal structure of compound Z222979552 in complex with Mpro.
One interesting feature of the inhibitors described here is that they are non-covalent, in contrast to the vast majority of previously described Mpro inhibitors, including the PF-07321332 inhibitor, which form covalent bonds with the catalytic cysteine. The therapeutic significance of having a non-covalent Mpro inhibitor is unclear at this moment, but the possibility of developing such inhibitors to complement the covalent inhibitors can only offer more therapeutic opportunities.
Methods
Protein expression and purification of SARS-CoV-2 Mpro
The Mpro construct8 provided by Rolf Hilgenfeld was transformed into E. coli strain BL21-Gold (DE3) (Agilent). Transformed clones were picked to prepare pre-starter cultures in 2 mL YT medium with ampicillin (100 μg/ml), at 37 °C for 8 h. The pre-starter culture was then inoculated into fresh 120 mL YT medium with ampicillin (100 μg/ml) and incubated at 37 °C overnight. The next day, the starter culture was inoculated into 1,600 mL YT medium with ampicillin (100 μg/ml) and incubated at 37 °C until OD600 reached a value between 0.6 and 0.8. 1 mM isopropyl-D-thiogalactoside (IPTG) was then added to induce the overexpression of Mpro at 30 °C for 5 h. The bacteria were harvested by centrifugation at 8260 × g, 4 °C for 15 min, resuspended in Binding Buffer (25 mM BTP [pH 6.8]; 300 mM NaCl; 2 mM DTT; 1 mM EDTA; 3% DMSO) and then lysed using an Emulsiflex-C3 homogenizer (Avestin). The lysate was clarified by ultracentrifugation at 137,088 × g, 4 °C for 1 h and loaded onto a HisTrap FF column (Cytiva) using an Äkta protein purification system (Cytiva). When all the supernatant containing Mpro had passed through the column, the column was washed with 80 mL binding buffer to remove non-specifically bound proteins and then Mpro was eluted using an imidazole gradient (0–500 mM) in Binding Buffer. The Mpro fractions were concentrated using 3 kDa Amicon Ultra Centrifugal Filters (Merck Millipore) and the Mpro protein was further purified by size exclusion chromatography using a HiLoad Superdex 200 column (Cytiva) attached to a SMART protein purification system (Pharmacia).
Compounds
We initially selected for on-demand synthesis 485 compounds that were identified by two in silico screening studies as putative Mpro inhibitors; these compounds are referred to as parent compounds (Supplementary Tables 1–4). After evaluating the activity of the parent compounds in vitro, we selected an additional 686 compounds that were analogues of the few active parent compounds (Supplementary Tables 5–8). All the above compounds were purchased from Enamine, their purity was ≥ 90% and they were synthesized on-demand. The compounds were dissolved in DMSO at a concentration of 2 mM and were stored at -20 °C. GC376 was purchased from BPSBioscience.
SARS-CoV-2 Mpro protease activity assay
Mpro protease assays were performed in duplicate in Falcon 384-well optilux flat bottom, TC-treated microplates (Corning) in a final volume of 10 μL. Mpro, at a final concentration of 100 nM, was preincubated for 20 min at room temperature (RT) with the compounds in assay buffer (5 mM HEPES pH 7.5, 0.1 mg/mL BSA, 0.01% Triton, 2 mM DTT) under gentle agitation. The FRET substrate, HiLyte-Fluor488-ESATLQSGLRKAK(QXL520)-NH2 (Eurogentec), was then added at a final concentration of 500 nM and incubated for 2 min at RT with gentle agitation prior to the start of fluorescence measurement. Compounds and FRET substrate were dispensed with an acoustic liquid dispenser (Gen5-Acoustic Transfer System; EDC Biosystems). The fluorescence intensity was measured kinetically for 7 cycles, every 10 min at 22 °C, using a Spark 10 M microplate reader (Tecan) and excitation and emission wavelengths of 485 and 528 nm, respectively.
SARS-CoV-2 Mpro thermal shift binding assay
Thermal shift assays were performed in duplicate in LightCycler 480 multiwell plates 96, white (Roche) in a final volume of 20 μL. Mpro protease, at a final concentration of 1 μM, was preincubated for 20 min at room temperature (RT) under gentle agitation with the compounds (final concentration: 20 μM) in assay buffer (10 mM HEPES pH 7.5; 150 mM NaCl). Protein unfolding was monitored with 5X SYPRO Orange (Sigma) binding dye. Compounds and SYPRO Orange were dispensed with an acoustic liquid dispenser (Gen5-Acoustic Transfer System; EDC Biosystems). Fluorescence (excitation wavelength: 465 nm; emission wavelength: 580 nm) was measured over a temperature gradient ranging from 20 to 95 °C, with incremental steps of 0.05 °C/s and 11 acquisitions per °C. The melting curves and peaks were obtained using the melting temperature (Tm) calling analysis of the LightCycler 480 Software (release 1.5.1.62; Roche; https://lifescience.roche.com/en_ch/brands/realtime-pcr-overview.html#software).
Crystallization and model building
Purified Mpro was concentrated to 5.6 mg/mL and crystallized using the hanging vapor diffusion method. Plate-shaped crystals grew in star-like clusters within one week in drops mixed in a 1:1 ratio with reservoir solution (0.1 M Tris [pH 8.0], 25% (v/v) PEG-3350 and 0.2 M LiCl) at 293 K. For soaking and cryoprotection, the crystal clusters were moved into 2 μL reservoir solution supplemented with 25% ethylene glycol. Then, 0.4 μL inhibitor solution was added resulting in a final compound concentration of 10 mM. Soaking was allowed to proceed for 2 h, after which single crystals were broken off the clusters and flash-frozen in liquid nitrogen. Diffraction data was collected at the Swiss Light Source (SLS), beamline PXI (X06SA) with λ = 1.000 Å at T = 100 K. The data were processed using the XDS package35 up to a resolution of 2.5 Å. The structure was solved using Phaser35 from the Phenix suite (v.19.1.) with 6wqf as a search model. In Phenix, the model was iteratively refined using Phenix Refine36 with manual adjustments done in Coot6 (v.0.9.3). The three-dimensional model and molecular restraints of the ligand were generated using eLBOW7 from the Phenix suite37. According to the Ramachandran statistics for the final model 95.38% of residues are in favored regions, 3.96% are in allowed regions, and 0.77% of residues are outliers. All figures concerning structural data were prepared with OpenSource Pymol by Schrodinger (v.1.20; https://github.com/schrodinger/pymol-open-source).
Viral replication assays
Vero E6 cells (kindly provided by Doreen Muth, Marcel Müller and Christian Drosten, Charité, Berlin, Germany) were propagated in Dulbecco's modified EMEM (DMEM), supplemented with 10% heat-inactivated fetal bovine serum, 1% nonessential amino acids, 100 µg/mL streptomycin, 100 IU/mL penicillin and 15 mM HEPES at 37 °C in a humidified incubator with 5% CO2. SARS-CoV-2 (SARS-CoV-2/München-1.1/2020/929, kindly provided by Daniela Niemeyer, Marcel Müller and Christian Drosten) passage 1 was used for infection of the cells.
Two million Vero E6 cells were plated per well of a 96-well plate; 24 h later, the cells were infected with SARS-CoV-2/München-1.1/2020/929 passage 1 at an MOI of 0.1 for 1 h at 37 °C and then washed 3 times with PBS. Z222979552 and remdesivir (or respective volumes of DMSO) were added to cells in following concentrations: 0, 1, 2.5, 5, 10, 20 and 40 µM. 24 h post-infection infectious supernatant was serially diluted and the 50% tissue culture infectious dose (TCID50) per ml was determined 96 h later using the Spearman-Kärber algorithm, as previously described38. Cytotoxic effects of Z222979552, remdesivir or the corresponding DMSO volumes were determined using the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega).
Vero E6 cells infected with SARS-CoV-2 were fixed with 4% formalin and then permeabilized in PBS supplemented with 50 mM NH4Cl, 0.1% (w/v) Saponin and 2% (w/v) Bovine Serum Albumin. The cells were stained using a mouse monoclonal antibody against dsRNA (SCICONS, clone J2) and Alexa-Fluor 488-labeled donkey-anti mouse IgG (H + L) (JacksonImmuno) secondary antibody. Images were acquired with an EVOS FL Auto 2 Imaging System, using a 10 × objective lens and processed using Fiji software packages39 version 1.53j (https://imagej.net/software/fiji/downloads) and assembled with the FigureJ plugin40 version 1.36 (https://imagejdocu.list.lu/plugin/utilities/figurej/start).
Data availability
Atomic coordinates and structure factors for the crystal structure of Mpro in complex with Z222979552 have been deposited in the Protein Data Bank under accession code 7P2G.
References
Zhu, N. et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020).
Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020).
Chan, J. F. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. https://doi.org/10.1080/22221751.2020.1719902 (2020).
Chen, Y., Liu, Q. & Guo, D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 92, 418–423 (2020).
Ullrich, S. & Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377 (2020).
Wu, C. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10, 766–788 (2020).
Vandyck, K. & Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 49, 36–40 (2021).
Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368, 409–412 (2020).
Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331–1335 (2020).
Jin, Z. et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582, 289–293 (2020).
Jin, Z. et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 27, 529–532 (2020).
Vuong, W. et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 11, 4282 (2020).
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020).
Hoffman, R. L. et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 63, 12725–12747 (2020).
Owen, D. R. et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science https://doi.org/10.1126/science.abl4784 (2021).
Sacco, M. D. et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L.. Sci. Adv. 6, 1005 (2020).
Riva, L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 586, 113–119 (2020).
Günther, S. et al. X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372, 642–646 (2021).
Zaidman, D. et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 Mpro inhibitor. Cell Chem. Biol. https://doi.org/10.1016/j.chembiol.2021.05.018 (2021).
Kim, Y. et al. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 86, 11754–11762 (2012).
Kim, Y. et al. Broad-spectrum inhibitors against 3C-like proteases of feline coronaviruses and feline caliciviruses. J. Virol. 89, 4942–4950 (2015).
Qiao, J. et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science 371, 1374–1378 (2021).
de Vries, M. et al. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 2, 10084 (2021).
Gorgulla, C. et al. A multi-pronged approach targeting SARS-CoV-2 proteins using ultra-large virtual screening. iScience 24, 102021 (2021).
Ton, A., Gentile, F., Hsing, M., Ban, F. & Cherkasov, A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol. Inf. 39, e2000028 (2020).
Clyde, A. et al. High throughput virtual screening and validation of a SARS-CoV-2 main protease non-covalent inhibitor. bioRxiv https://doi.org/10.1101/2021.03.27.437323 (2021).
Douangamath, A. et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 11, 5047 (2020).
Srinivas, C. S. et al. DNA-encoded chemistry technology yields expedient access to SARS-CoV-2 Mpro inhibitors. PNAS 118, e2111172118 (2021).
Enamine REAL. Available online: https://enamine.net/library-synthesis/real-compounds.
Sterling, T. & Irwin, J. J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015).
Lo, M.-C. et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 332, 153–159 (2004).
Haas, E. J. et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: an observational study using national surveillance data. Lancet 397, 1819–1829 (2021).
Forman, R., Shah, S., Jeurissen, P., Jit, M. & Mossialos, E. COVID-19 vaccine challenges: What have we learned so far and what remains to be done?. Health Policy 125, 553–567 (2021).
Falsey, A. R. et al. SARS-CoV-2 neutralization with BNT162b2 vaccine dose 3. N. Engl. J. Med. 15, 10058 (2021).
Kabsch, W. X. D. S. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 125–132 (2010).
Adams, P. D. et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Moriarty, N. W., Grosse-Kunstleve, R. W. & Adams, P. D. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 65, 1074–1080 (2009).
George, V. G., Hierholzer, J. C. & Ades, E. W. Cell culture. In Virology Methods Manual (eds Mahy, B. W. J. & Kangro, H. O.) 3–24 (Academic Press, 1996).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Mutterer, J. & Zinck, E. Quick-and-clean article figures with FigureJ. J. Microsc. 252, 89–91 (2013).
Acknowledgements
The authors thank Rolf Hilgenfeld for providing the plasmid expressing Mpro in bacteria and the beam line scientists at the Swiss Light Source for help with collection of X-ray diffraction data. Funding for this project was provided by the Fondation Aclon and by overhead funds from the ERC Project ONIDDAC (Principal Investigator: T.D.H.).
Author information
Authors and Affiliations
Contributions
G.G.R. and M.O. performed the protease and thermal shift assays; S.R. determined the crystal structure of Mpro with bound inhibitor; A.K. performed the viral replication assays; S.B. and L.T. expressed and purified Mpro protein; C.G. and H.A. analyzed and provided in silico screening hits; P. M. and R. G. selected hits and analogues for on-demand synthesis; V.S.D., V.T., G.G.R. and T.D.H designed and supervised experiments; T.D.H. conceived the study; all authors contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rossetti, G.G., Ossorio, M.A., Rempel, S. et al. Non-covalent SARS-CoV-2 Mpro inhibitors developed from in silico screen hits. Sci Rep 12, 2505 (2022). https://doi.org/10.1038/s41598-022-06306-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06306-4
This article is cited by
-
Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR
Nature Reviews Drug Discovery (2024)
-
Computational approaches streamlining drug discovery
Nature (2023)
-
Structural analysis of the coronavirus main protease for the design of pan-variant inhibitors
Scientific Reports (2023)
-
SARS-CoV2 billion-compound docking
Scientific Data (2023)
-
Rapid planning and analysis of high-throughput experiment arrays for reaction discovery
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
