
Abstract
The disaccharide trehalose is essential for viability of Mycobacterium tuberculosis, which synthesizes trehalose de novo but can also utilize exogenous trehalose. The mycobacterial cell wall encompasses two permeability barriers, the cytoplasmic membrane and the outer mycolic acid-containing mycomembrane. The ABC transporter LpqY–SugA–SugB–SugC has previously been demonstrated to mediate the specific uptake of trehalose across the cytoplasmic membrane. However, it is still unclear how the transport of trehalose molecules across the mycomembrane is mediated. In this study, we harnessed the antimycobacterial activity of the analogue 6-azido trehalose to select for spontaneous resistant M. tuberculosis mutants in a merodiploid strain harbouring two LpqY–SugA–SugB–SugC copies. Mutations mediating resistance to 6-azido trehalose mapped to the proline–proline–glutamate (PPE) family member PPE51 (Rv3136), which has recently been shown to be an integral mycomembrane protein involved in uptake of low-molecular weight compounds. A site-specific ppe51 gene deletion mutant of M. tuberculosis was unable to grow on trehalose as the sole carbon source. Furthermore, bioorthogonal labelling of the M. tuberculosis Δppe51 mutant incubated with 6-azido trehalose corroborated the impaired internalization. Taken together, the results indicate that the transport of trehalose and trehalose analogues across the mycomembrane of M. tuberculosis is exclusively mediated by PPE51.
Similar content being viewed by others


Introduction
Tuberculosis (TB) is among the major infectious diseases that affects several million people every year. According to the World Health Organization TB Report 2019, there are about 1.5 million deaths every year due to TB1. TB is caused by Mycobacterium tuberculosis (Mtb), a human pathogen that belongs to the class of actinobacteria2. The Mtb cell envelope consists of (from inside to outside) the cytoplasmic membrane, the mycolyl-arabinogalactan-peptidoglycan layer (mAGP), and a capsular layer mainly comprising α-glucan polysaccharides. The envelope confers physical robustness and protection against physicochemical stress and is also important for the virulence of the bacteria3. The mycolic acids are long chain α-branched β-hydroxy fatty acids that are either covalently bound to arabinogalactan, which in turn is linked to peptidoglycan, or esterified to sugars such as trehalose to give rise to the glycolipids, trehalose monomycolates (TMM) or trehalose dimycolates (TDM)3. Many components in the biosynthetic pathways of mycolic acids represent lucrative drug targets. TDM, also known as cord factor, is essential for the growth and survival of Mtb4. TDM provides virulence, stimulates host immune responses, and contributes to the interception of phagosomal maturation inside macrophages5. The arabinogalactan-bound mycolates form the inner leaflet and the trehalose mycolates form the outer leaflet of a lipid bilayer-like structure known as the mycomembrane that bears structural resemblance to the outer membrane of Gram-negative bacteria. The mycomembrane represents an efficient permeability barrier that contributes to the high intrinsic resistance of Mtb towards many antibiotics6.
Trehalose is crucial for the composition of mycobacterial cell envelope. De novo biosynthesis of trehalose in mycobacteria occurs through the OtsA–OtsB2 and the TreY–TreZ pathway7,8,9. TMM is synthesized in the cytoplasm by 6-O-mycoloylation of trehalose catalysed by Pks13 and is then transported to the mycomembrane through the mycobacterial membrane protein large 3 (MmpL3) transporter10. In the mycomembrane, the mycolic acid moiety from one TMM molecule is either transferred to another TMM molecule forming TDM, or it is transferred to arabinogalactan polysaccharides forming arabinogalactan-linked mycolates. These transfers are catalysed by the Antigen 85 (Ag85A, Ag85B, and Ag85C) complex leading to the concomitant release of free trehalose3. The free trehalose molecules are then recycled back to the cytoplasm through the LpqY–SugA–SugB–SugC ABC transporter located in the cytoplasmic membrane11. In addition to intrinsic de novo formation, Mtb can also utilize exogenous trehalose. Some flexibility regarding substrate specificity in the enzymatic machinery involved in trehalose uptake and metabolism also allows internalization of several synthetically altered trehalose analogues in a LpqY–SugA–SugB–SugC dependent manner and their subsequent metabolism and incorporation into the mycomembrane. These altered trehalose analogues might harbor structural modifications that allow conjugation with fluorescence probes employing biorthogonal chemistry12,13. However, while the LpqY–SugA–SugB–SugC ABC transporter exclusively mediates the uptake of trehalose across the cytoplasmic membrane, the route of trehalose transport across the mycomembrane is still unclear.
In this study, we harnessed the growth inhibitory properties of 6-azido trehalose (6-TreAz), which has anti-mycobacterial activity at high concentration14, for the isolation of spontaneously resistant mutants to identify genes potentially involved in trehalose uptake and its metabolism. This led to the identification of a member of the proline-proline-glutamate (PPE) family proteins. The Mtb genome encodes a high number of proteins comprising characteristic conserved proline-glutamate (PE) and PPE repeat motifs in their N-terminal region. There are 99 PE proteins and 69 PPE proteins encoded in the genome of the laboratory strain of Mtb H37Rv. PE/PPE proteins have been reported to be involved in modulation of the host immune response and in interaction with other bacteria15,16. Many PE/PPE proteins are secreted or cell surface-exposed and are substrates for type VII secretion systems17,18, with which they have co-evolved19. Some studies have previously suggested a role of certain PE/PPE proteins in nutrient uptake in Mtb20. Very recently, it has been shown that PPE51 is an essential integral mycomembrane protein responsible for transport of nutrients such as glucose, maltose and glycerol, as well for certain low-molecular-weight experimental drugs across the mycomembrane21,22. In the present study, we investigated the mechanism of trehalose uptake in Mtb in an unbiased approach and discovered a role in this process of the PPE51 mycomembrane transporter.
Results
6-Azido trehalose inhibits Mtb growth at higher concentrations
TMM is produced by 6-O-mycolylation of trehalose in the cytoplasm and is transported to the mycomembrane, where it is used to generate TDM with release of free trehalose. Differently modified trehalose analogs can also be taken up by Mtb and can undergo mycolylation. These modified TDMs and TMMs can be metabolically incorporated into the mycomembrane12,13. Modifications can occur at different positions of the trehalose molecule. 6-Azido trehalose (6-TreAz) has an azide group (N3) at the sixth carbon position of one of the glucose moieties in trehalose and has been used to label Mtb species (Fig. 1A). At concentrations ranging from 25 to 150 µM, 6-TreAz can be used as a bioorthogonal marker to label mycobacterial cells after conjugation with fluorescent moieties via click chemistry13. However, when incubated with increased concentrations, i.e. > 1 mM, we found that it inhibits growth of Mtb wild-type strain H37Rv on 7H10 solid medium. To further analyze this growth inhibition, we also tested the Mtb ΔlpqY-sugC mutant that lacks the complete LpqY–SugA–SugB–SugC (Rv1235–1238) ABC transporter, which recycles back the free trehalose that is produced by the antigen 85 complex when using TMM as the substrate11. The ΔlpqY-sugC deletion strain was able to grow on 7H10 solid medium containing 1 mM 6-TreAz (Fig. 1B). This highlights that the antibacterial effect of 6-TreAz is uptake-dependent and that LpqY–SugA–SugB–SugC not only tolerates trehalose but also 6-TreAz as a substrate. The growth inhibition of wild-type strain H37Rv by 6-TreAz likely results from the depletion of TDM in the mycomembrane as Mtb could not conjugate an additional mycolic acid to TMM because the 6′ position is occupied by the azide group, as already hypothesized previously for Mycobacterium smegmatis23. A previous study suggested that 6-TreAz also inhibits the trehalose synthase TreS when Mtb is grown under nutrient-limited biofilm conditions24. However, since TreS has been shown to be fully dispensable for growth of Mtb under nutrient-proficient in vitro culture conditions8,25,26, it is unlikely that potential inhibition of TreS contributes relevantly to the growth inhibitory effect of 6-TreAz as observed in our study.

6-Azido trehalose (6-TreAz) inhibits Mtb growth at higher concentrations. (A) Structure of trehalose and 6-TreAz. (B) Growth inhibition of Mtb H37Rv wild-type (WT), the ΔlpqY-sugC mutant and the complemented mutant ΔlpqY-sugC pMV306(Kan)::Rv1235–1238 on Middlebrook 7H10 agar containing 1 mM 6-TreAz or just solvent control (1% DMSO = w/o 6-TreAz). Equal amounts of diluted cell suspensions of each strain were plated onto both types of agar and incubated for 3 weeks at 37 °C.
Spontaneous resistant mutants reveal a candidate responsible for extracellular trehalose uptake in Mtb
In order to identify genes potentially involved in transport and metabolism of trehalose in Mtb, we isolated spontaneous resistant mutants after growth on 6-TreAz-containing solid medium. In order to avoid isolating clones with loss-of-function mutations in the LpqY–SugA–SugB–SugC ABC transporter, which we have already shown to mediate 6-TreAz resistance, we employed a merodiploid strain that contains an additional copy of the lpqY-sugC gene cluster (Rv1235–1238) in its genome, which was generated by electroporating the integrative plasmid pMV306(Kan)::Rv1235–1238 into wild-type H37Rv (Supplementary Fig. 1). This additional copy of the gene cluster would compensate for any loss-of-function mutations in the endogenous locus and will minimize the chance of resistant mutants occurring from mutations in this gene cluster. After 4 weeks of incubation, 6-TreAz spontaneous resistant mutants were obtained at a frequency of 10–7 (Fig. 2). Whole genome sequencing of five selected clones revealed that independent non-synonymous mutations in ppe51 (Rv3136) have occurred in three 6-TreAz-resistant mutants (nucleotide 661T→C, Leu204Pro; 41 G→C, Arg14Pro; 280 G→A, Ala94Thr). The two other spontaneous resistant clones harboured a non-synonymous mutation in the eccC5 gene (Rv1783) (nucleotide 3278 C→A, Pro1093Gln). All clones except one ppe51 mutant harboured additional mutations in different genes involved in phthiocerol dimycocerosate (PDIM) biosynthesis (ppsA, ppsB, fadD26; Table 1). We also generated complemented strains for some of these spontaneous resistant mutants constitutively expressing wild-type copies of eccC5 or ppe51, respectively, from an integrative single-copy plasmid to check for their resistance on medium containing 6-TreAz, and we observed that the sensitivity of the resistant mutants was at least partially restored (Supplementary Fig. 2). These results indicate that the observed resistance phenotype could be clearly attributed to eccC5 or ppe51, respectively. PPE protein family members have been shown to participate in uptake of nutrients through Mtb cell envelope, and PPE51 has been referred to as a mycomembrane-associated protein27. From previous studies it has been shown that EccC5, an ATP-binding protein belonging to the FtsK/SpoIIIE-like protein family, is required for secretion of ESX-5 specific substrates28. Further, it was shown that numerous PE and PPE proteins are secreted in an ESX-5-dependent manner in Mycobacterium marinum18. These findings suggest that PPE51 could be a potential gateway for the uptake of exogenous trehalose across the mycomembrane. The observed mutation in EccC5 might impair the ESX-5-mediated secretion of PPE51 and/or its proper translocation and insertion into the mycomembrane.

Isolation of spontaneous 6-TreAz-resistant Mtb mutants. 10 µl aliquots of tenfold-serially diluted cell suspensions of Mtb H37Rv wild-type (WT), the merodiploid strain Mtb H37Rv pMV306::Rv1235–1238, the ΔlpqY-sugC gene deletion mutant, the complemented mutant ΔlpqY-sugC pMV306::Rv1235–1238 and spontaneous 6-TreAz-resistant mutant clones were spotted each onto Middlebrook 7H10 agar containing 1 mM 6-TreAz or just solvent control (1% DMSO = w/o 6-TreAz). Plates were incubated for 3 weeks at 37 °C. For genotypes of mutant clones 1–4 and 6 refer to Table 1.
Bacterial growth of a Mtb Δppe51 deletion mutant in minimal medium with glucose and trehalose supplementation
To further analyze the function of PPE51 in trehalose uptake, we generated a site-specific gene deletion mutant in ppe51 (Rv3136) using the specialized transduction method. The Δppe51 deletion strain was subjected to whole genome sequencing to confirm its genotype. In addition to the ppe51 gene deletion, the sequencing also revealed a second-site non-synonymous mutation in the hypothetical gene Rv2662 (242 G→A, leading to amino acid substitution C81Y) and a base insertion in the mycocerosic acid synthase gene mas causing a frame shift (+ 1 bp insertion (+ a) at nucleotide position 5560 out of 6336 bp). This mas gene is responsible for the synthesis of multi-methyl branched mycocerosic acids, which form a segment of the phthiocerol dimycocerosate (PDIM) structure. PDIM loss is very common during in vitro culturing of Mtb strains29. Although growth at 1 mM 6-TreAz was weaker as compared to the ΔlpqY-sugC deletion strain, the Δppe51 deletion mutant exhibited a clear resistance toward this azidosugar (Fig. 3), corroborating the phenotype of the spontaneous resistant mutants and proving the role of ppe51 in resistance toward 6-TreAz.

6-TreAz resistance of the Mtb Δppe51 deletion mutant. 10 µl aliquots of tenfold-serially diluted cell suspensions of Mtb H37Rv wild-type, the ΔlpqY-sugC gene deletion mutant and the Δppe51 gene deletion mutant were spotted onto Middlebrook 7H10 agar containing 1 mM 6-TreAz and incubated for 3 weeks at 37 °C. The Δppe51 gene deletion mutant is unable to utilize glucose as will be demonstrated in Fig. 4 and therefore exhibits a growth defect on the used solid medium that contains glycerol and glucose as main carbon sources.
In addition to the Δppe51 deletion strain, we also generated a corresponding complemented strain Δppe51 pMV361::ppe51 that constitutively expresses the wild-type ppe51 gene from an integrative single-copy plasmid and could therefore compensate for the loss of transporter function. Next, we analyzed the growth of the Δppe51, Δppe51 pMV361::ppe51 and the H37Rv wild-type strain in minimal medium supplemented with increasing concentrations (0–5 mM) of trehalose or glucose to study the effect on growth of Mtb. The wild-type and the Δppe51 pMV361::ppe51 complemented strain showed a gradual increase in growth with increase in the concentrations of carbon sources provided. The Δppe51 deletion strain was not able to grow on the minimal medium with or without supplementation of glucose or trehalose, which strongly suggests a proposed function of PPE51 as a transporter for both sugars (Fig. 4). In addition, we also measured growth of H37Rv wild-type, the Δppe51 deletion strain and the Δppe51 pMV361::ppe51 complemented mutant in 7H9 medium + 0.05% tyloxapol (referred to as 7H9 limited medium) containing either trehalose or glycerol, respectively. Cultures grown in 7H9 + 0.05% tyloxapol medium supplemented with 10% ADS and 0.5% glycerol (referred to as 7H9 complete medium) were used as positive control. The Δppe51 deletion strain did not grow in 7H9 limited medium containing trehalose but displayed normal growth in 7H9 complete medium. Complementation of this mutant restored growth similar to the wild-type strain (Fig. 5), demonstrating that the growth defect of the mutant was unequivocally attributed to the loss of ppe51. Previous studies have shown that a PDIM-proficient ppe51 deletion strain was unable to utilize glycerol as a carbon source, whereas a PDIM-deficient ppe51 deletion strain harboring a loss-of-function second-site mutation in fadD26 could grow on both glucose and glycerol as the sole carbon source22. In contrast to glucose and trehalose, we observed that our Δppe51 mutant was able to utilize glycerol as the sole carbon source when cultivated in 7H9 limited medium containing 0.5% glycerol (Fig. 5). This could result from an impaired formation or complete loss of PDIM due the observed spontaneous second-site frame shift mutation in the mas gene that might allow the Δppe51 deletion mutant to use glycerol for growth in a PPE51-independent manner. However, no growth of the Δppe51 deletion mutant was observed with trehalose as the sole carbon source under these conditions, indicating that trehalose was unable to enter the cells in a PPE51-independent manner despite the defect in PDIM biosynthesis.

Growth of the Mtb Δppe51 deletion mutant in minimal medium with glucose or trehalose as the sole carbon source. Cells of Mtb H37Rv wild-type (WT) (red bars), the Δppe51 mutant (green bars) and the complemented mutant Δppe51 pMV361::ppe51 (blue bars) were cultivated in minimal medium containing increasing concentrations (0–5 mM) of trehalose (A) or glucose (B), respectively. Growth was determined using the resazurin microplate assay and normalized to cells of Mtb H37Rv wild-type grown in Middlebrook 7H9 complete medium (= 100% growth) or medium only control (= 0% growth). Values are means of triplicates ± SD. Growth defects of the Δppe51 mutant observed at 5 mM concentration of both sugars compared to wild-type or the complemented mutant was statistically significant (p < 0.005 as determined using unpaired t test).

Growth of the Mtb Δppe51 deletion mutant in 7H9 limited medium with glycerol or trehalose as the sole carbon source. Cells of Mtb H37Rv wild-type (WT), the Δppe51 mutant and the complemented mutant Δppe51 pMV361::ppe51 were cultivated in 7H9 limited medium containing 0.5% (v/v) glycerol (blue bars), 10 mM trehalose (red bars) or no additional carbon source (brown bars). Strains grown in 7H9 complete medium served as positive controls (green bars). Growth was determined using the resazurin microplate assay and normalized to cells of Mtb H37Rv wild-type grown in 7H9 complete medium (= 100% growth) or medium only control (= 0% growth). Values are means of two independent experiments each performed in triplicates ± SD. Growth differences between the indicated groups were statistically significant (***p < 0.005 as determined using unpaired t test).
The Δppe51 deletion mutant shows reduced fluorescence intensity when labelled with 6-TreAz and AF488 DIBO
Mtb strains are amenable to labeling with various trehalose analogs employing click chemistry to study the roles of different glycoproteins, membrane lipids and glycan molecules in the cellular envelope13,30,31. Copper-free click chemistry is based on the principle of bioorthogonal labelling which involves alkyne-azide cycloaddition. Mtb cells cultivated with azide-functionalized metabolic substrates can be conjugated to a cyclooctyne-functionalized probe such as dibenzocyclooctynes (DIBO). In this study, to corroborate the defect in uptake of trehalose and trehalose analogues, Mtb wild-type (positive control), the ΔlpqY (Rv1235) gene deletion mutant (negative control) and the Δppe51 gene deletion mutant were grown in 7H9 medium containing 100 µM 6-TreAz and were subsequently labelled with Alexa Fluor 488 DIBO Alkyne. Mean fluorescence intensity (MFI) was measured for 50,000 cells via FACS. It was observed that the Δppe51 mutant had reduced MFI compared to wild-type and almost similar MFI when compared to that of the ΔlpqY deletion strain indicating that uptake of 6-TreAz in wild-type is dependent both on PPE51 and LpqY-SugABC (Fig. 6). In the absence of PPE51 or LpqY, 6-TreAz could not enter the cytoplasm and hence AF488 DIBO Alkyne could not conjugate with azide molecules. The secreted antigen 85 complex working in reverse with 6-TreAz and TDM as substrates, however, can potentially produce some 6-TreAz-containing TMM extracellularly in the cell wall compartment. This 6-TreAz-containing TMM is incorporated into the cell wall and be conjugated with AF488 DIBO Alkyne, which may explain the background labeling even in the absence of uptake (Fig. 6).

Metabolic labelling of Mtb strains employing click chemistry proves PPE51-dependent uptake of 6-TreAz. Mtb strains were incubated with 100 µM 6-TreAz for 3 days and subsequently conjugated with AF488 DIBO alkyne. Mean fluorescence intensity (MFI) was measured for 50,000 cells from each sample in each experiment. Data shown is the mean of three independent experiments ± SD. Differences in MFI between the indicated groups were statistically significant (***p < 0.005 as determined using unpaired t test). The high background in the controls that were not incubated with 6-TreAz is presumably due to non-specific association of the secondary DIBO-fluorophore.
6-TreAz resistant mutants show no cross-resistance against 2-TreAz
It was recently reported that not only 6-TreAz but also 2-azido trehalose (2-TreAz) exhibits moderate anti-mycobacterial activity against M. smegmatis23. Therefore, to test whether 2-TreAz also inhibits growth of Mtb H37Rv, we grew wild-type strain and our spontaneous resistant mutants on 7H10 solid medium containing up to 2 mM of 2-TreAz. However, we observed only partial growth inhibition even at the highest tested concentration, indicating that 2-TreAz has a much weaker effect on growth of Mtb compared to 6-TreAz. Importantly, although the effect of 2-TreAz was rather weak, the 6-TreAz-resistant mutants did not show any improved growth compared to the wild-type (Supplementary Fig. 3). This demonstrates that the observed mutations do not mediate cross-resistance to 2-TreAz. When spontaneous 6-TreAz-resistant mutants were tested for growth on trehalose as the sole carbon source, we found that all tested clones were still capable of utilizing this sugar (Supplementary Fig. 4). This demonstrates that despite the subtle differences in chemical structure, the observed mutations in PPE51 obviously specifically impair uptake of 6-TreAz but not of unmodified trehalose. Thus, it is possible that these mutations may also allow discrimination between 6-TreAz and 2-TreAz. The definitive role of PPE51 in uptake and the antibacterial activity of 2-TreAz could be interrogated in a Δppe51 gene deletion mutant. Unfortunately, due to limited compound availability, we could not test susceptibility of the Δppe51 gene deletion mutant towards 2-TreAz yet to definitively assess the role of PPE51 in uptake and the antibacterial activity of 2-TreAz.
Discussion
Although the complete genome sequence of the Mtb wild-type strain H37Rv was revealed in 1998, many of its genes have not been fully functionally characterized yet. One such group of genes are the PE/PPE family of proteins. Initially, a set of PE/PPE proteins were known to contribute to the evasion of host immunity and replication in human macrophages15. It is very recently that these proteins gained attention in relevance to uptake of various molecules across the mycomembrane. In our study, we investigated growth inhibition of Mtb by the azide-functionalized sugar 6-TreAz. Inhibition of mycobacterial growth by 6-TreAz was previously observed with Mycobacterium aurum14. It was shown that this compound inhibited bacterial growth at 200 µg/ml on solid medium and also suppressed mycolyltransferase activity of the antigen 85 complex by 60% at a concentration of 100 µg/ml. It also resulted in lesser synthesis of TMMs and TDMs14. This compound also caused growth inhibition against planktonic cells of M. smegmatis at higher concentrations (500 µM) and displayed a significant anti-biofilm activity at 50 µM23,24. In the present study, we examined the anti-mycobacterial activity of the compound against Mtb H37Rv wild-type strain and observed growth inhibition at 1 mM. Although this activity is much too weak to provide a basis for antimycobacterial drug development, it offers a selectable tool for studying trehalose transport and metabolism.
Very recently, Mtb was shown to be inhibited by agrichemical compounds such as 3,3-bis-di(methyl sulfonyl) propionamide (3bMP1) and carbohydrate derivatives with thio group such as thio-glycoside. Spontaneous resistant mutants raised against these anti-mycobacterial compounds were characterized by whole genome sequencing and revealed PPE51 as a determinant of resistance. Subsequent biochemical characterization of PPE51 in these studies demonstrated that PPE51 is required for transport of low-molecular weight molecules such as the studied anti-mycobacterial compounds but also for carbohydrates such as glucose, glycerol, maltose and lactose21,22. However, information about the role of PPE51 in transport of trehalose was not available so far. Given the central importance of trehalose for viability of Mtb, it is important to understand the uptake of exogenous trehalose across the mycomembrane. In our study, we harnessed the antimycobacterial property of the analogue 6-TreAz as a surrogate to identify resistance determinants that might reveal genes involved in transport of trehalose across the mycomembrane in an unbiased manner. Mutations occurred in ppe51, eccC5 and in genes involved in PDIM biosynthesis. Our experiments regarding growth on selected carbon sources, genetic complementation and click-chemistry labelling studies employing 6-TreAz and AF488DIBO clearly revealed that trehalose uptake across the mycomembrane is strictly mediated through PPE51, whereas the LpqY-SugABC ABC transporter is responsible for transport across the cytoplasm membrane.
The role of PDIM biosynthesis in relation to the essentiality of PPE51 was already observed by Wang et al. during their effort to generate a ppe51 gene deletion in Mtb. They showed that the function of PPE51 is strictly essential for viability of Mtb during growth on carbohydrates unless loss of PDIM biosynthesis apparently enables the PPE51-independent uptake across the mycomembrane22. While a PDIM-proficient ppe51 deletion strain was unable to utilize either glycerol or glucose as the sole carbon source, a PDIM-deficient ppe51 deletion strain harboring a loss-of-function second-site mutation in fadD26 could grow on both glucose and glycerol as the sole carbon source. In contrast, our Δppe51 mutant harbouring a second-site frame shift mutation in the mas gene could not utilize trehalose and glucose but was able to grow on glycerol as the sole carbon source. This could result from different mutations (fadD26 gene in Wang et al. vs. mas gene in our study) that might have affected PDIM biosynthesis to different degrees. We speculate that the frame shift mutation did not abolish mas gene function completely in our Δppe51 mutant, leading to at least some residual PDIM formation that rendered the barrier function of the mycomembrane largely intact towards carbohydrates under the tested culture conditions, with exception of glycerol.
Korycka-Machała et al. reported that the uptake of other disaccharides such as maltose and lactose is also mediated through PPE51, and they also noticed that some isolated resistant mutants harbored mutations in eccC5, similar to our findings21. Recently, it has been demonstrated that EccC5 is part of the ESX-5 secretion apparatus28, and that numerous PE and PPE proteins are secreted in an ESX-5-dependent manner in Mycobacterium marinum18. Although PPE51 has not yet been formally established as a specific ESX-5 substrate, the findings by Korycka-Machała et al. and our own data suggest that the ESX-5 apparatus is involved in secretion of PPE51 and/or its proper translocation and insertion into the mycomembrane. The observed mutations in eccC5 might impair secretion of protein substrates including PPE51 by the ESX-5 secretion apparatus, thus leading to the uptake defect that confers resistance towards 6-TreAz or thioglycosides.
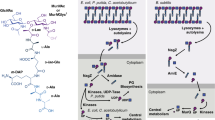
Taken together, our findings suggest that PPE51 serves as the gateway for the uptake of trehalose and trehalose analogs across the mycomembrane in addition to glucose, glycerol and other disaccharides. Figure 7 represents a simplified model of our current knowledge about the transport and metabolism of trehalose (analogues) in the different subcellular compartments of Mtb.

Simplified scheme summarizing transport and metabolism of trehalose and trehalose mycolates in Mtb. Exogenous trehalose is translocated across the mycomembrane via the PPE51 transporter. The LpqY-SugABC transporter allows the uptake of trehalose through the cytoplasmic membrane and recycles free trehalose that is released during synthesis of trehalose dimycolates (TDM) from trehalose monomycolates (TMM) by the mycoloyl transferases of the Antigen 85 complex (Ag85). The Ag85 also catalyzes the synthesis of arabinogalactan-mycolates from TMM, which is not depicted here for the sake of simplicity. Intracellularly, mycolates are conjugated to trehalose to yield TMM involving the enzymes Pks13 and CmrA, whereas the MmpL3 transporter mediates the secretion of TMM. IM inner membrane, AG arabinogalactan.
Materials and methods
Bacterial strains and growth conditions
All Mtb strains were grown at 37 °C either in Middlebrook 7H9 complete medium (Middlebrook 7H9 supplemented with 10% (v/v) ADS enrichment (5%, w/v, bovine serum albumin fraction V; 2%, w/v, glucose; 0.85%, w/v, sodium chloride), 0.5% (v/v) glycerol and 0.05% (v/v) tyloxapol) or on solidified 7H10 agar supplemented with 10% (v/v) ADS and 0.5% (v/v) glycerol unless otherwise stated. For the selection of appropriate strains, hygromycin (50 mg/l), apramycin (40 mg/l) and kanamycin (40 mg/l) were added. List of strains used for this study is provided in Supplementary Table 1. For testing growth on sole-carbon sources, Mtb strains were also grown in minimal medium [per liter: 0.15 g l-Asparagine × H2O, 0.5 g (NaH4)2SO4, 1 g KH2PO4, 2.5 g Na2HPO4, 50 mg ferric ammonium citrate, 0.5 g MgSO4 × 7 H2O, 0.5 mg CaCl2, 0.1 mg ZnSO4, 0.05% (v/v) tyloxapol, pH 7.0] and in Middlebrook 7H9 limited medium [containing only Middlebrook 7H9 and 0.05% (v/v) tyloxapol].
Construction of a merodiploid strain and generation of targeted gene deletion in Mtb
A merodiploid strain with an additional copy of the trehalose recycling transporter LpqY–SugABC (Rv1235–1238) in the Mtb genome was generated by electroporating the integrative plasmid pMV306(Kan)::Rv1235–123811 (1000 Ω, 25 µF, 2500 V) into Mtb H37Rv wild-type strain. This additional copy of the gene cluster compensates for any loss of Rv1235–1238 gene functions in spontaneous resistant mutants.
The Mtb Δppe51 mutant was generated by allelic exchange using specialized transduction as described previously32,33. Briefly, the regions flanking ppe51 (Rv3136) were amplified using Phusion DNA polymerase (New England Biolabs) using the primer pairs ppe51-LL-Van91I + ppe51-LR-Van91I and ppe51-RL-Van91I + ppe51-RR-Van91I (Supplementary Table 2), respectively, and cloned into an allelic exchange vector containing a hygromycin selection marker. The PacI digested vector was cloned into shuttle phasmid phAE159 and electroporated into M. smegmatis (1000 Ω, 25 µF, 2500 V). The shuttle phasmid produces phages containing recombinant DNA in M. smegmatis at the permissible temperature of 30 °C. High-titer phage lysates were used to transduce Mtb cells for gene deletion and replacement by the hygromycin selection marker (Supplementary Fig. 5). Transductants were obtained on selection plates containing hygromycin (50 mg/l) after incubation for 3–4 weeks at the non-permissive temperature of 37 C.
Mtb genomic DNA extraction and whole genome sequencing
The wild-type strain, spontaneous resistant mutants and the ppe51 deletion mutant were cultured in 7H9 complete medium and the genomic DNA was extracted using CTAB-lysozyme method. Briefly, the cultures were pelleted and incubated with 9:1 mixture of GTE (50 mM glucose, 25 mM Tris–HCl, 10 mM EDTA) and lysozyme solution (10 mg/ml) overnight at 37˚C. Next day, 150 µl of 2:1 solution of 10% SDS and 10 mg/ml proteinase K was added and incubated for 30 min at 55 °C. Then, 200 µl of 5 M NaCl was added and mixed gently. 160 µl of preheated CTAB (cetrimide-hexadecyltrimethylammonium bromide in water) was added and mixed gently followed by incubation for 10 min at 65 °C. Then an equal volume of 24:1 (v/v) chloroform/isoamyl alcohol was added and mixed vigorously and then centrifuged for 5 min. The aqueous layer was then transferred to fresh microcentrifuge tube and the previous step was repeated to get rid of residual organic phase. 560 µl of 70% isopropanol was then added to the aqueous layer for precipitating the genomic DNA. This step was followed by incubation for 5 min and then centrifugation for 10 min. The pellet was then washed with 70% ethanol, air dried and resuspended in water.
Genomic DNA samples used for genome sequencing were quantified by photometric measurement using a NanoDrop One device (Thermo Fisher Scientific Inc.) and quality measured by capillary electrophoresis using the Fragment Analyzer and the ‘High Sensitivity genomic DNA Assay’ (Agilent Technologies, Inc.). Library preparation was performed according to manufacturer’s protocol using ‘TruePrep DNA Library Prep Kit V2 for Illumina (1 ng) (Vazyme Biotech Co.; Ltd). Libraries were normalized to 4 nM and pooled and subsequently sequenced at the BMFZ, Heinrich Heine University, on a MiSeq system (Illumina Inc) with a read setup of 2 × 251 bp. The reads were assembled using a comparative genome assembly method, using Mtb H37RvMA as a reference (GenBank accession GCA_000751615.1)34. Mean depth of coverage ranged from 69x to 114x.
Genetic complementations
For complementation purposes, the genes ppe51 and eccC5 were amplified by PCR using the primer pairs ppe51-5′-PacI + ppe51-3′-HindIII and eccC5-5′-PacI + eccC5-3′-HindIII, respectively (Supplementary Table 2), and cloned into the integrative single-copy plasmid pMV361 containing an apramycin resistance marker providing constitutive heterologous gene expression from the HSP60 promoter. The plasmids were transformed into Mtb cells via electroporation (1000 Ω, 25 µF, 2500 V). Transformants were selected on agar plates containing apramycin (40 mg/l). During transformation into cells of spontaneous 6-TreAz-resistant mutants generated in the merodiploid Mtb H37Rv strain harbouring the integrative plasmid pMV306(Kan)::Rv1235–1238, the plasmid-encoded integrase mediates excision of the integrated plasmid pMV306(Kan)::Rv1235–1238 and replacement by pMV361(Apra)::ppe51 or pMV361(Apra)::eccC5, respectively, during selection on apramycin and absence of kanamycin.
Resazurin microplate assay (REMA) for growth quantification
Trehalose and glucose dependent growth of Mtb strains was quantified using the resazurin microplate assay. Cells were grown in liquid medium containing various concentrations of the indicated carbon sources in a total volume of 100 μl in 96-well round-bottom microtiter plates and incubated at 37 °C. After 5 days, resazurin solution (10 μl/well from 100 μg/ml stock, Sigma-Aldrich) was added to the cells and incubated for 16 h at room temperature for reduction of resazurin to resorufin by aerobic respiration of metabolically active cells. Next, Mtb cells were inactivated by incubation with formalin (10%, v/v, final concentration) for 30 min at room temperature. Subsequently, fluorescence was measured using a microplate reader (TECAN) (excitation 560 nm, emission 590 nm).
Metabolic labelling of Mtb
All Mtb strains were grown in Middlebrook 7H9 complete medium until OD600 nm ~ 0.8. 50 µl from the stock culture was added to 950 µl Middlebrook 7H9 complete medium containing 100 µM 6-TreAz in a 1.5 ml screw-cap tube and incubated for 3 days at 37 °C. After 3 days, the cells were centrifuged at 13,000 rpm for 5 min and the supernatant was discarded. The cells were then washed twice with PBS containing 0.5% (w/v) bovine serum albumin (PBSB) by centrifugation. After the washing step, the cells were incubated with 1 ml from 1:500 dilution of 1 mM Alexa Fluor 488 DIBO Alkyne stock solution in DMSO (ThermoFisher Scientific) at room temperature for 45 min in dark. Then, cells were washed again with PBSB twice and inactivated with 4% paraformaldehyde for 60 min at room temperature. The inactivated cells were washed with PBS and the final pellet was resuspended in 100 µl PBS. The cells were then prepared for measuring Mean fluorescence intensity (MFI) using flow cytometry. Flow cytometry was performed on BD LSRFortessa™ cell analyzer. Samples were gated according to forward and side scatter to exclude large cellular aggregates. MFI data was collected for 50,000 cells for each sample and processed using FlowJo (BD Biosciences).
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary information files.
References
WHO. World Health Organisation—Global Tuberculosis Report 2020 (WHO, 2020).
Koch, A. & Mizrahi, V. Mycobacterium tuberculosis. Trends Microbiol. 26, 555–556. https://doi.org/10.1016/j.tim.2018.02.012 (2018).
Jankute, M., Cox, J. A., Harrison, J. & Besra, G. S. Assembly of the mycobacterial cell wall. Annu. Rev. Microbiol. 69, 405–423. https://doi.org/10.1146/annurev-micro-091014-104121 (2015).
Warrier, T. et al. Antigen 85C inhibition restricts Mycobacterium tuberculosis growth through disruption of cord factor biosynthesis. Antimicrob. Agents Chemother. 56, 1735–1743. https://doi.org/10.1128/AAC.05742-11 (2012).
Ryll, R., Kumazawa, Y. & Yano, I. Immunological properties of trehalose dimycolate (cord factor) and other mycolic acid-containing glycolipids—a review. Microbiol. Immunol. 45, 801–811. https://doi.org/10.1111/j.1348-0421.2001.tb01319.x (2001).
Dulberger, C. L., Rubin, E. J. & Boutte, C. C. The mycobacterial cell envelope—a moving target. Nat. Rev. Microbiol. 18, 47–59. https://doi.org/10.1038/s41579-019-0273-7 (2020).
Kalscheuer, R. & Koliwer-Brandl, H. Genetics of mycobacterial trehalose metabolism. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.MGM2-0002-2013 (2014).
Miah, F. et al. Flux through trehalose synthase flows from trehalose to the alpha anomer of maltose in mycobacteria. Chem. Biol. 20, 487–493. https://doi.org/10.1016/j.chembiol.2013.02.014 (2013).
Tropis, M. et al. The crucial role of trehalose and structurally related oligosaccharides in the biosynthesis and transfer of mycolic acids in Corynebacterineae. J. Biol. Chem. 280, 26573–26585. https://doi.org/10.1074/jbc.M502104200 (2005).
Xu, Z., Meshcheryakov, V. A., Poce, G. & Chng, S. S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 114, 7993–7998. https://doi.org/10.1073/pnas.1700062114 (2017).
Kalscheuer, R., Weinrick, B., Veeraraghavan, U., Besra, G. S. & Jacobs, W. R. Jr. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 107, 21761–21766. https://doi.org/10.1073/pnas.1014642108 (2010).
O’Neill, M. K., Piligian, B. F., Olson, C. D., Woodruff, P. J. & Swarts, B. M. Tailoring trehalose for biomedical and biotechnological applications. Pure Appl. Chem. 89, 1223–1249. https://doi.org/10.1515/pac-2016-1025 (2017).
Swarts, B. M. et al. Probing the mycobacterial trehalome with bioorthogonal chemistry. J. Am. Chem. Soc. 134, 16123–16126. https://doi.org/10.1021/ja3062419 (2012).
Belisle, J. T. et al. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276, 1420–1422. https://doi.org/10.1126/science.276.5317.1420 (1997).
Brennan, M. J. The enigmatic PE/PPE multigene family of mycobacteria and tuberculosis vaccination. Infect. Immun. https://doi.org/10.1128/IAI.00969-16 (2017).
Delogu, G., Brennan, M. J. & Manganelli, R. PE and PPE genes: A tale of conservation and diversity. Adv. Exp. Med. Biol. 1019, 191–207. https://doi.org/10.1007/978-3-319-64371-7_10 (2017).
Ates, L. S., Houben, E. N. G. & Bitter, W. Type VII secretion: A highly versatile secretion system. Microbiol. Spectr. https://doi.org/10.1128/microbiolspec.VMBF-0011-2015 (2016).
Ates, L. S. et al. Essential role of the ESX-5 secretion system in outer membrane permeability of pathogenic mycobacteria. PLoS Genet. 11, e1005190. https://doi.org/10.1371/journal.pgen.1005190 (2015).
Gey van Pittius, N. C. et al. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol. Biol. 6, 95. https://doi.org/10.1186/1471-2148-6-95 (2006).
Mitra, A., Speer, A., Lin, K., Ehrt, S. & Niederweis, M. PPE surface proteins are required for heme utilization by Mycobacterium tuberculosis. MBio https://doi.org/10.1128/mBio.01720-16 (2017).
Korycka-Machala, M. et al. PPE51 is involved in the uptake of disaccharides by Mycobacterium tuberculosis. Cells https://doi.org/10.3390/cells9030603 (2020).
Wang, Q. et al. PE/PPE proteins mediate nutrient transport across the outer membrane of Mycobacterium tuberculosis. Science 367, 1147–1151. https://doi.org/10.1126/science.aav5912 (2020).
Wolber, J. M. et al. The trehalose-specific transporter LpqY-SugABC is required for antimicrobial and anti-biofilm activity of trehalose analogues in Mycobacterium smegmatis. Carbohydr. Res. 450, 60–66. https://doi.org/10.1016/j.carres.2017.08.003 (2017).
Lee, J. J. et al. Transient drug-tolerance and permanent drug-resistance rely on the trehalose-catalytic shift in Mycobacterium tuberculosis. Nat. Commun. 10, 2928. https://doi.org/10.1038/s41467-019-10975-7 (2019).
Kalscheuer, R. et al. Self-poisoning of Mycobacterium tuberculosis by targeting GlgE in an alpha-glucan pathway. Nat. Chem. Biol. 6, 376–384. https://doi.org/10.1038/nchembio.340 (2010).
Koliwer-Brandl, H. et al. Metabolic network for the biosynthesis of intra- and extracellular alpha-glucans required for virulence of Mycobacterium tuberculosis. PLoS Pathog. 12, e1005768. https://doi.org/10.1371/journal.ppat.1005768 (2016).
Baker, J. J. & Abramovitch, R. B. Genetic and metabolic regulation of Mycobacterium tuberculosis acid growth arrest. Sci. Rep. 8, 4168. https://doi.org/10.1038/s41598-018-22343-4 (2018).
Di Luca, M. et al. The ESX-5 associated eccB-EccC locus is essential for Mycobacterium tuberculosis viability. PLoS One 7, e52059. https://doi.org/10.1371/journal.pone.0052059 (2012).
Domenech, P. & Reed, M. B. Rapid and spontaneous loss of phthiocerol dimycocerosate (PDIM) from Mycobacterium tuberculosis grown in vitro: Implications for virulence studies. Microbiology (Reading) 155, 3532–3543. https://doi.org/10.1099/mic.0.029199-0 (2009).
Backus, K. M. et al. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 7, 228–235. https://doi.org/10.1038/nchembio.539 (2011).
Parker, H. L., Tomas, R. M. F., Furze, C. M., Guy, C. S. & Fullam, E. Asymmetric trehalose analogues to probe disaccharide processing pathways in mycobacteria. Org. Biomol. Chem. 18, 3607–3612. https://doi.org/10.1039/d0ob00253d (2020).
Bardarov, S. et al. Specialized transduction: An efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology (Reading) 148, 3007–3017. https://doi.org/10.1099/00221287-148-10-3007 (2002).
Jain, P. et al. Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. MBio 5, e01245-e11214. https://doi.org/10.1128/mBio.01245-14 (2014).
Ioerger, T. R. et al. Variation among genome sequences of H37Rv strains of Mycobacterium tuberculosis from multiple laboratories. J. Bacteriol. 192, 3645–3653. https://doi.org/10.1128/JB.00166-10 (2010).
Acknowledgements
M.R.B.S., R.K. and M.J. would like to thank the Jürgen Manchot foundation and the MOI graduate school for funding. B.M.S. acknowledges financial support from the National Institutes of Health (Grant number R15 AI117670). We thank Ernest Adankwah, Miguel Lopez Venegas and Jean De Dieu Harelimana for help with the FACS analyses. Computational support of the Zentrum für Informations- und Medientechnologie, especially the HPC team (High Performance Computing) at the Heinrich-Heine University is acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
R.K. designed and supervised the study. M.R.B.S. and H.K.B. performed the experiments. M.R.B.S. and R.K. analyzed the data. B.S. and J.A.S. provided reagents. M.J. processed data from flow cytometry analysis. T.R.I. analyzed genome sequencing data. M.R.B.S. and R.K. wrote the manuscript with edits from all coauthors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Babu Sait, M.R., Koliwer-Brandl, H., Stewart, J.A. et al. PPE51 mediates uptake of trehalose across the mycomembrane of Mycobacterium tuberculosis. Sci Rep 12, 2097 (2022). https://doi.org/10.1038/s41598-022-06109-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06109-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
