
Abstract
This study clarifies the role of refugia and landscape permeability in the formation of the current genetic structure of peoples of the Caucasus. We report novel genome-wide data for modern individuals from the Caucasus, and analyze them together with available Paleolithic and Mesolithic individuals from Eurasia and Africa in order (1) to link the current and ancient genetic structures via landscape permeability, and (2) thus to identify movement paths between the ancient refugial populations and the Caucasus. The ancient genetic ancestry is best explained by landscape permeability implying that human movement is impeded by terrain ruggedness, swamps, glaciers and desert. Major refugial source populations for the modern Caucasus are those of the Caucasus, Anatolia, the Balkans and Siberia. In Rugged areas new genetic signatures take a long time to form, but once they do so, they remain for a long time. These areas act as time capsules harboring genetic signatures of ancient source populations and making it possible to help reconstruct human history based on patterns of variation today.
Similar content being viewed by others


Introduction
The Caucasus is a mountainous region located on the dividing line between Europe and Asia, between the Black and Caspian seas. In spite of relatively small geographic extent and mostly temperate climate, the diversity of natural landscapes, plant and animal species, and cultivars is unusually high in the Caucasus1. Thanks to this diversity the Caucasus is one of the global biodiversity hotspots that also contain considerable linguistic diversity, accounting for most of all languages on Earth2,3. The Caucasus provided some of the globe’s important refugia, where most of the terrestrial plants and animals (incl. humans) survived during a series of glacial maxima, and their current distribution largely reflects post-glacial expansion from these refugia1. These glacial refugia and the barriers to migration played an important role in human evolution, and generated much of the human genetic and ethno-linguistic patterns found in the world today4,5,6,7. The Caucasus contributed about half of the genetic ancestry of Yamnaya8—that is, pastoralists of the Pontic-Caspian steppe, who date from the Late Copper Age to the Early Bronze Age and made profound demographic and cultural impacts on much of Eurasia e.g. by spreading genomes, Indo-European languages and horse-riding9,10,11,12,13,14. It is hypothesized that Proto-Indo-European was generated from admixture between archaic languages from the Caucasus and Proto-Uralic languages in the Pontic-Caspian steppe15,16. Thus, the Caucasus has played an important role in the formation of past and present Eurasian genetic and cultural diversity.
As research technologies advance and more samples are obtained, studies have been unveiling more of the genetic variation of Caucasian populations in relation to geography and various indicators of ethnicity7,17,18,19,20,21,22,23,24. Autosomal genome and mtDNA variations in the Caucasus appear relatively homogenous, while the Y-chromosome diversity clearly shows geographic heterogeneity distinguishing the eastern from western Caucasians7,17,24. This east–west cline in the Caucasus was also shown for autosomal genome variation when Georgians were divided into sub-ethnic groups22. The studies of genome-wide autosomal profiles as well as the mitochondrial and Y chromosomal haplogroups support that there is genetic continuity in the present-day southern Caucasus stretching back at least 13,000 years to the Late Upper Palaeolithic8,25. In the North Caucasus this continuity was disrupted due to post-bronze age admixture with populations from the Eurasian Steppe26. At present genetic differentiation between ethnic/subethnic groups in the Caucasus correlates with landscape permeability to human movement—as determined by terrain ruggedness, forest cover, and snow cover—rather than ethnic or linguistic boundaries22.
Tarkhnishvili et al.22 hypothesized that the genetic makeup of the modern Caucasians was largely shaped up by human dispersal from few distinct glacial refugia in the last glacial period and the early Holocene, followed by less gene flow between the populations of the Greater Caucasus than between those of the rest of the Caucasus, where the populations have undergone substantial admixture in historical time. Understanding the nature of human movement from refugial populations was the primary motivation behind this study. Here we measure genome-wide genetic affinity between modern populations of the Caucasus and ancient populations of hunter-gatherers, test whether the genetic affinity between these populations is determined by geographic features, and infer major dispersal paths from refugial populations into the Caucasus. The results of this study allow us to reconstruct an integral picture of the peopling of the Caucasus from the last glacial period through the early Holocene.
Methods
Sampling and genotyping
We collected hair and cheek swab samples from 77 men from geographically and linguistically distinct groups of the Caucasus: Kartvelian speakers from Georgia and Turkey, Northeast Caucasian speakers and Turkic speakers from the Russian Federation and Armenian speakers from Georgia’s southern province of Javakheti, descendants of the families displaced from Mush and Erzurum provinces of eastern Turkey in the early nineteenth century (Table 1, Fig. 1). To maximize the representativeness of the genetic signature of each population, the samples were collected from locals with no ancestors from outside of the respective ethnic/geographic population over the last three generations. DNA was extracted from follicles of 10–12 male chest hairs and cheek swab samples. Extraction was performed using Qiagen DNeasy Blood and Tissue kit, following the manufacturer’s recommendations (Qiagen, Valencia, CA, USA). The DNA samples were genotyped for 693,719 autosomal and 17,678 X-chromosomal SNPs by Family Tree DNA (FTDNA—Gene By Gene, Ltd, Houston, TX, www.familytreedna.com).

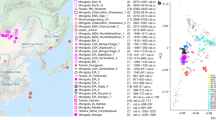
The distribution of the study populations: averaged centroids of ancient populations (uniquely colored points in the main map, see Table 2 for details) and hubs of the modern Caucasian populations (identified in the inset map, see Table 1 for details). Glacial human refugia extracted from Gavashelishvili and Tarkhnishvili5 are shaded in purple. The map is generated using QGIS Desktop 3.10.6-A Coruña (https://qgis.org).
Our dataset of modern Caucasian genotypes was supplemented with published 10 modern Mbuti (Supplementary Table S1) and 122 Upper Paleolithic-Mesolithic human genotypes, retrieved as a part of 1240 K dataset from David Reich’s Lab website, Harvard University (https://reich.hms.harvard.edu/downloadable-genotypes-present-day-and-ancient-dna-data-compiled-published-papers; see Supplementary Table S2 for details). The ancient genotypes were selected such that they either dated from the LGM or fell within the glacial refugia identified by Gavashelishvili and Tarkhnishvili5. We did so in order to maximize the genetic signature of potential refugial populations in our analysis. We divided the ancient genotypes into 2000-year-long intervals, and then grouped each of these intervals into geographic units (hereafter ancient populations, Table 2, Fig. 1). The modern and ancient genotypes were merged using PLINK 1.9 (PLINK 1.9: www.cog-genomics.org/plink/1.9/27.
Ethics statement
The research team members, through their contacts in the studied communities, inquired whether locals would voluntarily participate in genetic research that would help clarify the genetic makeup of the Caucasus. A verbal agreement was made with volunteer donors of DNA samples, according to which the results would be communicated, electronically or in hard copy, with participants individually. Participants were informed that, upon the completion of the lab work, the research would be published without mentioning the names of sample donors. Those who agreed provided us with the envelopes containing their chest hairs or cheek swab samples, with the birthplace of their ancestors (last three generations) written on the envelope or a piece of paper. In accordance with the preferences of the sample donors, the agreement was verbal and not written. The envelopes and papers are stored as evidence of voluntary provision of the samples and the related information. Analysis of data was done anonymously, using only location and ethnic information; only the first and third authors of the manuscript had access to names associated with the samples. Therefore, this study was based on noninvasive and nonintrusive sampling (volunteers provided hair and swab samples they collected themselves), and the information destined for open publication does not contain any personal information. The study methodology and the procedure of verbal consent was discussed in detail with and approved by the members of the Ilia State University Commission for Ethical Issues before the field survey started, and the commission decided that formal ethical approval was not needed for conducting this study. This is confirmed in a letter from the commission chairman, a copy of which has been provided to the journal editor as part of the submission process.
Genetic affinity and geography
First, we measured genetic affinity between the modern Caucasian populations, and between the modern populations and the ancient populations of hunter-gatherers, and then tested whether the genetic affinity between these populations was determined by geographic features. Data were mapped using QGIS Desktop 3.10.6-A Coruña, whereas graphs were created using the “ggplot2” package28 in R version 3.5.229.
To evaluate genetic affinities and structure of the modern populations, we used Wright’s fixation index (Fst), inbreeding coefficient, admixture analysis and the principal component analysis (PCA). For these procedures we filtered the raw SNP genotypes in PLINK 1.9, first removing all SNPs with the minor allele frequency < 0.05, followed by LD pruning of markers that exceeded the pairwise correlation threshold of r2 > 0.3, calculated in windows of 50 bp size and 10 bp steps (–maf 0.05 –indep-pairwise 50 10 0.3). Since all individuals in our dataset possess a single copy of the X-chromosome, we did not expect any differential ploidy bias, and SNPs on the X were treated similarly to those on the autosomes. Fst pairwise values were calculated using the smartpca program of EIGENSOFT30 with default parameters, inbreed: YES, and fstonly: YES. The relationship between the modern populations based on Fst values was visualized by constructing a neighbor-joining tree using the “ape” package31 in R version 3.5.2. The average and standard deviation of the inbreeding coefficient for each population was calculated using “fhat2” estimate of PLINK 1.9. The LD pruned genotypes were used in ADMIXTURE 1.3.032, performed in unsupervised mode in order to infer the population structure from the modern individuals. The number of clusters (k) was varied from 2 to 7 and the fivefold cross-validation error was calculated for each k33. We conducted principal components analysis in the smartpca program of EIGENSOFT30, using default parameters and the lsqproject: YES and numoutlieriter: 0 options. Eigenvectors of principal components were inferred with the modern populations from the Caucasus, while the ancient populations were then projected onto the PCA plots. We also assessed the relatedness between sampled individuals using kinship coefficients estimated by KING34.
To quantify genetic affinities between the modern and ancient populations, we used the programs qp3Pop and qpDstat in the ADMIXTOOLS suite (https://github.com/DReichLab35 for f3- and f4-statistics, respectively. f3-statistics of the form f3(X,Y,Outgroup) measure the amount of shared genetic drift of populations X and Y after their divergence from an outgroup. We used an ancient population and a modern Caucasian population for X, Y and Mbuti as an outgroup. f4-statistics of the form f4(Outgroup,Test;X,Y) show if population Test is equally related to X and Y or shares an excess of alleles with either of the two. In the f4-statistic calculation we used Mbuti for Outgroup, a modern population of the Caucasus for Test, and X and Y for contemporaneous ancient populations. This meant that f4 < 0 indicated higher genetic affinity between the test population and X, while f4 > 0 indicated higher genetic affinity between the test population and Y.
To quantify geographic features, we derived least-cost paths and measured least-cost distances (LCD) between the modern and ancient populations using the Least Cost Path Plugin for QGIS. The computation of LCD considers a friction grid that is a raster map where each cell indicates the relative difficulty (or cost) of moving through that cell. A least-cost path minimizes the sum of frictions of all cells along the path, and this sum is the least-cost distance (LCD). For impedance to human movement and expansion, we used 15 geographic features (Table 3). All gridded geographic features (i.e. raster layers) were resampled to a resolution of 1 km using the nearest-neighbor assignment technique. All possible subsets of the 15 geographic features, that did not cancel out each other, were used to calculate different variables of LCD. We assumed that most human movements occurred during climate warming events when the earth’s surface was not dramatically different from that of today, and hence used the current data of the geographic features.
Linking genetic affinity and geography
Generalized additive models (GAMs) were used to fit the outgroup f3-statistic to time and variously calculated LCD between the modern and ancient populations using the “mgcv” package36 in R version 3.5.2. Time between the modern and ancient populations was measured in BP (years before present, defined by convention as years before 1950 CE). We used GAMs because without any assumptions they are able to find nonlinear and non-monotonic relationships. GAMs were fitted using a Gamma family with a log link function. Penalized thin plate regression splines were used to represent all the smooth terms. The restricted maximum likelihood (REML) estimation method was implemented to estimate the smoothing parameter because it is the most robust of the available GAM methods36.
Model and variable selection were performed by exploring LCD, time BP and the interaction term. The predictive power of the models was evaluated through a tenfold cross-validation. The cross-validation of many models was handled through R’s parallelization capabilities37,38. The best model was selected by the mean squared error of the cross-validation. Akaike’s Information Criterion (AIC) is generally used as a means for model selection. However, we preferred cross-validation for model selection because AIC a priori assumes that simpler models with the high goodness of fit are more likely to have the higher predictive power, while cross-validation without any a priori assumptions measures the predictive performance of a model by efficiently running model training and testing on the available data.
We additionally validated the effect of different subsets of geographic features by assessing the relationship between statistically significant values of f4-statistic (i.e. |Z|> 3) and each subset. The relationship between f4-statistic of the form of f4(Outgroup,Test;X,Y) and geographic features was determined by measuring the agreement between the negative/positive signs of f4-statistic and the difference in LCD (LCD.D) for each pair of contemporaneous ancient populations X and Y. LCD.D was calculated as (LCD1–LCD2), where LCD1 was least-cost distance between the test population and X, and LCD2 was least-cost distance between the test population and Y. LCD.D < 0 indicated less least-cost distance between Test and X, while LCD.D > 0 indicated less least-cost distance between Test and Y. So, the same sign of f4 and LCD.D values indicated agreement between geographic proximity and genetic affinity. We used Cohen’s kappa39 to measure the agreement.
In order to test if geographic features (Table 3) accounted for present-day genetic differentiation in the Caucasus, we measured the relationship between Fst and LCD across the modern populations using the Mantel test in the “vegan” package40 in R version 3.5.2. In addition, we checked whether contribution from ancient samples was related to today’s genetic differentiation. To do so, we calculated median of f3-statistic of ancient populations of each geographic grouping (e.g. the following 6 populations made up one group: Balkans 39,950–41,950 BP, Balkans 37,950–39,950 BP, Balkans 31,950–33,950 BP, Balkans 9950–11,950 BP, Balkans 7950–9950 BP, Balkans 5950–7950 BP). Then we measured the manhattan distance of f3 median values of all combinations of the geographic groupings between the modern populations and compared the results to Fst and LCD using the Mantel test.
Results
12 sampled individuals were inferred to be 3rd and 4th degree relatives, while the rest were unrelated: two 3rd degree relatives from CHCHN, four 4th degree relatives from KHEVS, two 4th degree relatives from LAZ, two 4th degree relatives from MSKH and two 4th degree relatives from TUSH. Our sample of modern populations showed near-zero levels of inbreeding (Table 4). Average Fst between the modern populations in the Caucasus was 0.00951, with the maximum value of 0.027 between KHEVS and MSKH (Table 5). There was no significant differentiation between Kartvelian speakers AJAR, SMG, IMR, KRT and KAKH (Z < 3). The rest were significantly different from these and each other. ADMIXTURE analyses on the modern data distinguished KHEVS and TUSH from the rest across all K values (Supplementary Fig. S1). K = 2 had the smallest of the cross-validation errors that increased with K. The ADMIXTURE plots suggested a cline running from southwest and south (LAZ, MSKH) to north and northeast (CHCHN, KHEVS, TUSH).
The first and second PCA axes (Supplementary Fig. S2) showed a clear gradient from northeastern Anatolia (Kartvelian-speaking Laz communities) to the northern (Balkar, Karachay and Chechen communities) and northeastern (Kartvelian-speaking Tush and Khevsur) Caucasus. Populations occurring south of the main ridge of the Greater Caucasus were more closely related to each other than the northern populations were to each other. The northern populations showed two distinct clusters: the northeastern cluster of Kartvelian speakers (KHEVS, TUSH) and the northern cluster of Northeast-Caucasian and Turkic speakers (CHCHN, BLK, KRCH). Projecting the ancient populations onto the first two PCA axes generated a similar gradient, where the ancient populations of Anatolia and Levant were positioned opposite of the ancient Siberia and Europe. These gradients had the ancient populations of Levant-Anatolia and the modern populations of NE Anatolia at one end, and the ancient populations of Siberia and Europe and the modern populations of northern Caucasus at the other end. The ancient Caucasian hunter-gatherers fell within the variation of present-day Kartvelian speakers SVN and SMG. Ancient Anatolians and Levantines fell within the variation of southern Kartvelian speakers (MSKH) and Armenian speakers (ARM). Ancient Siberians and Europeans were closer to populations north of the Greater Caucasus main ridge (Chechens, Balkars and Karachays).
Generally, modern Caucasian populations had more genetic affinity with those ancient populations that were closer in time and space, and this relationship well explained the genetic difference between the modern populations (Figs. 2, 3, Supplementary Figs. S2–S5). This affinity was highest with the early post-glacial populations of the Caucasus, Anatolia and the Balkans. The ancient Caucasian ancestry was the highest in Kartvelian-speaking groups peaking in Imeretians (IMR), Svans (SVN) and Megrelians (SMG) in western Georgia—that is, the populations in the closest geographic proximity to the findings of the ancient Caucasian hunter-gatherers (aka CHG). Anatolian ancestry was the highest in southern and south-western Kartvelian-speaking Meskhs and Lazs (MSKH, LAZ) and Indo-European-speaking Armenians (ARM) in southern Georgia and north-eastern Turkey, respectively. Balkan and Siberian ancestry peaked in groups occurring north of the main ridge of the Greater Caucasus, namely in Kartvelian-speaking Tushs (TUSH) in north-eastern Georgia, Northeast-Caucasian-speaking Chechens (CHCHN) and Turkic-speaking Balkars (BLK) in the Russian Federation.

Genetic affinity (f3) between modern Caucasians and ancient populations.

Genetic affinity (f3) between modern populations of the Caucasus and ancient populations (labeled in different colors), plotted against least-cost distance for different 2000-year-long periods before present (BP). Pearson correlation coefficients (r) show relationships between f3 and linear least-cost distance. The smoothing lines are applied using the GAM-REML method.
Genetic affinity of f3-statistic between the modern Caucasians and ancient populations was best explained by the interaction of LCD and mean time BP between these populations (Table 6). In the best model LCD implied that (1) human movement was impeded by terrain ruggedness (TRI), (2) the Bosporus-Dardanelles and the English Channel did not act as barriers, (3) swamps, glaciers and desert were not full barriers, but permeable at the highest cost of the cost grid, and (4) riversides in desert and rivers were permeable at TRI values—i.e., the cost grid was the combination of geographic features 2, 4, 6, 8, 10, 12, 14 and 15 identified in Table 3. Genetic affinity generally increased as time BP and LCD decreased (Fig. 3, Supplementary Figs. S3 and S4). This cost grid generated LCD.D that had the greatest measure of agreement with genetic affinity based on f4-statistic (Kappa = 0.59, p-value = 2.02e−14, Fig. 4). There was overall negative correlation between f3-statistic and LCD (r = − 0.207, p < 0.0001), and between f3-statistic and time BP (r = − 0.540, p < 0.0001). Semi-partial correlation between f3 and LCD (controlled for the influence of time BP on f3) was − 0.297 (p < 0.0001). Correlation between LCD and genetic affinity were highly significant for a period of 5950–15,950 BP (r = − 0.909, p < 0.0001). During the glacial peak (17,950–25,950 BP) the correlation decreased (r = − 0.032, p = 0.8412). Further back in time (i.e. time BP > 25,950) the correlation was mostly not significant.

Genetic affinity (f4) between modern populations of the Caucasus and pairs of contemporaneous ancient populations. The numbers show the significant Z-values of f4 (i.e. |Z|> 3). Z < 0 indicates higher genetic affinity between the modern population and the ancient population at left of the pair on the Y-axis, while Z > 0 indicates higher genetic affinity between the modern population and the ancient population at right. Asterisk indicates disagreement between the genetic affinity and geographic proximity based on the least-cost distance.
Our model had disagreement between genetic affinity and geographic proximity (as a function of landscape permeability) to ancient populations dating from 13,950 to 15,950 BP and 37,950–39,950 BP (Figs. 3 and 4). All modern samples of the Caucasus had more affinity with Apennine 13,950–15,950 BP than with C. Europe 13,950–15,950 BP that was at closer least-cost distance, and all modern samples northeast of Laz and Armenians had more affinity with Anatolia 13,950–15,950 BP than with C. Europe 13,950–15,950 BP that was at closer least-cost distance. There was more affinity of our sample of modern Caucasians with ancient E. Asian individual than with a Balkan individual 37,950–39,950 BP that was at closer least-cost distance.
Our model generated least-cost paths from Eurasian and African ancient populations to the current Caucasian populations (Fig. 5). All least-cost paths from ancient populations of Europe, Siberia and East Asia reached populations south of the Greater Caucasus through the East European Plain, the north-western Caspian coast and the flood plain of the River Mtkvari (aka Kura) (Supplementary Fig. S6). All ancient populations occurring south of the Greater Caucasus reached populations north of the Greater Caucasus through either the western Caspian coast or across the Bosporus-Dardanelles and through the East European Plain (Supplementary Fig. S6). Least-cost paths from the ancient population of Anatolia to the Caspian watershed of the Caucasus ran across the Bosporus-Dardanelles and through the East European Plain, the western Caspian coast and the flood plain of the River Mtkvari (Supplementary Fig. S6). Least-cost paths from Africa, Levant, Anatolia and Alborz reached the southern Caucasus either across the Pontic Mountains or through the flood plains of the Rivers Araxes and Mtkvari. Least-cost paths from Africa converged in the Levant before reaching the Caucasus.

Migration pathways that best explain genetic affinity between modern Caucasians and ancient humans, showing least-cost paths from Eurasian and African ancient populations (shown in the main map, see Table 2 for details) to the current Caucasian populations (identified in the inset map, see Table 1 for details). The least-cost paths imply that (1) human movement is impeded by terrain ruggedness (TRI), (2) the Bosporus-Dardanelles and the English Channel do not act as barriers, (3) swamps, glaciers and desert are not full barriers, but permeable at the highest of the cost grid values, and (4) riversides in desert are permeable at TRI values. It is assumed that most human movements occurred during climate warming events when the earth’s surface was not dramatically different from that of today, and hence the current data of the geographic features are used in the calculation of the least-cost paths. The map is generated using QGIS Desktop 3.10.6-A Coruña (https://qgis.org).
Fst had the strongest correlation with LCD derived from the same geographic features that explained best f3 and f4 statistics (Mantel Test: r = 0.402, Monte-Carlo simulated p-value = 0.0295 based on 1,000,000 replicates; Supplementary Fig. S7). Neighbor-Joining tree based on Fst (Supplementary Fig. S8) clustered together (1) Kartvelian-speaking populations from western Georgia and north-eastern Turkey, (2) Kartvelian and Armenian speakers from southern Georgia and eastern Turkey, (3) Northeast-Caucasian and Turkic speakers from the north Caucasus, and (4) Kartvelian speakers from the northeastern Greater Caucasus being the most genetically distinct from the others. Sampled Kartvelian-speaking populations from central (KRT) and eastern Georgia (KAKH) were most admixed. Additional Mantel tests showed that Fst was significantly correlated with difference in contribution from a combination of 4 ancient populations: Anatolia, Balkans, Caucasus, Siberia (r = 0.2906, p = 0.04033). The difference in contribution from these ancient populations was also significantly correlated with LCD (r = 0.3701, p = 0.003187). Partial Mantel test controlling for LCD made the correlation between Fst and ancient contribution insignificant (r = 0.1668, p = 0.15608). Controlling for ancient contribution significantly reduced the correlation between Fst and LCD (r = 0.311, p = 0.1). The Mantel tests suggested that LCD and ancient ancestry explained much of the present-day genetic variation in the Caucasus.
Discussion
Our results suggest that the current populations of the Caucasus bear detectable ancestry from Caucasian, Anatolian and Balkan hunter-gatherers. Caucasian hunter-gatherers (CHG) are the major gene contributors to the modern Caucasian populations. The proportion of CHG alleles is the highest in modern populations that live in close proximity to the archaeological sites in western Georgia, where the bones of CHG were discovered8, and gradually decrease away from this area, being replaced mostly with alleles of ancient Anatolians and Europeans. These archaeological sites fall within the Colchic refugium where humans survived a series of glacial maxima including the LGM in the Caucasus5. Our model shows that in early post-glacial time the migration of hunter-gatherers from elsewhere into the Caucasus intensified. Ancient Anatolian alleles are most frequent in the genomes of modern people from southern Georgia and eastern Turkey (i.e. Georgians from Meskheti province, Laz and Armenians), while ancient European or Balkan alleles are most common in modern populations of the North Caucasus, suggesting that European hunter-gatherers migrated to the Caucasus across the East European Plain rather than through Asia Minor. Remarkably, ancient genomes from Levant (Natufian culture) and Atlas Mountains (northwestern Africa), have the highest allele share with the same modern populations as ancient Anatolians, which suggests some admixture between these populations before Anatolian population started to expand to the Caucasus. The similarity of genetic gradients of the ancient and modern Caucasus in our PCA plot once again confirms genetic continuity spanning from the Late Upper Paleolithic until today8,25. Our study explicitly explains the reasons behind this phenomenon.
Traces of different ancient populations (migration sources) in modern communities of the Caucasus decrease with (i) geographic distance weighted by physical barriers to dispersal, and (ii) the age of the ancient population. This signal is sufficiently strong to differentiate between the modern populations by their affinity to individual ancient populations. Our analyses suggest that the expansion of the refugial populations of Upper Paleolithic and Mesolithic hunter-gatherers was greatly impeded by rugged terrain, swamps, glaciers and desert, and the genetic legacy of these hunter-gatherers in the modern Caucasus is well explained by landscape permeability between the refugial human populations. The relationship between the ancient genetic signature and landscape permeability is stronger if the refugial populations are closer in time to the present-day Caucasus, suggesting that the genetic signature associated with a particular ancestral area fades away as a result of gene drift and later admixture events.
Although our model performed highly significantly, it failed to explain disagreement between genetic affinity and geographic proximity (as a function of landscape permeability) to ancient populations dating from 13,950–15,950 BP and 37,950–39,950 BP (Figs. 3 and 4). All modern samples of the Caucasus had more affinity with Apennine 13,950–15,950 BP than with geographically closer C. Europe 13,950–15,950 BP, and all modern samples northeast of Laz and Armenians had more affinity with Anatolia 13,950–15,950 BP than with geographically closer C. Europe 13,950–15,950 BP. This is probably due to early post-glacial admixture of Anatolian hunter-gatherers with Caucasian and European hunter-gatherers, and that of East Asian hunter-gatherers with European hunter-gatherers at this period41,42. These admixture events, coupled with our landscape permeability model, suggest that ancient Anatolians carrying the genetic signature of Caucasian hunter-gatherers would contribute more to Apennine hunter-gatherers through Bosporus-Dardanelles and the Balkans than to C. European ones, while C. European ones would get more genes through Eurasian Steppes from East Asian hunter-gatherers. These events of admixture from east to west, that may be facilitated by technological advances or advantages through natural selection (e.g. resistance to lethal infectious diseases), make our sample of modern populations more closely related to the ancient populations from Anatolia and the Apennine than to the ancient population from C. Europe.
Our sample of ancient populations dating from 37,950 to 39,950 BP were each represented by a single individual from the Balkans and E. Asia. More affinity of our sample of modern Caucasians with an ancient E. Asian individual than with a geographically closer Balkan individual 37,950–39,950 BP could be explained by two scenarios: either this Balkan individual was a vagrant from elsewhere, which was shown not to have contributed to any present-day populations43 or between when anatomically modern humans arrived in Europe approximately 45,000 years ago44 and the LGM, climate was the mildest around 37,950–39,950 BP45,46 allowing more gene flow between western and eastern Eurasia. This gene flow is supported by the fact that this individual from the Balkans was more closely related to East Asians than Paleolithic and Mesolithic Europeans47, while a contemporaneous individual from western Russia was more closely related to later Europeans than to East Asians48. At glacial maxima human source populations were highly fragmented, had no or limited gene flow with each other and survived in glacial refugia, from where the populations would expand during benign climates and shrink back to during harsh climates5. Thus, our model better explains the genetic legacy of ancient populations in the modern Caucasus, if there is more genetic dissimilarity between these ancient populations than within them.
Even today genetic affinity (Fst) between modern populations of the Caucasus significantly correlates with least-cost distance between the hubs of these populations, which is in line with previous studies based on limited sample and genetic markers22. The genetic affinity between the modern populations has the strongest response to the least-cost distance weighted by the same geographic features that account best for the genetic affinity between the ancient and the modern populations. However, without considering the past metapopulation picture, landscape permeability between present-day populations fails to explain the considerable fraction of variation in current genetic structure in the Caucasus.
The derived least-cost paths suggested that both the Greater Caucasus, the Lesser Caucasus and the Pontic Mountains considerably impeded the inferred human movements through the Caucasus (Figs. 1, 5, Supplementary Fig. S6). The Greater Caucasus posed the major barrier to human movements that is in agreement with the north–south cline in genetic variation (this study26,49. The genetic differentiation was the strongest between the populations from south and north of the Greater Caucasus main ridge, which once again indicates that the Greater Caucasus has been an important physical barrier for human dispersal since the last glacial period. However, there were also noticeable differences between populations from the same side of this mountain range, suggesting their different exposure to incoming migrants through heterogenous landscape. The western flank of the Caspian Sea appeared to be more permeable for human movement than the eastern flank of the Black Sea. None of the least-cost paths from Europe, Siberia and East Asia ran through the eastern flank of the Black Sea. The eastern flank of the Black Sea is walled by the Greater Caucasus to the north and the Lesser Caucasus and the Pontic Mountains to the south, while the western flank of the Caspian Sea is largely open. This difference between the eastern and western edges of the Caucasus explains the east–west cline in Y-chromosome and autosomal genome variations in the Caucasus7,17,22,24.
We were not able to sample other ethnic groups inhabiting the Caucasus. These groups would have provided more information about the genetic structure of the region. However, our sample was sufficient to test our hypotheses. Publications to date have measured the gnome-wide ancestry of ancient populations in many of the present-day communities on earth. However, to the best of our knowledge, this study is the first of its kind to estimate the relationship of genome-wide genetic affinity between modern and ancient populations to landscape permeability as a function of different geographic features, and infer major dispersal paths from refugial populations. Our model can help to time and map the dispersal and migration paths of prehistoric events not only in relation to the Caucasus but also elsewhere. Presence of significant negative correlation between the weighted geographic distance from the ancient source of a distinct genetic signature and genetic affinity with this source can be seen as evidence of migration via a particular path and at a particular time.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files). Any additional data related to this study are available from the corresponding author on request.
References
Tarkhnishvili, D. Historical Biogeography of the Caucasus (Nova Science Publishers Inc., 2014).
Gorenflo, L. J., Romaine, S., Mittermeier, R. A. & Walker-Painemilla, K. Co-occurrence of linguistic and biological diversity in biodiversity hotspots and high biodiversity wilderness areas. PNAS 109(21), 8032–8037 (2012).
Myers, N., Mittermeier, R. A., Mittermeier, C. G., da Fonseca, G. A. B. & Kent, J. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (2000).
Cavalli-Sforza, L. L. Genes, peoples, and languages. Proc. Natl. Acad. Sci. U. S. A. 94, 7719–7724 (1997).
Gavashelishvili, A. & Tarkhnishvili, D. Biomes and human distribution during the last ice age. Glob. Ecol. Biogeogr. 25, 563–574 (2016).
Stewart, J. R. & Stringer, C. B. Human evolution out of Africa: the role of refugia and climate change. Science 335, 1317–1321 (2012).
Tarkhnishvili, D., Gavashelishvili, A., Murtskhvaladze, M., Gabelaia, M. & Tevzadze, G. Human paternal lineages, languages and environment in the Caucasus. Hum. Biol. 86, 113–130 (2014).
Jones, E. R. et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nat. Commun. 6, 8912. https://doi.org/10.1038/ncomms9912 (2015).
Allentoft, M. E. et al. Population genomics of Bronze Age Eurasia. Nature 522, 167–172. https://doi.org/10.1038/nature14507;pmid:26062507 (2015).
Anthony, D. The Horse, the Wheel, and Language: How Bronze-Age Riders from the Eurasian Steppes Shaped the Modern World (Princeton University Press, 2007).
Gimbutas, M. Primary and secondary homeland of the Indo-Europeans. J. Indo-Eur. Stud. 13, 185–202 (1985).
Guimaraes, S. et al. Ancient DNA shows domestic horses were introduced in the southern Caucasus and Anatolia during the Bronze Age. Sci. Adv. 6, eabb0030 (2020).
Haak, W. et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature 522, 207–211. https://doi.org/10.1038/nature14317;pmid:25731166 (2015).
Narasimhan, V. M. et al. The formation of human populations in South and Central Asia. Science 365, eaat7487. https://doi.org/10.1126/science.aat7487 (2019).
Anthony, D. Archaeology, genetics and language in the steppes: A comment on Bomhard. J. Indo-Eur. Stud. 47(1–2), 175–198 (2019).
Bomhard, A. R. The origins of proto-Indo-European: The Caucasian substrate hypothesis. J. Indo-Eur. Stud. 47, 9–124 (2019).
Balanovsky, O. et al. Parallel evolution of genes and languages in the Caucasus region. Mol. Biol. Evol. 28, 2905–2920 (2011).
Karafet, T. et al. Coevolution of genes and languages and high levels of population structure among the highland populations of Daghestan. J. Hum. Genet. 61, 181–191. https://doi.org/10.1038/jhg.2015.132 (2016).
Nasidze, I. et al. Mitochondrial DNA and Y-chromosome variation in the Caucasus. Ann. Hum. Genet. 68, 205–221 (2004).
Nasidze, I. et al. Testing hypotheses of language replacement in the Caucasus: Evidence from the Y-chromosome. Hum. Genet. 112, 255–261 (2003).
Schonberg, A., Theunert, C., Li, M., Stoneking, M. & Nasidze, I. Highthroughput sequencing of complete human mtDNA genomes from the Caucasus and West Asia: High diversity and demographic inferences. Eur. J. Hum. Genet. 19, 988–994 (2011).
Tarkhnishvili, D., Gavashelishvili, A., Murtskhvaladze, M. & Latsuzbaia, A. Landscape complexity in the caucasus impedes genetic assimilation of human populations more effectively than language or ethnicity. Hum. Biol. 88(4), 287–300 (2016).
Yardumian, A. et al. Genetic diversity in Svaneti and its implications for the human settlement of the Highland Caucasus. Am. J. Phys. Anthropol. 164(4), 837–852. https://doi.org/10.1002/ajpa.23324 (2017).
Yunusbayev, B. et al. The Caucasus as an asymmetric semipermeable barrier to ancient human migrations. Mol. Biol. Evol. 29(1), 359–365 (2012).
Skourtanioti, E. et al. Genomic history of neolithic to bronze age Anatolia, Northern Levant, and Southern Caucasus. Cell 181, 1158–1175 (2020).
Wang, C. et al. Ancient human genome-wide data from a 3000-year interval in the Caucasus corresponds with eco-geographic regions. Nat. Commun. https://doi.org/10.1038/s41467-018-08220-8 (2019).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. (2018). http://www.R-project.org.
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Paradis, E. & Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528 (2019).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 12, 246. https://doi.org/10.1186/1471-2105-12-246 (2011).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26(22), 2867–2873 (2010).
Patterson, N. et al. Ancient admixture in human history. Genetics 192(3), 1065–1093. https://doi.org/10.1534/genetics.112.145037 (2012).
Wood, S. N. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. R. Stat. Soc. Ser. B (Stat. Methodol.) 73, 3–36. https://doi.org/10.1111/j.1467-9868.2010.00749.x (2011).
Weston, S. foreach: Provides foreach looping construct for R. R package version 1.4.4. (2017). http://CRAN.R-project.org/package=foreach.
Weston, S. doParallel: foreach parallel adaptor for the “parallel” package. R package version 1.0.14. (2018). https://cran.r-project.org/web/packages/doParallel/index.html.
Cohen, J. A coefficient of agreement for nominal scales. Educ. Psychol. Meas. 20, 37–47 (1960).
Oksanen J. et al. vegan: Community Ecology Package. R package version 2.5-7. (2020). https://CRAN.R-project.org/package=vegan.
Feldman, M. et al. Late Pleistocene human genome suggests a local origin for the first farmers of central Anatolia. Nat. Commun. 10, 1218. https://doi.org/10.1038/s41467-019-09209-7 (2019).
Fu, Q. et al. The genetic history of Ice Age Europe. Nature 534(7606), 200–205. https://doi.org/10.1038/nature17993 (2016).
Yang, et al. 40,000-Year-old individual from Asia provides insight into early population structure in Eurasia. Curr. Biol. 27, 3202–3208 (2017).
Gamble, C., Davies, W., Pettitt, P. & Richards, M. Climate change and evolving human diversity in Europe during the last glacial. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 359, 24–53. https://doi.org/10.1098/rstb.2003.1396.discussion253-244 (2004).
EPICA Community Members. Eight glacial cycles from an Antarctic ice core. Nature 429, 623–628 (2004).
Petit, J. R. et al. Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica. Nature 399, 429–436 (1999).
Fu, Q. et al. An early modern human from Romania with a recent Neanderthal ancestor. Nature 524(7564), 216–219. https://doi.org/10.1038/nature14558 (2015).
Seguin-Orlando, A. et al. Paleogenomics. Genomic structure in Europeans dating back at least 36,200 years. Science 346, 1113–1118 (2014).
Jeong, C. et al. The genetic history of admixture across inner Eurasia. Nat. Ecol. Evol. 3(6), 966–976 (2019).
Acknowledgements
We would like to thank Giorgi Janashvili, Anzori Gogotidze and Ozan Sari for helping during field work with collecting samples, and Mehmet Somel for discussion of the results and allowing us the access to computational resources at Middle East Technical University in Ankara.
Author information
Authors and Affiliations
Contributions
A.G. and D.T. conceived this study and wrote the manuscript. A.G. and A.Y. designed, performed the analyses and interpreted the results with contributions from D.T. Sample collection was coordinated by A.G., D.T., M.M., I.A. and C.K. and M.M. conducted DNA extraction and storage. A.G. and D.T. supervised the work. All authors approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gavashelishvili, A., Yanchukov, A., Tarkhnishvili, D. et al. Landscape genetics and the genetic legacy of Upper Paleolithic and Mesolithic hunter-gatherers in the modern Caucasus. Sci Rep 11, 17985 (2021). https://doi.org/10.1038/s41598-021-97519-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97519-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
