
Abstract
Inherited retinal dystrophies (IRDs) constitute one of the most heterogeneous groups of Mendelian human disorders. Using autozygome-guided next-generation sequencing methods in 17 consanguineous pedigrees of Iranian descent with isolated or syndromic IRD, we identified 17 distinct genomic variants in 11 previously-reported disease genes. Consistent with a recessive inheritance pattern, as suggested by pedigrees, variants discovered in our study were exclusively bi-allelic and mostly in a homozygous state (in 15 families out of 17, or 88%). Out of the 17 variants identified, 5 (29%) were never reported before. Interestingly, two mutations (GUCY2D:c.564dup, p.Ala189ArgfsTer130 and TULP1:c.1199G > A, p.Arg400Gln) were also identified in four separate pedigrees (two pedigrees each). In addition to expanding the mutational spectrum of IRDs, our findings confirm that the traditional practice of endogamy in the Iranian population is a prime cause for the appearance of IRDs.
Similar content being viewed by others


Introduction
Inherited retinal dystrophies/degenerations (IRDs) are Mendelian disorders of the eye that affect approximately 1 in 1500 people worldwide and constitute a major cause of inherited blindness1. Mainly characterized by the progressive death of photoreceptor cells in the retina, IRDs present a high degree of genetic and phenotypic heterogeneity2. So far, mutations in over 270 genes have been associated with various forms of IRDs (RetNet: https://sph.uth.edu/retnet/; accessed April 17, 2020); however, this list is constantly increasing. Sequencing of all exons and exon–intron boundaries of these genes has successfully contributed to the understanding of the genetic etiology of 50–70% of patients3,4,5. Since a larger proportion of IRDs are caused by recessive mutations6, next-generation sequencing (NGS) coupled with homozygosity mapping has further accelerated detection of candidate variants in consanguineous pedigrees3.
Close-kin marital unions have long been practiced in the Iranian population as a cultural feature7. In spite of a significant decline in consanguinity in Iran in recent years, prevalence of recessive genetic disorders is still high in the country, possibly due to the presence of marked population stratification that favors intra-community marriages7,8. As a result, increased genomic homozygosity in specific societies or ethnic groups leads to the clinical observation of the effect of rare founder mutations7,9. The present study was performed with the aim of characterizing genetically a cohort of 17 consanguineous Iranian families with IRDs.
Methods
Enrollment of families and DNA extraction
This study was approved by the Ethics Committees of all our respective Institutions (the Ethikkommission Nordewest- and Zentralschweiz, the Ethics Committee of Mashhad University of Medical Sciences, the Ethics Commission of the Noncommunicable Diseases Research Center of Fasa University of Medical Sciences, and the Ethics Commission of the Canton de Vaud) and adhered to the principles of the Declaration of Helsinki. All individuals participating in this study were Iranian residents, who agreed in contributing to this study by signing a written informed consent form. Patients were clinically evaluated by local ophthalmologists and their medical records were maintained at their respective hospitals. Approximately 5.0 ml peripheral blood was collected using a EDTA K2 golden vac disposable vacuum blood collection tube (Zhejiang Gondong Medical Technology, China) or were mixed with EDTA anticoagulant (Merck KGaA, Darmstadt, Germany) after sample collection. DNA was extracted from peripheral blood leukocytes following standard protocols. Quantitative assessment of DNA was made using a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, USA), whereas integrity was evaluated by running the DNA samples on a 1% agarose gel. Pedigrees were drawn with the help of HaploPainter10.
Genetic analyses
Exome capture and library preparation was performed on one affected individual per family using the SureSelect Human All Exon v6 kit (Agilent, Santa Clara, CA, USA) and the HiSeq Rapid PE Cluster Kit v2 (Illumina, San Diego, CA, USA), from 2 μg genomic DNA. Whole-exome sequencing (WES) was performed at the Institute of Genomics of the University of Tartu (Estonia) using an Illumina HiSeq (HSQ-700358) instrument. Bioinformatic analyses were performed as described previously11. Briefly, raw reads were mapped to the human reference genome (hg19/GRCh37) using the Novoalign software (V3.08.00, Novocraft Technologies). Next, Picard (version 2.14.0-SNAPSHOT) was used to remove duplicate reads and Genome Analysis Toolkit (GATK) (version 3.8) was used to perform base quality score recalibration on both single-nucleotide variants and insertion–deletions. A VCF file with the variants was generated by HaplotypeCaller. They were annotated according to a specific in-house pipeline using mainly ANNOVAR software12 (Oct 2019 release) and splicing predictors: spliceAI13, MaxEntScan14, and Ada and RF scores from dbscSNV15. Then, DNA variants were filtered to have less than 1% allelic frequency in ExAC16, gnomAD17, 1000 Genomes18, and GME (GME Variome http://igm.ucsd.edu/gme/index.php). Variants were then retained according to their predicted impact on protein sequence and splicing of preRNA, according to ANNOVAR RefSeq annotation (missense, stopgain, frameshift or non-frameshift indels, or canonical splicing) and splicing predictors (MaxEntScan [minimal change of 6], spliceAI [minimal score of 0.2] and dbscSNV-ADA [minimal score of 0.2]).
Autozygosity mapping was performed on WES data with AutoMap19. Nomenclature of all the variants was confirmed through VariantValidator20. Clinical significance of the variants was evaluated with the help of publicly available databases such as ClinVar21, the Human Gene Mutation Database (HGMD)22 and Varsome23, as well as according to their frequency in available databases, e.g. the Genome Aggregation Database (gnomAD)17. Seven online in-silico methods were used to predict the pathogenicity of all variants. The online in-silico tools used included MutationTaster24, Mutation Assessor24, Polymorphism Phenotyping v2 (PolyPhen-2)25, Likelihood Ratio Test (LRT)26, Sorting Intolerant from Tolerant (SIFT)27, PROVEAN28, and Combined Annotation Dependent Depletion (CADD)29. Furthermore, all candidate variants were compared with data from Iranome, a database containing information from 800 exomes from individuals belonging to eight major ethnic groups in Iran (http://www.iranome.ir/; accessed on April 19, 2020). Finally, Sanger sequencing was performed to validate all potentially pathogenic variants and to establish their causality through strict genotype–phenotype co-segregation within the available family members.
Results
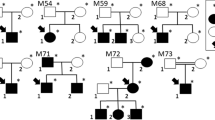
Following WES analysis in 17 probands of Iranian descent, 16 of which were the direct offspring of consanguineous unions (Fig. 1), we identified 17 distinct genetic variants in 11 genes linked to inherited retinal diseases (Tables 1 and 2). Of the 17 pedigrees, two families each were linked to disease-causing variants in CNGA3, GUCY2D, IQCB1, RDH12, RP1, and TULP1 genes, while only one family was found with causative variants in either USH1G, ABCA4, NMNAT1, CRB1, or BBS2 genes. The mutational spectrum across these 11 genes comprised 7 missense variants, 4 nonsense variants, 4 small insertion-deletions (Indels) or duplications leading to frameshifts, one canonical splice site variant and one synonymous variant with effect on splicing. As expected, most of the variants in our study were found in a homozygous state (in 15 families out of 17, or 88%). Compound heterozygosity was detected in two families (F009 and IRN_070, Tables 1, 2). Except for one homozygous allele in the RP1 gene (NM_006269.1:c.788-1G > A) in family IRN_039, all other homozygous pathogenic variants were found in genes that were located inside a so-called run of homozygosity (ROH), generally spanning more than one megabase (Mb) in size.


Pedigrees showing genotype–phenotype co-segregation for all detected variants. m, variant identified; + , wild-type allele.
Overall, 29% (5 out of 17 different alleles) of the variants reported here were previously unpublished. Similarly, 8 of the total 17 distinct alleles had no gnomAD entry, while the others were all very rare, with no occurrence of homozygous individuals in the gnomAD database (Table 3). Interestingly, we identified two variants, each segregating homozygously in two separate pedigrees: GUCY2D (NM_000180.3:c.564dup, p.Ala189ArgfsTer130; families IRN_001 and IRN_042) and TULP1 (NM_003322.3:c.1199G > A, p.Arg400Gln; families F007 and F034).
All 7 missense variants with their predicted pathogenicity scores are listed in Table 3. While 6 of them were previously known to be pathogenic, including NMNAT1 (p.Glu257Lys)30, CNGA3 (p.Arg283Gln)31, CRB1 (p.Cys201Arg)32, TULP1 (p.Arg400Gln)33, RDH12 (p.Arg239Trp)34, and CNGA3 (p.Arg277His)35, the remaining one, NMNAT1 (p.Ile174Phe), was novel. Additionally, segregation analysis within the available family members using Sanger sequencing revealed strict genotype–phenotype correlation for all nonsynonymous variants.
Furthermore, we identified four already published nonsense pathogenic variants in our cohort, including RDH12 (p.Gly127Ter)36, CRB1 (p.Trp1293Ter)37, IQCB1 (p.Arg489Ter)38, and IQCB1 (p.Arg502Ter)38. Similarly, we identified three novel frameshift variants, in addition to the previously known frameshift in the ABCA4 gene (p.Leu296CysfsTer4)39. Novel frameshift variants comprised GUCY2D (p.Ala189ArgfsTer130), RP1 (p.Val196GlyfsTer33), and USH1G (p.Glu332ThrfsTer53).
Lastly, we detected a novel canonical splice site variant in the RP1 gene (NM_006269.1:c.788-1G > A) in family IRN_039, and a known pathogenic synonymous change predicted to alter splicing in the BBS2 gene (NM_031885.3:c.471G > A;p.Thr157 =) in family IRN_06540.
Discussion
Consanguinity is a major risk factor for the occurrence of rare recessive Mendelian disorders, yet it is a long-lived social practice in many Asian and African countries. In Iran, the second most populated country in the Middle East, 37.4% of all marriages are between consanguineous partners. Of these, 19.3% occur between first cousins and 18.1% involve second cousins7.
In this work, we used consanguinity as a means to facilitate identification of mutations in IRD cases, through an autozygome-guided NGS approach. Consistent with the high level of consanguinity displayed by the Iranian population, we observed a recessive inheritance pattern in all our cases, with the largest majority of them carrying indeed homozygous pathogenic variants in known IRD genes. With only one exception, all genes carrying homozygous pathogenic variants resided inside runs of homozygosity, thus supporting earlier studies that highlighted the importance of homozygosity mapping in consanguineous families41,42,43,44,45,46. Nevertheless, compound heterozygous patients were also identified, with mutations in CRB1 and NMNAT1. Interestingly, these patients (from families F009, and IRN_070, respectively) also had relatively lower values of overall genomic homogeneity (197, and 71 Mb, respectively, over an average of 280.0 Mb in the cohort as a whole). The appearance of compound heterozygosity in the Iranian population is not unprecedented, and an earlier study suggested that CRB1 is a commonly mutated gene in Iranian patients with non-syndromic IRDs43,47. Interestingly, our cohort did not include any instance of variants in USH2A, although mutations in this gene are considered to be among the most frequent causes of Usher syndrome or non-syndromic retinitis pigmentosa (RP)48.
The mutational spectrum in our cohort comprised 1 synonymous (with predicted effect on splicing), 1 splice change, 7 missense, 4 nonsense, and 4 frameshift variants. To establish pathogenicity of the novel missense variants we heavily relied on data from existing literature and the ACMG guidelines. Lastly, we assessed the status of each variant by comparing them with the Iranome database, to filter out common variants specific to Iranian population.
Unlike missense substitutions, the majority of nonsense and frameshift DNA changes can be considered as bona fide deleterious mutations, since they mostly constitute loss-of-function (pLoF) alleles in genes where this pathogenicity mechanism is well known (criteria PVS1 of ACMG guidelines). We therefore classified all of them as such, based on this feature and the fact that they were all either absent or present at an extremely low frequency in the gnomAD database.
We also found a synonymous change in the BBS2 gene (c.471G > A, p.Thr157 =) co-segregating with Bardet-Biedl syndrome in one family (IRN_065) and reported in three previous studies40,49,50. Due to the high nucleotide conservation and its localization at an exon–intron boundary, it is possible that the c.471G > A substitution may impair the correct splicing of BBS2 pre-mRNA. Indeed, all splicing predictors tested (AdaBoost and RandomForest from dbscSNC, MaxEntScan, and spliceAI) indicated a high impact on splicing and disruption of the 5’ site. Our findings thus provide additional support to the potential pathogenicity of this apparently neutral variant. It is worthwhile to mention here that the majority of the previously reported patients with the p.Thr157 = mutation originated from Middle Eastern countries, such as Lebanon and Iran49,50. Although this variant perfectly co-segregates with disease in family IRN_065 and has been described in previous reports in association with Bardet-Biedl syndrome40,49,50, there is still a chance that it could represent a benign DNA change, detected in homozygosity in our patients by virtue of their ethnical origin. Additional functional studies are needed to definitely confirm its pathogenic role in syndromic IRD.
Since geographic isolation and consanguinity-driven genomic homozygosity lead to the enrichment of rare founder mutations in specific societies or ethnic groups7,9,51,52, the presence of such mutations in our cohort of patients from related families is not surprising. Similar to other reports53,54,55, we identified two mutations that were present in more than one pedigree. The first, p.Ala189ArgfsTer130 in GUCY2D, was shared by two families originating from the Fars province in the Southwest of Iran and was found in a common ROH of 3.0 Mb with an identical haplotype. The second was a homozygous missense variant (p.Arg400Gln) in the TULP1 gene, detected in two families from the Razavi Khorasan province, in Northeastern Iran, again in a common ROH of 21.5 Mb with an identical haplotype. This latter variant has been previously reported in an Indian family in a homozygous state33.
In summary, this work extends current knowledge about the genetic landscape of IRDs in Iran and, in line with previous studies, supports the evidence that homozygosity mapping is an effective tool for uncovering rare genomic variants in consanguineous pedigrees with rare recessive disorders. Most importantly, we hope that our data would contribute to better molecular diagnosis and access to future gene therapy trials in Iran.
References
Hanany, M., Rivolta, C. & Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. U S A 117, 2710–2716 (2020).
Wright, A. F., Chakarova, C. F., Abd El-Aziz, M. M. & Bhattacharya, S. S. Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 11, 273–284 (2010).
Sharon, D. et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 41, 140–149 (2020).
Ellingford, J. M. et al. Molecular findings from 537 individuals with inherited retinal disease. J. Med. Genet. 53, 761–767 (2016).
Weisschuh, N. et al. Genetic architecture of inherited retinal degeneration in Germany: A large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 41, 1514–1527 (2020).
Hartong, D. T., Berson, E. L. & Dryja, T. P. Retinitis pigmentosa. Lancet 368, 1795–1809 (2006).
Hosseini-Chavoshi, M., Abbasi-Shavazi, M. J. & Bittles, A. H. Consanguineous marriage, reproductive behaviour and postnatal mortality in contemporary Iran. Hum. Hered. 77, 16–25 (2014).
Bittles, A. H. & Black, M. L. Evolution in health and medicine Sackler colloquium: Consanguinity, human evolution, and complex diseases. Proc. Natl. Acad. Sci. U S A 107(Suppl 1), 1779–1786 (2010).
Bittles, A.H. Consanguinity, genetic drift, and genetic diseases in populations with reduced numbers of founders. in Vogel and Motulsky's Human Genetics. 4th Edn. 507–528 (2010).
Thiele, H. & Nürnberg, P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics 21, 1730–1732 (2005).
Royer-Bertrand, B. et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci. Rep. 5, 17154 (2015).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535-548e24 (2019).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Jian, X., Boerwinkle, E. & Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 42, 13534–13544 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Quinodoz, M. et al. AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 12, 518 (2021).
Freeman, P. J., Hart, R. K., Gretton, L. J., Brookes, A. J. & Dalgleish, R. VariantValidator: Accurate validation, mapping, and formatting of sequence variation descriptions. Hum. Mutat. 39, 61–68 (2018).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Stenson, P. D. et al. Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 21, 577–581 (2003).
Kopanos, C. et al. VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980 (2019).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 7(7), 20 (2013).
Chun, S. & Fay, J. C. Identification of deleterious mutations within three human genomes. Genome Res. 19, 1553–1561 (2009).
Sim, N. L. et al. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40, W452–W457 (2012).
Choi, Y. & Chan, A. P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019).
Falk, M. J. et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 44, 1040–1045 (2012).
Kohl, S. et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 19, 257–259 (1998).
Talib, M. et al. CRB1-associated retinal dystrophies in a Belgian cohort: Genetic characteristics and long-term clinical follow-up. Br. J. Ophthalmol. https://doi.org/10.1136/bjophthalmol-2020-316781 (2021).
Singh, H. P., Jalali, S., Narayanan, R. & Kannabiran, C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Invest. Ophthalmol. Vis. Sci. 50, 4065–4071 (2009).
Thompson, D. A. et al. Retinal degeneration associated with RDH12 mutations results from decreased 11-cis retinal synthesis due to disruption of the visual cycle. Hum. Mol. Genet. 14, 3865–3875 (2005).
Wissinger, B. et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am. J. Hum. Genet. 69, 722–737 (2001).
Perrault, I. et al. Retinal dehydrogenase 12 (RDH12) mutations in leber congenital amaurosis. Am. J. Hum. Genet. 75, 639–646 (2004).
Hanein, S. et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum. Mutat. 23, 306–317 (2004).
Estrada-Cuzcano, A. et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest. Ophthalmol. Vis. Sci. 52, 834–839 (2011).
Zernant, J. et al. Analysis of the ABCA4 gene by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 52, 8479–8487 (2011).
Wang, F. et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: Identification of a novel genotype-phenotype correlation and clinical refinements. Hum. Genet. 133, 331–345 (2014).
Wakeling, M. N. et al. Homozygosity mapping provides supporting evidence of pathogenicity in recessive Mendelian disease. Genet. Med. 21, 982–986 (2019).
Li, L. et al. Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 consanguineous Pakistani families. Invest. Ophthalmol. Vis. Sci. 58, 2218–2238 (2017).
Ghofrani, M. et al. Homozygosity mapping and targeted Sanger sequencing identifies three novel CRB1 (crumbs homologue 1) mutations in Iranian retinal degeneration families. Iran Biomed. J. 21, 294–302 (2017).
Maria, M. et al. Homozygosity mapping and targeted Sanger sequencing reveal genetic defects underlying inherited retinal disease in families from Pakistan. PLoS ONE 10, e0119806 (2015).
Beryozkin, A. et al. Identification of mutations causing inherited retinal degenerations in the Israeli and Palestinian populations using homozygosity mapping. Invest. Ophthalmol. Vis. Sci. 55, 1149–1160 (2014).
Alkuraya, F. S. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum. Genet. 132, 1197–1211 (2013).
Tayebi, N. et al. Targeted next generation sequencing reveals genetic defects underlying inherited retinal disease in Iranian families. Mol. Vis. 25, 106–117 (2019).
Lenassi, E. et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 23, 1318–1327 (2015).
Fattahi, Z. et al. Mutation profile of BBS genes in Iranian patients with Bardet-Biedl syndrome: Genetic characterization and report of nine novel mutations in five BBS genes. J. Hum. Genet. 59, 368–375 (2014).
Deveault, C. et al. BBS genotype-phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Hum. Mutat. 32, 610–619 (2011).
Rehman, A.U. et al. Exploring the genetic landscape of retinal diseases in North-Western Pakistan reveals a high degree of autozygosity and a prevalent founder mutation in ABCA4. Genes (Basel) 11 (2019).
Ravesh, Z. et al. Advanced molecular approaches pave the road to a clear-cut diagnosis of hereditary retinal dystrophies. Mol. Vis. 24, 679–689 (2018).
Mohseni, M. et al. Identification of a founder mutation for Pendred syndrome in families from northwest Iran. Int. J. Pediatr. Otorhinolaryngol. 78, 1828–1832 (2014).
Fallahi, J. et al. Founder effect of KHDC3L, p.M1V mutation, on Iranian patients with recurrent hydatidiform moles. Iran J. Med. Sci. 45, 118–124 (2020).
Mojbafan, M., Bahmani, R., Bagheri, S. D., Sharifi, Z. & Zeinali, S. Mutational spectrum of autosomal recessive limb-girdle muscular dystrophies in a cohort of 112 Iranian patients and reporting of a possible founder effect. Orphanet. J. Rare Dis. 15, 14 (2020).
Acknowledgements
We would like to thank all of the participating families. We are also grateful to the Swiss Confederation for the award of a PhD fellowship to AUR, to Mashhad University of Medical Sciences for supporting part of the work, in the framework of the PhD thesis of AS, to the Swiss National Science Foundation for grant # 176097 to CR, and to the Fondation Guillaume Gentil for support to ASF.
Author information
Authors and Affiliations
Contributions
A.U.R., Z.R. and C.R. wrote the manuscript. A.U.R., N.B., and F.C. performed all experimental work, including DNA extraction, Sanger validation of WES results and segregation analysis. M.Q., N.B., and V.G.P. analyzed WES and SNP genotype data. A.S., M.M., N.S., A.P., A.G.A., S.G., M.P., and M.P. recruited patients. A.S.F. and C.R. designed the study. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rehman, A.U., Sepahi, N., Bedoni, N. et al. Whole exome sequencing in 17 consanguineous Iranian pedigrees expands the mutational spectrum of inherited retinal dystrophies. Sci Rep 11, 19332 (2021). https://doi.org/10.1038/s41598-021-98677-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98677-3
This article is cited by
-
Generation of Zebrafish Models of Human Retinitis Pigmentosa Diseases Using CRISPR/Cas9-Mediated Gene Editing System
Molecular Biotechnology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
