
Abstract
Cystoseira sensu lato (Class Phaeophyceae, Order Fucales, Family Sargassaceae) forests play a central role in marine Mediterranean ecosystems. Over the last decades, Cystoseira s.l. suffered from a severe loss as a result of multiple anthropogenic stressors. In particular, Gongolaria barbata has faced multiple human-induced threats, and, despite its ecological importance in structuring rocky communities and hosting a large number of species, the natural recovery of G. barbata depleted populations is uncertain. Here, we used nine microsatellite loci specifically developed for G. barbata to assess the genetic diversity of this species and its genetic connectivity among fifteen sites located in the Ionian, the Adriatic and the Black Seas. In line with strong and significant heterozygosity deficiencies across loci, likely explained by Wahlund effect, high genetic structure was observed among the three seas (ENA corrected FST = 0.355, IC = [0.283, 0.440]), with an estimated dispersal distance per generation smaller than 600 m, both in the Adriatic and Black Sea. This strong genetic structure likely results from restricted gene flow driven by geographic distances and limited dispersal abilities, along with genetic drift within isolated populations. The presence of genetically disconnected populations at small spatial scales (< 10 km) has important implications for the identification of relevant conservation and management measures for G. barbata: each population should be considered as separated evolutionary units with dedicated conservation efforts.
Similar content being viewed by others

Introduction
Marine forests (i.e. seascapes dominated by habitat-forming seaweeds1) are among the most productive ecosystems in temperate rocky coasts, enhancing biodiversity, ecosystem functioning and habitat complexity2,3. However, multiple anthropogenic stressors interacting at both local and global scales increasingly threaten these iconic ecosystems4. As a result, a severe decline of large seaweeds abundance has been observed across temperate coastal systems over the last decades5,6,7,8. In particular, Cystoseira sensu lato (Class Phaeophyceae, Order Fucales, Family Sargassaceae), which forms the most extensive macroalgae forest in the Mediterranean Sea9,10, have recently suffered from irreversible loss11, likely underestimated as much of it may have gone unnoticed12,13.
Facing the dramatic decline of Cystoseira s.l. populations throughout the Mediterranean Sea, Cystoseira-formed habitats are under the protection of the Resolution 4 of the Bern Convention (Council of Europe 2010), EU Habitats Directive (The Council of the European Union 1992) and are listed in the European red list of habitats14. The resulting need of setting conservation actions for Cystoseira s.l. has recently fostered the development of ecological restoration programs: they aim to increase the effectiveness of conservation strategies of Cystoseira s.l., by notably including fostering natural recovery, reforesting lost areas, and/or enhancing the performance of existing human infrastructures as substrata for colonisation15,16,17,18,19,20,21,22. In addition, restoration actions also include local measures to control the drivers of canopy loss (e.g. excess of sediments and/or nutrients5, habitat destruction23,24), such as improving water quality25 or restoring habitat locally12. Such actions intend to increase the natural recovery rate of disturbed populations of Cystoseira s.l., expected to be very low, notably due to their life cycle comparable to a one-phase life cycle. Indeed, although Cystoseira s.l. display a biphasic life cycle, alternating a haploid phase with a diploid phase, the gametophytic stage, i.e. the haploid phase, is highly reduced and retained in the conceptacles, so that plants superficially regenerate only from sporophytes26. In addition, relatively large and heavy eggs and zygotes tend to rapidly sink just after being released27. As a result, Cystoseira likely display very limited dispersal abilities28,29,30, hampering the natural recovery of the populations.
In parallel to encouraging restoration actions, expanded research efforts regarding Cystoseira s.l. species distribution31,32 and ecology10,30,33,34,35 constantly improve scientific-based conservation plans. The diversity and phylogenetic relationships among the Cystoseira taxa from the eastern Atlantic and Mediterranean have also very recently been reappraised36,37,38. Nevertheless, knowledge of the genetic diversity and connectivity of natural populations further needs to be improved in order to assess the potential resilience of disturbed populations. Knowledge of the genetic variation in a study species could also help designing new restorations actions that are “future proofed” against rapidly changing conditions, such as assisted gene flow, or habitat restorations in sites according to the spatial pattern of genetic variation observed, i.e. prioritizing sites where genetic diversity would be seriously eroded.
On the one hand, genetic diversity, defined as the diversity of the basic units of hereditary information (genes) within a species, is one of the three biological diversity components, receiving increased consideration in IUCN planning and actions14. Beyond its significant effect on a number of ecological processes, including population recovery from disturbance and community structure39,40,41,42,43, genetic diversity is required for populations to evolve in response to ubiquitous, natural environmental change44. On the other hand, population connectivity, defined as the exchange of individuals among geographically separated subpopulations, plays a fundamental role in local and metapopulation dynamics, community dynamics and structure, and the resiliency of populations45. It ensures the genetic cohesion of a biological species over its distribution range46 because migration is the evolutionary force that spreads genetic diversity among populations, leading to relatively similar allele frequencies and counterbalancing the negative effect of genetic drift on genetic diversity47.
Severe and sudden reductions in population size are expected to result in a loss of genetic diversity and increased inbreeding44. In species restricted to small and isolated remnant populations, notably due to the fragmentation or destruction of their habitat, these threats can be worsened, further reducing the ability of populations to cope with environmental changes and hence increasing their risk of local extinction48. Nonetheless, the risk of population extinction can be reversed by migration processes49, making population connectivity a key feature for the recovery of the demographic structure and genetic diversity of disturbed populations. To avoid the extirpation of disturbed populations, scientific-based genetic management measures are therefore warranted50. As such, inferring population connectivity is a first step as the degree of connectivity among geographic areas sets the scale at which conservation strategies for marine species need to be applied51.
Population connectivity can be inferred from the analysis of the genetic differentiation among populations52. Population genetics thus offer a conceptual framework that provides increasingly appropriate statistical analysis methods to determine intra-population genetic variation, assess genetic connectivity in marine environment and infer demography history of populations48,53. In Cystoseira s.l., high levels of genetic structure indicative of low population connectivity have been observed in several species11,13,54,55,56,57,58 in accordance with their expected poor dispersal abilities.
In the Mediterranean Sea, Gongolaria barbata (Stackhouse) Kuntze, formerly known as Cystoseira barbata (Stackhouse) C. Agardh then Treptacantha barbata (Stackhouse)59, is one of the widely distributed Mediterranean Cystoseira s.l., thriving in shallow sheltered coasts and lagoons36. Local populations of G. barbata have recently suffered from spectacular regression in several areas of the Mediterranean Sea6,15,60,61, as well as in the Black Sea, where it was formerly a dominant species62,63. This species is severely threatened both by land-based and marine human impacts, such as the artificialization of the coastline which likely appears more important in sheltered bays than in exposed shores. The decrease in water quality, including pollution, eutrophication and increase in sediment loads, provides an additional threat whose effects are particularly important in enclosed bays with low water renewal. As a result, this species is now only found in few localities across its distribution range64. The combination of geographic isolation of remnant populations, recent decreases in abundance and expected restricted dispersal abilities in G. barbata call for an urgent exploration of its genetic status.
The only genetic work involving G. barbata samples from several locations showed that Black Sea mitochondrial DNA (mtDNA) sequences of the cytochrome oxidase subunit 1 (COI) gene systematically differed by one nucleotide from Mediterranean sequences62, indicating restricted gene flow between these two seas. In addition, very low intra-specific diversity was found among the Adriatic Sea, Menorca, Sicily and Black Sea samples. However, as the mtDNA lacks the necessary resolution to provide efficient detection of genetic structure65, the use of nuclear makers with higher polymorphism level is warranted. Microsatellite markers would likely unravel the population genetic diversity and structure in G. barbata, thanks to their codominance and high level of polymorphism properties66.
In the present study, we investigated the genetic diversity and structure of 15 relict populations sampled in the Ionian, the Adriatic and the Black Seas based on highly polymorphic markers specifically developed for the study species. This will inform whether the natural recovery of G. barbata depleted populations will strongly be compromised without enhanced restoration actions.
Results
A total of 503 individuals from 15 sampled populations (Fig. 1; Table 1) was amplified using the set of twelve microsatellite markers newly developed; 383 individuals successfully amplified at six markers at least, with up to two alleles per marker. Out of these 383 genotyped individuals, eight pairs of individuals (2.1%) were presumed to be clones. These were observed across several distinct sites (two pairs in site 5, in site 9 and in site 12; one pair in site 7 and in site 8). In addition, these pairs of identical individuals were labeled with sequential—or very close—numbers. As we can not rule out that the same individual was sampled twice, we decided that one unique genotype per clone was retained for further analysis. This resulted in a final dataset composed of 375 unique genotypes using twelve microsatellite markers.

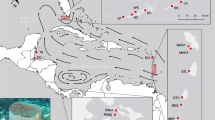
Sampling locations of Gongolaria barbata and the global Sea surface circulation (grey arrows). Each study site is labeled as follow: (A) in the Ionian Sea: 1—Porto Cesareo—Isola Scoglio (Italy), 2—Porto Cesareo (Italy), (B) in the Adriatic Sea: 3—Conero (Italy), 4—Kornati (Croatia), 5- and 6—Boka Kotorska (Montenegro), and (C) in the Black Sea: 7—Costinesti (Romania), 8—Kaliakra (Bulgaria), 9–11—Ropotamo Kiten (Bulgaria), 12—Sile (Turkey), 13—Sinop (Turkey), 14—Karadagand (Ukraine) and 15—Tarhankut (Ukraine). GPS coordinates are provided in Table 1. The map was created using the R package mapdata105,106,107.
Quality control of the newly developed markers
The quality and variability of the microsatellite markers were tested for population genetic studies of G. barbata based on a sampling comprising 15 sites in three Seas, the Ionian, the Adriatic and the Black Seas. All loci were found to be polymorphic (5–39 alleles, Table 2), with Hs estimates varying from 0.242 (Cysbar2) to 0.736 (Cysbar5). FIS values ranged from − 0.710 (Cysbar6) to 0.501 (Cysbar3), with significant deviations from Hardy–Weinberg Equilibrium observed for seven loci (Cysbar3–Cysbar6, Cysbar9–Cysbar11). Missing data were present in all subsamples and in ten out of the 12 microsatellite markers (Supporting Information SI1), likely attributed to the presence of null alleles. Of the twelve microsatellite markers tested, MicroChecker suggested the presence of null alleles in one to five subsamples (out of 15) for seven loci (Cysbar1 to Cysbar5, Cysbar10 and 11). However, using unilateral exact binomial tests, heterozygosity deficiencies were likely explained by the presence of null alleles in only Cysbar3 (p-value = 0.133). MicroChecker did not detect any stuttering or short allele dominance (SAD), however, the regression approaches detected significant SAD in Cysbar5 (ρFIS = − 0.530, p-value = 0.0002; ρFST = − 0.591, p-value = 0.00003). Stuttering was not detected following De Meeûs et al.67 approach.
The analysis of 375 G. barbata individuals genotyped with twelve microsatellite markers showed significant linkage disequilibrium between two loci, cysbar6 and cysbar12, after Benjamini and Yekutieli adjustment. We removed three problematic microsatellite markers, Cysbar3 and Cysbar5 for which technical issues were observed, and Cysbar6, in linkage disequilibrium with Cysbar12, from the final dataset.
Once removing these three markers (N = 375 individuals, nine microsatellite markers, 6.7% missing data, SI1), we analyzed the stability of FIS and FST from one locus to the other following the determination key proposed by De Meeûs68. A strong and significant heterozygote deficiency was observed with f = 0.066 (p-value = 10–3). The FIS variation across loci was moderate (Fig. 2), but a high FST variation was observed among loci, the Pearson correlation between FIS and FST was not significant (r(FIS,FST) = 0.257 p-value = 0.397) and the standard error of FIS (StrdErrFIS = 0.022) was two times lower than for FST (StrdErrFST = 0.046). A strong and significant correlation was however observed between the number of missing data and FIS values (ρ = 0.733, p-value = 0.016). According to the determination key proposed by De Meeûs68, null alleles may have persisted in the dataset, despite a careful attention paid to minimize them (cf. analyses aforementioned), which would explain these results. This hypothesis cannot be ruled out since null homozygotes (i.e. non-amplifying locus of an individual, while all other loci successfully amplify in this individual) were still present in the final dataset. Heterozygote deficiencies may also be explained by demographic causes, such as a Wahlund effect.

Variation of Weir and Cockerham120 estimation of FIS (f) and FST (θ) for each of the twelve microsatellite markers and across loci in Gongolaria barbata sampled in the Ionian, Adriatic and Black Seas (a and b, respectively). The variation of Weir and Cockerham120 estimation of FIS (f) and FST (θ) for the nine microsatellites used in the study within the Ionian and Adriatic Seas (in pink) and within the Black Sea (in blue) was also provided (c and d, respectively). For each locus, 95% confidence intervals were obtained by jackknife over subsamples and by bootstrap over loci. The averaged f and θ across loci were computed based on nine microsatellite markers, i.e. removing the three markers shaded in grey. The horizontal dashed line represents the 0 value. This figure was created using the R software107.
A similar level of genetic diversity among samples
Estimates of allelic richness, gene diversity and departure from Hardy–Weinberg equilibrium, for each study site are presented in Table 1. Allelic richness (Ar) ranged from 1.9 (site 14) to 3.5 (site 3), and expected heterozygosity (HS) from 0.272 (site 14) to 0.611 (site 11). Ar and HS did not differ significantly among subsamples (Ar: p-value = 0.17; HS: p-value = 0.20).
Departure from Hardy–Weinberg equilibrium was found in six study sites (sites 3–5, 7 and 13).
Multi-Locus Heterozygosity (MLH) ranged from 0 (sites 6, 7, 12 and 13) to 8 (sites 1, and 7). MLHs were normally distributed (Shapiro test p-values > 0.5) and no difference in MLH distribution was observed (ANOVA p-value > 0.5). Identity disequilibrium, i.e. a measure of departure from random associations of homozygosity between loci, was however identified in site 4, with the only significant g2 observed (Table 1), suggesting that few inbred individuals were present in this subsample. Selfing rate was estimated within each subsample and did not differ significantly from zero (p-values > 0.1 after Benjamini and Yekutieli adjustment).
Strong genetic structure at large spatial scale
High and significant genetic differentiation was observed among all subsamples across loci (θ = 0.375, p-value < 10–3, ENA corrected FST = 0.355, IC = [0.283, 0.440]). Based on a hierarchical analysis with three levels (level 1: Mediterranean and Black Seas, level 2: Ionian, Adriatic and Black Seas, level 3: all subsamples), we observed that the three levels influenced genetic structure at a 5% level (Flev1/Total = 0.137, IC = [− 0.033, 0.322]); Flev2/lev1 = 0.279, IC = [0.196, 0.350]); Flev3/lev2 = 0.142, IC = [0.144, 0.250]); FInd/lev3 = 0.066, IC = [0.027, 0.109]).
The Structure analysis revealed a net differentiation between the Mediterranean and the Black Seas samples (Fig. 3a), in line with the strong and significant heterozygote deficiencies observed, partly explained by the presence of null allele, but also likely explained by a Wahlund effect, which justified a clustering approach. This result is congruent with the Principal Component Analysis along the first axis (PCA; SI2) and the genetic distances (Table 3). Note that different values of K were explored (from two to 18) and K = 2 provided the best meaningful result, not only using the Evanno et al.69 method (ΔKK=2 = 4558.9, ΔKK>2 < 1.5), but also by crosschecking these results with PCA and FST estimates. Further increases in K were provided in SI2.

Genetic population structure of Gongolaria barbata based on nine microsatellite markers. Individual Bayesian ancestry proportions were determined using Structure with K = 2 across G. barbata distribution range (a), with K = 3 (b) within the Ionian and Adriatic Seas and with K = 3 (c) within the Black Sea. Black lines separate each study site. The clusters identified are distinguished by pink and blue colors, standing for the Mediterranean and Black Sea sites, respectively and referring to Fig. 1. Each individual is depicted as a vertical bar with colors distinguishing its ancestries to the clusters identified. Individuals’ assignment was visualized using the R software106.
These two lineages, i.e. Mediterranean samples and Black Sea samples, required being analyzed separately.
Genetic structure within the Ionian and Adriatic Seas
A strong and significant heterozygote deficiency was observed with f = 0.103 (p-value < 10–3). High and significant genetic differentiation was observed among all subsamples across loci (θ = 0.313, p-value < 10–3, ENA corrected FST = 0.278, IC = [0.208, 0.341]). The FIS variation across loci was higher than the variation observed among all subsamples (Fig. 2), the Pearson correlation between FIS and FST was not significant (r(FIS,FST) = − 0.133 p-value = 0.733), and the standard error of FIS (StrdErrFIS = 0.066) was 1.5 higher than for FST (StrdErrFST = 0.043). No correlation was observed between the number of missing data and FIS values (ρ = − 0.017, p-value = 0.517).
Further, within the Ionian and Adriatic Seas, a strong structuration was revealed by the clustering analyses. Again, different values of K were explored (from two to ten, results provided in SI3) and K = 3 (Fig. 3b) provided the best meaningful results (ΔKK=3 = 1551.4, ΔKK=4 = 112.6, ΔKK=2, 5–10 < 1, but also by crosschecking these results with PCA and FST estimates). Geographic groups comprised (i) sites 1–2, (ii) sites 3–4 and (iii) sites 5–6. A similar genetic structure was detected by PCAs, provided in SI3.
Genetic structure within the Black Sea
A moderate but significant heterozygote deficiency was observed with f = 0.052 (p-value = 10–3). High and significant genetic differentiation was observed among all subsamples across loci (θ = 0.169, p-value < 10–3, ENA corrected FST = 0.163, IC = [0.111, 0.207]). The FIS variation across loci was moderate (Fig. 2), the Pearson correlation between FIS and FST was not significant (r(FIS,FST) = 0.311, p-value = 0.300), and the standard error of FIS (StrdErrFIS = 0.040) was c.a. two times higher than for FST (StrdErrFST = 0.026). No correlation was observed between the number of missing data and FIS values (ρ = − 0.167, p-value = 0.666).
Genetic structure within the Black Sea was not depicted as clearly as in the Mediterranean Sea, revealing higher levels of gene flow among sampled sites. Based on the clustering analyses, at K = 3 (Fig. 3c; ΔKK=3 = 117. 44), (i) sites 14 and 15, (ii) sites 7–8 and 10, and (iii) site 13 showed clear differentiation among them, though some individuals were genetically assigned to the alternative genetic backgrounds (Fig. 3c). Further, sites 9–12 showed mixed ancestry to three genetic backgrounds, suggesting some local gene flow. In addition to mixed ancestries observed in sites 9–12, some individuals were assigned to one of the two genetic backgrounds with an ancestry rate higher than 0.8. Further increases in K did not result in new meaningful clusters (ΔKK < 11, except ΔKK=2 = 101.3, results provided in SI4). Clustering from PCA (provided in SI4) appeared similar to clustering revealed by Structure.
Within sea estimates of dispersal
Significant correlations were observed between genetic differentiation and oceanographic distances among sites, either using the shortest path, i.e. the shortest distance between two sites, or the longest path, i.e. following the coastlines, in the Black Sea, but only using the shortest path in the Adriatic Sea (Fig. 4). In the Adriatic, we observed a slope b = 0.121, which, once translated, suggested a relatively small neighborhood size Nb = 8 individuals and an immigration from neighbors within the same sea of Nem = 1.32 individual per generation and subpopulation. With an averaged Ne = 78.55, and based on an estimated number of populations between 10 and 100 in the Adriatic Sea, the estimates of effective population density ranged from 0.006 to 0.06 individuals/km2, i.e. very sparse populations, and the dispersal (δ) was estimated between 186 and 587 m, i.e. very restricted dispersal. In the Black Sea, based on the longest path that better explained the regressions, the slope b = 0.0238 was translated into a neighborhood size Nb = 42 individuals and an immigration from neighbors within the same sea of Nem = 6.69 individuals per generation and subpopulation. With an averaged Ne = 50.76, and based on an estimated number of populations between 100 and 1000 in the Black Sea, the estimates of effective population density ranged between 0.014 and 0.140 individuals/km2, i.e. very sparse populations, though higher than in the Adriatic, and the dispersal (δ) was estimated between 129 and 408 m, i.e. very restricted dispersal as estimated in the Adriatic.

Genetic Isolation by distance in Gongolaria barbata in the Adriatic and Black Seas based on the shortest (diamond) and the longest path (circle). Distances displayed are site pairwise FST (y-axis) and natural logarithm of oceanographic distances (x-axis). IBD was performed without the sampling site 11 in the Black Sea. This figure was created using the R software107.
Discussion
In the present study, we investigated the genetic diversity and structure of fifteen remnant populations of G. barbata sampled in three seas, the Ionian, the Adriatic and the Black Sea, aiming to evaluate whether the natural recovery of disturbed populations may occur without the help of restoration actions.
The use of microsatellites for conservation genetic purposes has undoubtedly be a major advance, providing several advantages over allozymes as nuclear markers65,70. But a variety of potential pitfalls need to be carefully considered when using microsatellites, especially regarding null alleles67,68,71,72. The determination key68 applied on the microsatellite markers specifically developed for G. barbata allowed us interpreting data with heterozygote deficits and discriminating technical to demographic causes. Facing G. barbata population decline, low level of intra-population genetic variation would be expected due to recent demographic bottlenecks in wild populations. Interestingly, the polymorphism observed in G. barbata is on the same range than that observed not only in other Cystoseira species54,57,61, but also, in other Fucales73,74,75. Across the fifteen G. barbata sampled sites, we found significant heterozygote deficiency, which is common in Cystoseira54,56,58. However, in most previous published studies, markers with potential technical issues were not removed nor discussed, excluding the possibility of direct comparisons among species regarding the possible causes explaining these deviations.
Here, the technical issues of the microsatellite markers were ruled out and allele frequencies corrected to consider the presence of null alleles, leaving the origin of the significant deviations from HW expectations to be biological, such as inbreeding or related to a Wahlund effect. Most Fucales are monoecious, bearing hermaphroditic conceptacles so that self-fecundation may occur63,76,77,78. Because all Cystoseira s.l. are considered to be self-compatible hermaphrodites with extremely restricted gamete dispersal, inbreeding may likely arise from the mating among relatives or selfing. In T. elegans, based on allele frequencies corrected considering the presence of null alleles, departures from HWE equilibrium in all three Catalan populations were interpreted as resulting from inbreeding79. Likewise, in E. amentacea, based on allele frequencies corrected considering the presence of null alleles, departures from HWE are likely explained by selfing or between neighboring individuals that are expected to be more related on average55. In the present study, however, departures from HWE in G. barbata cannot be explained solely by the presence of inbred individuals: inbreeding was detected in a single site located in the Adriatic Sea. In addition, we did not detect selfing from the analyses of the multilocus heterozygosity distributions. In Cystoseira s.l., fertilization is oogamous, with thousands of biflagellate male gametes meeting female gametes in the vicinity of hermaphroditic conceptacles grouped within terminal receptacles. This reproduction mode strongly suggests that selfing may occur in these species if gametes are self-compatible. But, we could not find solid evidence of self-compatibility in the literature, or direct citation of an experimental study specifically testing for it55,56,80, a common assumption that should therefore deserve further investigation. Given this, mating among relatives would be more likely, without solid evidence of self-compatibility in Cystoseira s.l., facilitated by the very low dispersal of the fertilized eggs29, leading to clustering of related individuals55. This said, deviations from HW expectations may also be attributed to a Wahlund effect: given the very limited dispersal abilities of the species29,30, mixing of populations that differ in allele frequencies would result in significant heterozygote deficiencies in the pooled population, a scenario that is very likely, especially if more related individuals settle close to each other so that populations are made of a sum of different families.
One of the most expected results was certainly the strong genetic structure observed across the three seas, with the convergence of all methods towards nearly no gene flow between the Black Sea and the Ionian and Adriatic Seas. Strongly differentiated populations have been observed not only in other Cystoseira s.l. species overall their distribution ranges54, but also in other canopy-forming seaweeds81,82,83. The genetic distinction of the Black Sea populations is very likely explained by a lack of exchange of migrants through the Bosphorus strait, consistent with previous finding based on mtDNA markers62. A very similar structuration pattern was found over the same sampled sites in Tritia neritea, a gastropod characterized by a direct cycle without a pelagic larval stage84. The restricted exchange of water masses and the difference in water density salinity and temperature between the water masses of the Mediterranean and the Black Seas across the Bosphorus strait has already been reported as a main driver of genetic structure in various species84,85,86,87,88,89. Besides the environmental gradients, transition zones are often zones of secondary contact of two lineages, i.e. a zone of contact between lineages previously isolated into glacial refugia90. Current data make it difficult to establish the historical demographic contexts of the observed genetic divergence, the identification of any barriers to gene flow that promote/maintain genetic isolation, as well as the exact spatial segregation of the two main genetic clusters in G. barbata. Given the observed differentiation between the Mediterranean and Black Seas populations, both using nuclear and mitochondrial markers62, and the inter-basins context, this pattern is likely resulting from divergence without gene flow. The extent to which these two genetic backgrounds are reproductively isolated deserves further attention which could be addressed, for instance, with a study covering the entire range distribution of the species, and especially covering the contact zones to identify barrier to gene flow91. The presence of reproductively isolated lineages would prevent the success of genetic rescue if the populations crossed were found too divergent to reproduce. Genome-wide data will likely help to delineate the presence of reproductively isolated lineages and test the hypothesis of one or several evolutionary species92. Similar and high genetic diversity was nevertheless observed in these two genetic backgrounds, supporting long-term regional persistence of large populations rather than recent expansions and isolation by distance, which are often associated with loss of diversity in fucoids93.
In addition to the strong differentiation between the Mediterranean and Black Seas observed in G. barbata, genetic analyses revealed high genetic structure at fine spatial scale, indicative of limited population connectivity, even between sampled sites separated by few kms. Such levels of genetic differentiation are in the range of that observed among populations of Cystoseira s.l. studied over the similar spatial scales (i.e. within seas) using microsatellite markers11,55,56,79, suggesting similar dispersal abilities of G. barbata to other Cystoseira s.l. The estimated dispersal distance per generation in G. barbata was extremely small, likely ranging from 129 to 587 m across the Adriatic and the Black Sea. Although these estimated dispersal distances should be carefully interpreted94,95, these estimations were nonetheless comparable to distances estimated from spatial autocorrelation analyses in E. amentacea55, and in agreement with estimated distances from field observations29. These estimated dispersal distance were likely smaller than the distance between two consecutive suitable habitat patches (shallow sheltered coasts and lagoons), enabling the migration to reverse the negative effect of genetic drift. Our results suggest that, so far, G. barbata isolated populations did not seem to suffer from inbreeding depression, and displayed intra-population genetic variation similar to other Fucales; genetic and demographic monitoring should be pursued to address the most appropriate conservation issues, e.g. assisted gene flow, minimizing non-genetic threats, or in case of small inbred populations, genetic rescue with highly divergent populations50.
Pairwise genetic differentiation among sampled sites were significantly correlated with geographic distances in the Black Sea and Adriatic. Such isolation-by-distance (IBD) is common in macroalgae, whatever their algal division, habitat and life cycle96, and usually observed in Fucales97,98,99. However, in E. amentacea, genetic structure was not significantly related to geographic distances; this relationship was improved when dispersal probability across available rocky habitat was estimated by simulating stepping-stone directional transport mediated by ocean currents55,58. Considering the very low dispersal life history of E. amentacea, dispersal in this species would be facilitated by possible transport in floating rafts. Indeed, rafting is a relatively frequent and documented mechanism of marine dispersal allowing fragments of thalli to be transported entangled in floating rafts of other algae100. In G. barbata, relative long dispersal could also occur through drifting of fertile individuals or ramification, bearing fertile receptacles, thanks to their aerocysts, helping in preserving inter-population genetic variation. Greater connectivity was, for instance, observed in C. compressa compared to E. selaginoides and E. amentacea, which was ascribed to the presence of aerocysts, providing a buoyancy property that E. selaginoides and E. amantacea do not possess54. Dispersal as fertilized zygotes has also been observed in Cystoseira s.l. under strong hydrodynamic events58,100 but would not allow such long-distance dispersal. Such properties would enhance connectivity among suitable habitat patches in G. barbata, especially within the Black Sea as compared to the Adriatic Sea. Indeed, while differences in the range of geographic distances among sampled populations may partially explain this result, this finding could also reflect: (1) the presence of more free-living individuals within the Black Sea as compared to the Adriatic, (2) the higher homogeneity of water masses in the Black Sea; the oceanographic circulation is composed of a counter-rotating (cyclonic) Rim Current surrounded by many predominantly anticyclonic coastal eddies101 while the three cyclonic gyres (the north, the central and the southern Adriatic sub-gyres) and the physical and biogeochemical features operating in the Adriatic Sea creates natural barriers for dispersion102 and (3) larger effective population size in the Black Sea, in line with higher abundances in the Black Sea than in the Mediterranean Sea, these three hypothesis being non-exclusive. Nevertheless, despite potential occasional long-distance gene flow through rafting or drifting, strong genetic differentiation among populations together with homozygotes deficiencies suggest very restricted connectivity among natural populations of G. barbata, a result coherent among Cystoseira s.l. studies54,55,56,58. G. barbata populations are likely connected via neighboring subpopulations in the form of stepping stones dispersal, as supported by significant IBD. Considering the limited dispersal estimates found here, our results call into question the natural recovery of disturbed populations if suitable patches of habitat are more than 1 km far apart.
The restricted connectivity observed among G. barbata subpopulations suggests that each population represents a different evolutionary significant unit, giving the highest conservation priority to this threatened species103. Conservation should be designed to preserve the distinctive genetic diversity here observed. However, conservation efforts must be complemented by a reflection considering all the management units and their exchanges through gene flow, e.g. genetic rescue by assisted gene flow, to counteract genetic diversity erosion within each evolutionary significant unit. Other active strategies should be considered to restore the connectivity between these currently isolated populations, fostering the natural recovery of degraded populations (e.g. limiting the regression causes) or actively restoring lost populations15,17,19,20. Regarding the very restricted dispersal abilities, more genetic data on Cystoseria based on a sampling not only covering the large but also the very fine-scale distribution ranges and across time are needed to conserve the iconic Mediterranean and Black Seas marine forests and upscale reasoned ecological restoration programs.
Methods
Sampling collection and DNA extraction
Cystoseira sensu lato corresponds to three monophyletic clades recognized as three different genera62,104. The two clades outside the Cystoseira sensu stricto clade were recently better redefined, so that the formerly-known Cystoseira barbata was first renamed Treptancha barbata104 then Gongolaria barbata62. Gongolaria barbata samples were collected by snorkeling in 2013 in 15 sites along the coasts of the Ionian, Adriatic and Black Seas, (Fig. 1; Table 1). Individuals were collected at least 50 cm away from each other, and only the apical part of a primary ramification (ten centimeters) was collected and preserved in dried silica gel for future DNA analysis. The map (Fig. 1) was created using the R package mapdata105,106,107.
Genomic DNA was extracted from 10 mg of lyophilized tissue using DNeasy 96 Plant Kit (Qiagen) following manufacturer’s instructions.
Microsatellite development
Genomic DNA of 15 individuals from different locations in the Mediterranean and Black Seas was sent to the Ecogenics GmBH company (http://www.ecogenics.ch), commissioned to identify and develop a set of twelve Single Sequence Repeats (SSRs), i.e. microsatellite markers. Briefly, size selected fragments from genomic DNA were enriched for SSR content by using magnetic streptavidin beads and biotin-labeled CT and GT repeat oligonucleotides. The SSR-enriched library was analyzed on a Roche 454 platform using the GS FLX Titanium reagents. A total of 24,770 reads were sequenced with an average length of 474 base pairs. Of these, 3008 reads (12.2%) contained a microsatellite insert with a tetra- or a trinucleotide of at least six repeat units, or a dinucleotide of at least ten repeat units. Suitable primer design was possible in 1997 reads. Twelve markers (0.6%) showed a correct amplification and variability by polymerase chain reactions (PCR).
Following PCR optimization and polymorphism analysis of each of these twelve primer pairs, a multiplex procedure was performed using Multiplex Manager108 and four combinations of multiplex were tested. Based on banding patterns checked on agarose and polyacrylamide sequencing gels (2% and 8%, respectively), two multiplex reactions of six microsatellites with forward primers labeled with ABI fluorescent dyes (Table 2) showed clear banding patterns and were selected for routine genotyping on a capillary sequencer.
Microsatellite genotyping
Individuals were genotyped using these twelve microsatellite markers newly developed as follow: PCR amplifications were done in a final volume of 25 µl containing 5–15 ng of genomic DNA, 1X Master Mix (Type-it kit, Qiagen) and 0.1 μM of each primer. DNA amplification started with a 5 min denaturation at 95 °C, followed by 30 cycles comprising a denaturation of 30 s at 95 °C, a hybridization of 90 s at 60 °C and an elongation of 30 s at 72 °C and finished with a last elongation step at 60 °C for 30 min. PCR products were genotyped using an ABI 3730XL capillary sequencer at Macrogen Europe Inc. (Amsterdam, the Netherlands). Alleles were scored using GeneMapper v. 4.0 (Applied Biosystems) using an internal size standard (Genescan LIZ‐500, Applied Biosystems) and manually checked. Genotypes of every individual were checked twice with identical results.
Testing the dataset quality
Before further analysis, we checked for clonal propagation across all samples by estimating the probability of identity (PI, i.e. the probability that two independently sampled individuals within a mating population share an identical multilocus genotype by chance) using GenAlEx v6.502109. With a PI < 1.8 10–5 per population, any identical genotypes found in the dataset were presumed to be clones. These clones could either belong to a unique genet or be two representatives of the same clone that have spread. To discriminate between these alternative hypotheses, we carefully checked the sampling site(s) of the two individuals presumed as clones and their label numbering, used as a proxy of the distance between these individuals.
For each pair of loci, linkage disequilibrium (LD) was tested using the G-based test implemented in Fstat 2.9.4110, which was proven to be the most powerful approach to combine tests over all subsamples111. The 66 tests conducted (12loci × (12–1)/2) were not independent and p-values were corrected using the Benjamini and Yekuteli procedure112 to obtain FDR (false discovery rate) corrected individual p-values.
MicroChecker software113 was used to detect the presence of null alleles and eventual scoring errors (due to stuttering and/or large allele dropout/short allele dominance). We reported the null allele frequencies estimated using the Bookfield's second method provided by MicroChecker, and computed (1) null alleles frequencies expected under Hardy–Weinberg Equilibrium, and (2) the number of null alleles expected based on these frequencies and the number of individuals genotyped. We then tested the adjustment between observed and expected numbers of missing data67,71 with a unilateral exact binomial (H1: there is less missing data observed than expected). Additionally, we tested for genotyping errors due to the occurrence of short allele dominance (SAD) using unilateral (ρ < 0) Spearman's rank correlation tests between allele size and FIS. These tests were also performed with FIT, aiming to increase the power of statistical tests71. Genotyping errors due to the occurrence of stuttering were tested following De Meeûs et al.67.
Analyses of genetic diversity
Allele frequencies, the average number of alleles (Nall), allelic richness (Ar), gene diversity (HS) and the fixation index (FIS) were estimated for each study site using Fstat 2.9.4110. Briefly, Ar is the expected number of alleles in a random subsample of size g drawn from the population114,115. This rarefaction method allows evaluation of the expected number of different alleles among equal sized samples drawn from several different populations. Genetic diversities between two groups were compared using bilateral tests with 10,000 randomizations implemented in Fstat 2.9.4110. We tested for heterozygote deficiency across loci and subsamples using the U test112 implemented in Genepop 4.7.5116.
To test for possible inbreeding occurring within each sample, individual Multi-Locus Heterozygosity (MLH) was calculated for each individual. In a population with variance in inbreeding, inbred individuals are less heterozygous (i.e. lower MLH) than non-inbred ones. Inbreeding variance generates Identity Disequilibria (ID)—i.e. correlations in homozygosity across loci, a measure of departure from random associations between loci117. IDs were measured for each sampling site by calculating the g2 parameter and its standard deviation using the RMES software118. The g2 parameter measures the excess of double heterozygotes at two loci relative to the expectation under a random association, standardized by average heterozygosity, providing a measure of genetic association and inbreeding variance in the population, free of technical biases. To test for the significance of g2, random re-assortments of single-locus heterozygosities among individuals were tested using 1000 iterations. Selfing rate within each subsample was also estimated using RMES118. To test whether the selfing rate significantly differed from 0, both the unconstrained and constrained likelihood options were run. We then computed Δdev = 2 * (lnl(unconstrained) − lnl(constrained)) and compared it to a chi-square with one degree of freedom118.
Analyses of genetic structure
First, we estimated population genetic structure through the approach developed in De Meeûs68 based on the Wright's fixation indices119, FIS, which measures inbreeding of individuals relative to inbreeding of subsamples, FST, which measures inbreeding of subsamples relative to total inbreeding, and FIT, which measures inbreeding of individuals relative to total inbreeding. In other words, FIS measures the deviations form Hardy–Weinberg equilibrium (i.e. local panmixia), FST measures genetic differentiation between subsamples, and FIT reflects the combination of both. Briefly, this approach is based on the examination of the F-statistics stability across loci and subsamples to interpret data with heterozygosity deficiencies. Heterozygosity deficiencies can arise from null alleles, stuttering, short allele dominance, allele dropouts (i.e. technical problems), Wahlund effect (the juxtaposition of several groups with different allele frequencies), and/or selfing and sib-mating. By examining the (1) missing data in the dataset (across loci and subsamples), (2) variations of FIS and FST across loci, (3) standard error of jackknife over loci for FIS (StrdErrFIS) and FST (StrdErrFST), and (4) the correlation between FIS and FST, De Meeûs68 proposed a determination key to help discriminating demographic from technical causes.
Wright's F-statistics were estimated through Weir and Cockerham's unbiased estimators120, i.e. f, θ and F for FIS, FST and FIT estimators, respectively. We computed the 95% confidence intervals of F-statistics using the StrdErrFIS and StrdErrFST computed by jackknife over subsamples, for each locus, and we used the 95% confidence intervals computed by bootstrap over loci. Correlations between FIS and FST were measured using Pearson’s correlation coefficients in R107. All these analyses (parameters estimates, jackknife, bootstrap) were performed using Fstat 2.9.4110 and R107.
We further tested a correlation between FIS and the number of missing data71: the significance of correlations was tested with a unilateral (ρ > 0) Spearman's rank correlation test in R107.
In addition to the aforementioned approach, genetic structure among sampling sites was depicted using more classical tools, aiming to look whether geographic differentiation exists. We used three different approaches: (1) FST estimates (2) Principal Component Analyses (PCA) computed on the matrix of genotypes, and (3) an individual-based Bayesian clustering method. Comparing results from analyses using different statistical approaches allows us to make solid assumptions about our data: it allowed us to cross check the outputs of a model-based approach with strong priors and hypotheses (HW equilibrium, no linkage between markers) as implemented in Structure 2.3.4121,122, with methods based either on distance with few (nearly no) assumptions on data (i.e. PCA) or on distance matrices among sets of individuals that fall within predefined population samples. Estimates of FST were calculated using FreeNA with the ENA correction72. To test for genetic isolation among subsamples, we used the G-based test over all loci. The PCAs were carried out using the R package adegenet 1.4–2123. The individual-based Bayesian clustering analysis was performed with the software Structure 2.3.4121,122. For each value of K (ranging from 1 to 18), 30 replicate chains of 200,000 Markov Chain Monte Carlo iterations were run after discarding 25,000 burn-in iterations. An admixture model with correlated allele frequencies was applied with a priori information on sample origin. For each value of K, 30 output files were produced by Structure 2.3.4 with slightly different ancestry proportions; using Clumpp124, we determined individual ancestry proportions (q-values; i.e. assignment probabilities) that best matched across all replicate runs for each K value. We visualized individuals’ assignment in the R software107.
We estimated and tested hierarchical F-statistics using the HierFstat package125 with three levels, the Mediterranean vs. the Black Seas subsamples (level 1), the Ionian, Adriatic and Black Seas (level 2), and all subsamples (level 3).
Estimates of larval dispersal
Finally, we tested for the presence of genetic Isolation-By-Distance (IBD) by investigating the relationship between using Cavalli-Sforza and Edward’s chord distance126 (Table 3) and the natural logarithm of oceanographic distances (Ln(Dgeo)127. Pairwise oceanographic distance between sampled sites was estimated within each sea as the shortest oceanographic distance according to the ‘Vincenty (ellipsoid)’ method using the geosphere package128. However, as the shortest path does not reflect the sea hydrodynamics129,130, pairwise oceanographic distances between sampled sites were also estimated following the coastlines, calculated by summing all the straight lines connecting each cape on a map at 1:500,000 on QGis 3.18. For the Black Sea, which is circular, having the choice between following the movement against the clock or clockwise to calculate the coastline distance between two populations, only the shortest distance was kept.
Regressions between genetic and geographic distances as described above were investigated within the Adriatic on the one hand and within the Black Sea (excluding subsample 11) on the other hand, using Fstat110. With significant IBDs, the IBD slope allows estimating the neighborhood Nb = 1/b and the number of immigrants from neighbors Nem = 1/(2πb) = 4πDeσ2, with De the effective population density and σ2 the average of squared axial distances between adults and their parents131,132. To estimate De and σ2, we first estimated the effective population size Ne within each Sea (Adriatic and Black Seas): we measured the non-random association of alleles at different loci within F1 population133 using a bias correction134,135 implemented in NeEstimator v2 software136. The Pcrit was adjusted following recommendations135 and random mating was applied. Estimating Ne allows computing effective population densities De, as the averaged Ne multiplied by the number of subsamples within a sea divided by the surface of the sea. The number of populations in the Adriatic Sea was estimated as follow: (1) Gongolaria barbata stands for 10% of the Cystoseira s.l. observed in the Mediterranean Sea32, (2) in the Adriatic, ca. 100 Cystoseira s.l. populations were recorded, (3) we estimated that 10% would be Gongolaria barbata, so that the number of populations in the Adriatic would be close to 10. Despite this estimation, we thought very low, we decided to test different number of populations, ranging from 10 to 100. In the Black Sea, the number of populations was estimated to be 10063, but we also tested different number of populations, ranging from 10 to 100. Once De computed, a rough proxy of dispersal (δ), in order of magnitude, can be computed as δ ≈ 2 (1/(4πDe × b))−1/2 (i.e. Nem = 4πDeσ2 < = > δ ≈ 2 (1/(4πDe × b))-1/2126).
Data availability
Microsatellite data has been deposited at DRYAD: https://doi.org/10.5061/dryad.g4f4qrfqs.
References
Wernberg, T. & Filbee-Dexter, K. Missing the marine forest for the trees. Mar. Ecol. Prog. Ser. 612, 209–215 (2019).
Thompson, R. C., Wilson, B. J., Tobin, M. L., Hill, A. S. & Hawkins, S. J. Biologically generated habitat provision and diversity of rocky shore organisms at a hierarchy of spatial scales. J. Exp. Mar. Biol. Ecol. 202, 73–84 (1996).
Christie, H., Jørgensen, N. M. & Norderhaug, K. M. Bushy or smooth, high or low; importance of habitat architecture and vertical position for distribution of fauna on kelp. J. Sea Res. 58, 198–208 (2007).
Steneck, R. S. et al. Kelp forest ecosystems: Biodiversity, stability, resilience and future. Environ. Conserv. 29, 436–459 (2002).
Strain, E. M. A., Thomson, R. J., Micheli, F., Mancuso, F. P. & Airoldi, L. Identifying the interacting roles of stressors in driving the global loss of canopy-forming to mat-forming algae in marine ecosystems. Glob. Change Biol. 20, 3300–3312 (2014).
Mineur, F. et al. European seaweeds under pressure: Consequences for communities and ecosystem functioning. J. Sea Res. 98, 91–108 (2015).
Krumhansl, K. A. et al. Global patterns of kelp forest change over the past half-century. PNAS 113, 13785–13790 (2016).
Straub, S. C. et al. Resistance, extinction, and everything in between—The diverse responses of seaweeds to marine heatwaves. Front. Mar. Sci. 6, 763 (2019).
Cheminée, A. et al. Nursery value of Cystoseira forests for Mediterranean rocky reef fishes. J. Exp. Mar. Biol. Ecol. 442, 70–79 (2013).
Piazzi, L. et al. Biodiversity in canopy-forming algae: Structure and spatial variability of the Mediterranean Cystoseira assemblages. Estuar. Coast. Shelf Sci. 207, 132–141 (2018).
Thibaut, T., Pinedo, S., Torras, X. & Ballesteros, E. Long-term decline of the populations of Fucales (Cystoseira spp. and Sargassum spp.) in the Albères coast (France, North-western Mediterranean). Mar. Pollut. Bull. 50, 1472–1489 (2005).
Gianni, F. et al. Conservation and restoration of marine forests in the Mediterranean Sea and the potential role of marine protected areas. Adv. Oceanogr. Limnol. 4, 83–101 (2013).
Blanfuné, A., Boudouresque, C. F., Verlaque, M. & Thibaut, T. The fate of Cystoseira crinita, a forest-forming Fucale (Phaeophyceae, Stramenopiles), in France (North Western Mediterranean Sea). Estuar. Coast. Shelf Sci. 181, 196–208 (2016).
Gubbay, S. et al. European Red List of Habitats. Part 1. Marine habitats. Luxembourg: Publications Office of the European Union (2016).
Perkol-Finkel, S., Ferrario, F., Nicotera, V. & Airoldi, L. Conservation challenges in urban seascapes: Promoting the growth of threatened species on coastal infrastructures. J. Appl. Ecol. 49, 1457–1466 (2012).
Falace, A., Kaleb, S., Fuente, G. D. L., Asnaghi, V. & Chiantore, M. Ex situ cultivation protocol for Cystoseira amentacea var. stricta (Fucales, Phaeophyceae) from a restoration perspective. PLoS ONE 13, e0193011 (2018).
Gianni, F., Bartolini, F., Airoldi, L. & Mangialajo, L. Reduction of herbivorous fish pressure can facilitate focal algal species forestation on artificial structures. Mar. Environ. Res. 138, 102–109 (2018).
Gianni, F. et al. Optimizing canopy-forming algae conservation and restoration with a new herbivorous fish deterrent device. Restor. Ecol. 28, 750–756 (2020).
Verdura, J., Sales, M., Ballesteros, E., Cefalì, M. E. & Cebrian, E. Restoration of a canopy-forming alga based on recruitment enhancement: Methods and long-term success assessment. Front. Plant Sci. 9, 1832 (2018).
Fuente, G. D. L., Chiantore, M., Asnaghi, V., Kaleb, S. & Falace, A. First ex situ outplanting of the habitat-forming seaweed Cystoseira amentacea var. stricta from a restoration perspective. PeerJ 7, e7290 (2019).
Tamburello, L. et al. Are we ready for scaling up restoration actions? An insight from Mediterranean macroalgal canopies. PLoS ONE 14, e0224477 (2019).
Medrano, A. et al. From marine deserts to algal beds: Treptacantha elegans revegetation to reverse stable degraded ecosystems inside and outside a No-Take marine reserve. Restor. Ecol. 28, 632–644 (2020).
Chryssovergis, F. & Panayotidis, P. Évolution des peuplements macrophytobenthiques le long d’un gradient d’eutrophisation. Oceanol. Acta 18, 649–658 (1995).
Sales, M., Cebrian, E., Tomas, F. & Ballesteros, E. Pollution impacts and recovery potential in three species of the genus Cystoseira (Fucales, Heterokontophyta). Estuar. Coast. Shelf Sci. 92, 347–357 (2011).
Díez, I., Santolaria, A., Secilla, A. & Gorostiaga, J. M. Recovery stages over long-term monitoring of the intertidal vegetation in the ‘Abra de Bilbao’ area and on the adjacent coast (N. Spain). Eur. J. Phycol. 44, 1–14 (2009).
Bringloe, T. T. et al. Phylogeny and evolution of the brown algae. Crit. Rev. Plant Sci. 39, 281–321 (2020).
Guern, M. Embryologie de quelques espèces du genre Cystoseira Agardh 1821 (FUCALES). Vie et Milieu 649–680 (1962).
Dudgeon, S., Kübler, J. E., Wright, W. A., Vadas, R. L. & Petraitis, P. S. Natural variability in zygote dispersal of Ascophyllum nodosum at small spatial scales. Funct. Ecol. 15, 595–604 (2001).
Mangialajo, L. et al. Zonation patterns and interspecific relationships of fucoids in microtidal environments. J. Exp. Mar. Biol. Ecol. 412, 72–80 (2012).
Capdevila, P. et al. Recruitment patterns in the Mediterranean deep-water alga Cystoseira zosteroides. Mar. Biol. 162, 1165–1174 (2015).
Assis, J. et al. A fine-tuned global distribution dataset of marine forests. Sci. Data 7, 119 (2020).
Fabbrizzi, E. et al. Modeling macroalgal forest distribution at Mediterranean scale: Present status, drivers of changes and insights for conservation and management. Front. Mar. Sci. 7, 20 (2020).
Benedetti-Cecchi, L., Tamburello, L., Maggi, E. & Bulleri, F. Experimental perturbations modify the performance of early warning indicators of regime shift. Curr. Biol. 25, 1867–1872 (2015).
Bulleri, F., Benedetti-Cecchi, L., Ceccherelli, G. & Tamburello, L. A few is enough: A low cover of a non-native seaweed reduces the resilience of Mediterranean macroalgal stands to disturbances of varying extent. Biolical Invasions 19, 2291–2305 (2017).
Rindi, L., Bello, M. D., Dai, L., Gore, J. & Benedetti-Cecchi, L. Direct observation of increasing recovery length before collapse of a marine benthic ecosystem. Nat. Ecol. Evol. 1, 1–7 (2017).
Draisma, S. G. A., Ballesteros, E., Rousseau, F. & Thibaut, T. DNA sequence data demonstrate the polyphyly of the genus Cystoseira and other Sargassaceae genera (Phaeophyceae). J. Phycol. 46, 1329–1345 (2010).
Bruno de Sousa, C. et al. Improved phylogeny of brown algae Cystoseira (Fucales) from the Atlantic-Mediterranean region based on mitochondrial sequences. PLoS ONE 14, e0210143 (2019).
Jódar-Pérez, A. B., Terradas-Fernández, M., López-Moya, F., Asensio-Berbegal, L. & López-Llorca, L. V. Multidisciplinary analysis of Cystoseira sensu lato (SE Spain) suggest a complex colonization of the Mediterranean. J. Mar. Sci. Eng. 8, 961 (2020).
Hughes, A. R. & Stachowicz, J. J. Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. PNAS 101, 8998–9002 (2004).
Reusch, T. B. H. & Hughes, A. R. The emerging role of genetic diversity for ecosystem functioning: Estuarine macrophytes as models. Estuaries and Coasts J ERF 29, 159–164 (2006).
Reusch, T. B. H., Ehlers, A., Hämmerli, A. & Worm, B. Ecosystem recovery after climatic extremes enhanced by genotypic diversity. PNAS 102, 2826–2831 (2005).
Ehlers, A., Worm, B. & Reusch, T. B. H. Importance of genetic diversity in eelgrass Zostera marina for its resilience to global warming. Mar. Ecol. Prog. Ser. 355, 1–7 (2008).
Hughes, A. R., Inouye, B. D., Johnson, M. T. J., Underwood, N. & Vellend, M. Ecological consequences of genetic diversity. Ecol. Lett. 11, 609–623 (2008).
Frankham, R., Ballou, J. D. & Briscoe, D. A. Introduction to Conservation Genetics (Cambridge University Press, 2002) https://doi.org/10.1017/CBO9780511808999 .
Cowen, R., Gawarkiewicz, G., Pineda, J., Thorrold, S. & Werner, F. Population connectivity in marine systems: An overview. Oceanography 20, 14–21 (2007).
Mayr, E. Animal Species and Evolution. Animal Species and Evolution (Harvard University Press, 2013).
Kimura, M. The Neutral Theory of Molecular Evolution (Cambridge University Press, 1983) https://doi.org/10.1017/CBO9780511623486 .
Frankham, R. Conservation genetics. Annu. Rev. Genet. 29, 305–327 (1995).
Lacy, R. C. Loss of genetic diversity from managed populations: Interacting effects of drift, mutation, immigration, selection, and population subdivision. Conserv. Biol. 1, 143–158 (1987).
Frankham, R. et al. Genetic Management of Fragmented Animal and Plant Populations (Oxford University Press, 2017).
Planes, S., Jones, G. P. & Thorrold, S. R. Larval dispersal connects fish populations in a network of marine protected areas. PNAS https://doi.org/10.1073/pnas.0808007106 (2009).
Pineda, J., Hare, J. A. & Sponaugle, S. Larval transport and dispersal in the coastal ocean and consequences for population connectivity. Oceanography 20, 22–39 (2007).
Caughley, G. Directions in conservation biology. J. Anim. Ecol. 63, 215–244 (1994).
Buonomo, R. et al. Predicted extinction of unique genetic diversity in marine forests of Cystoseira spp. Mar. Environ. Res. 138, 119–128 (2018).
Buonomo, R. et al. Habitat continuity and stepping-stone oceanographic distances explain population genetic connectivity of the brown alga Cystoseira amentacea. Mol. Ecol. 26, 766–780 (2017).
Bermejo, R. et al. Marine forests of the Mediterranean-Atlantic Cystoseira tamariscifolia complex show a southern Iberian genetic hotspot and no reproductive isolation in parapatry. Sci. Rep. 8, 10427 (2018).
Engelen, A. H. et al. A population genetics toolbox for the threatened canopy-forming brown seaweeds Cystoseira tamariscifolia and C. amentacea (Fucales, Sargassaceae). J. Appl. Phycol. 29, 627–629 (2017).
Thibaut, T. et al. Connectivity of populations of the seaweed Cystoseira amentacea within the Bay of Marseille (Mediterranean Sea): Genetic structure and hydrodynamic connections. crya 37, 233–255 (2016).
Guiry, M.D. & Guiry, G.M. AlgaeBase. World-wide electronic publication (National University of Ireland, 2021) http://www.algaebase.org (Accessed 21 Jan 2021).
Sales, M. & Ballesteros, E. Shallow Cystoseira (Fucales: Ochrophyta) assemblages thriving in sheltered areas from Menorca (NW Mediterranean): Relationships with environmental factors and anthropogenic pressures. Estuar. Coast. Shelf Sci. 84, 476–482 (2009).
Robvieux, P. et al. First characterization of eight polymorphic microsatellites for Cystoseira amentacea var. stricta (Fucales, Sargassaceae). Conserv. Genet. Resour. 4, 923–925 (2012).
Sadogurska, S. S., Neiva, J., Falace, A., Serrão, E. A. & Israel, Á. The genus Cystoseira s.l. (Ochrophyta, Fucales, Sargassaceae) in the Black Sea: Morphological variability and molecular taxonomy of Gongolaria barbata and endemic Ericaria crinita f. bosphorica comb. nov. Phytotaxa 480, 1–21 (2021).
Bologa, A. S. & Sava, D. Progressive decline and present trend of Romanian Black Sea macroalgal flora. Cercetari Mar. 36, 31–60 (2006).
Irving, A. D., Balata, D., Colosio, F., Ferrando, G. A. & Airoldi, L. Light, sediment, temperature, and the early life-history of the habitat-forming alga Cystoseira barbata. Mar. Biol. 156, 1223–1231 (2009).
Allendorf, F. W. Genetics and the conservation of natural populations: Allozymes to genomes. Mol. Ecol. 26, 420–430 (2017).
Ellegren, H. Microsatellites: Simple sequences with complex evolution. Nat. Rev. Genet. 5, 435–445 (2004).
De Meeûs, T. et al. Deceptive combined effects of short allele dominance and stuttering: An example with Ixodes scapularis, the main vector of Lyme disease in the USA. bioRxiv https://doi.org/10.1101/622373 (2019).
De Meeûs, T. Revisiting FIS, FST, Wahlund effects, and null alleles. J. Hered. 109, 446–456 (2018).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Selkoe, K. A. & Toonen, R. J. Microsatellites for ecologists: A practical guide to using and evaluating microsatellite markers. Ecol. Lett. 9, 615–629 (2006).
Manangwa, O. et al. Detecting Wahlund effects together with amplification problems: Cryptic species, null alleles and short allele dominance in Glossina pallidipes populations from Tanzania. Mol. Ecol. Resour. 19, 757–772 (2019).
Chapuis, M.-P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631 (2007).
Engel, C. R., Brawley, S. H., Edwards, K. J. & Serrão, E. Isolation and cross-species amplification of microsatellite loci from the fucoid seaweeds Fucus vesiculosus, F. serratus and Ascophyllum nodosum (Heterokontophyta, Fucaceae). Mol. Ecol. Notes 3, 180–182 (2003).
Paulino, C. et al. Characterization of 12 polymorphic microsatellite markers in the sugar kelp Saccharina latissima. J. Appl. Phycol. 28, 3071–3074 (2016).
Coleman, M. A., Dolman, G., Kelaher, B. P. & Steinberg, P. D. Characterisation of microsatellite loci in the subtidal habitat-forming alga, Phyllospora comosa (Phaeophyceae, Fucales). Conserv. Genet. 9, 1015–1017 (2008).
Coleman, M. A. & Brawley, S. H. Are life history characteristics good predictors of genetic diversity and structure? A case study of the intertidal alga Fucus spiralis (heterokontophyta; Phaeophyceae). J. Phycol. 41, 753–762 (2005).
Coleman, M. A. & Brawley, S. H. Spatial and temporal variability in dispersal and population genetic structure of a rockpool alga. Mar. Ecol. Prog. Ser. 300, 63–77 (2005).
Engel, C. R., Daguin, C. & Serrão, E. A. Genetic entities and mating system in hermaphroditic Fucus spiralis and its close dioecious relative F. vesiculosus (Fucaceae, Phaeophyceae). Mol. Ecol. 14, 2033–2046 (2005).
Medrano, A. et al. Ecological traits, genetic diversity and regional distribution of the macroalga Treptacantha elegans along the Catalan coast (NW Mediterranean Sea). Sci. Rep. 10, 19219 (2020).
Engelen, A. H. et al. Periodicity of propagule expulsion and settlement in the competing native and invasive brown seaweeds, Cystoseira humilis and Sargassum muticum (Phaeophyta). Eur. J. Phycol. 43, 275–282 (2008).
Assis, J., Serrão, E. A., Claro, B., Perrin, C. & Pearson, G. A. Climate-driven range shifts explain the distribution of extant gene pools and predict future loss of unique lineages in a marine brown alga. Mol. Ecol. 23, 2797–2810 (2014).
Neiva, J. et al. Genes left behind: Climate change threatens cryptic genetic diversity in the canopy-forming seaweed Bifurcaria bifurcata. PLoS ONE 10, e0131530 (2015).
Coleman, M. A. & Kelaher, B. P. Connectivity among fragmented populations of a habitat-forming alga, Phyllospora comosa (Phaeophyceae, Fucales) on an urbanised coast. Mar. Ecol. Prog. Ser. 381, 63–70 (2009).
Boissin, E. et al. Chaotic genetic structure and past demographic expansion of the invasive gastropod Tritia neritea in its native range, the Mediterranean Sea. Sci. Rep. 10, 21624 (2020).
Olsen, J. L. et al. North Atlantic phylogeography and large-scale population differentiation of the seagrass Zostera marina L. Mol. Ecol. 13, 1923–1941 (2004).
Peijnenburg, K. T. C. A., Breeuwer, J. A. J., Pierrot-Bults, A. C. & Menken, S. B. J. Phylogeography of the planktonic chaetognath Sagitta setosa reveals isolation in European Seas. Evolution 58, 1472–1487 (2004).
Luttikhuizen, P. C., Campos, J., van Bleijswijk, J., Peijnenburg, K. T. C. A. & van der Veer, H. W. Phylogeography of the common shrimp, Crangon crangon (L.) across its distribution range. Mol. Phylogenet. Evol. 46, 1015–1030 (2008).
Wilson, A. B. & Eigenmann Veraguth, I. The impact of Pleistocene glaciation across the range of a widespread European coastal species. Mol. Ecol. 19, 4535–4553 (2010).
Riquet, F. et al. Parallel pattern of differentiation at a genomic island shared between clinal and mosaic hybrid zones in a complex of cryptic seahorse lineages. Evolution 73, 817–835 (2019).
Hewitt, G. M. Hybrid zones-natural laboratories for evolutionary studies. Trends Ecol. Evol. 3, 158–167 (1988).
Johannesson, K., Le Moan, A., Perini, S. & André, C. A Darwinian laboratory of multiple contact zones. Trends Ecol. Evol. https://doi.org/10.1016/j.tree.2020.07.015 (2020).
McCoy, S. J., Krueger-Hadfield, S. A. & Mieszkowska, N. Evolutionary phycology: Toward a macroalgal species conceptual framework. J. Phycol. 56, 1404–1413 (2020).
Neiva, J., Pearson, G. A., Valero, M. & Serrão, E. A. Fine-scale genetic breaks driven by historical range dynamics and ongoing density-barrier effects in the estuarine seaweed Fucus ceranoides L. BMC Evol. Biol. 12, 78 (2012).
Whitlock, M. C. & McCauley, D. E. Indirect measures of gene flow and migration: FST ≠1/(4 Nm + 1). Heredity 82, 117–125 (1999).
Lowe, W. H. & Allendorf, F. W. What can genetics tell us about population connectivity?. Mol. Ecol. 19, 3038–3051 (2010).
Durrant, H. M. S. et al. Implications of macroalgal isolation by distance for networks of marine protected areas. Conserv. Biol. 28, 438–445 (2014).
Engelen, A., Olsen, J., Breeman, A. & Stam, W. Genetic differentiation in Sargassum polyceratium (Fucales: Phaeophyceae) around the island of Curaçao (Netherlands Antilles). Mar. Biol. 139, 267–277 (2001).
Billot, C., Engel, C. R., Rousvoal, S., Kloareg, B. & Valero, M. Current patterns, habitat discontinuities and population genetic structure: The case of the kelp Laminaria digitata in the English Channel. Mar. Ecol. Prog. Ser. 253, 111–121 (2003).
Tatarenkov, A., Jönsson, R. B., Kautsky, L. & Johannesson, K. Genetic structure in populations of Fucus vesiculosus (phaeophyceae) over spatial scales from 10 m to 800 km. J. Phycol. 43, 675–685 (2007).
Susini, M.-L., Thibaut, T., Meinesz, A. & Forcioli, D. A preliminary study of genetic diversity in Cystoseira amentacea (C. Agardh) Bory var. stricta Montagne (Fucales, Phaeophyceae) using random amplified polymorphic DNA. Phycologia 46, 605–611 (2007).
Korotenko, K., Bowman, M. & Dietrich, D. High-resolution numerical model for predicting the transport and dispersal of oil spilled in the Black Sea. Terrest. Atmos. Oceanic Sci. J. 21, 123–136 (2010).
Barale, V., Schiller, C., Tacchi, R. & Marechal, C. Trends and interactions of physical and bio-geo-chemical features in the Adriatic Sea as derived from satellite observations. Sci. Total Environ. 353, 68–81 (2005).
Hauser, L. & Carvalho, G. R. Paradigm shifts in marine fisheries genetics: Ugly hypotheses slain by beautiful facts. Fish Fish. 9, 333–362 (2008).
Orellana, S., Hernández, M. & Sansón, M. Diversity of Cystoseira sensu lato (Fucales, Phaeophyceae) in the eastern Atlantic and Mediterranean based on morphological and DNA evidence, including Carpodesmia gen. emend. and Treptacantha gen. emend. Eur. J. Phycol. 54, 447–465 (2019).
Richard, B., A. & Wilks, A., R. Maps in S. AT\&T Bell Laboratories Statistics Research Report [93.2] (1993).
Richard, B., A. & Wilks, A., R. Constructing a Geographical Database. AT\&T Bell Lab-oratories Statistics Research Report [95.2] (1995).
R Core Team. R: A Language and Environment for Statistical Computing https://www.R-project.org/ (R Foundation for Statistical Computing, 2017).
Holleley, C. E. & Geerts, P. G. Multiplex Manager 1.0: A cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 46, 511–517 (2009).
Peakall, R. & Smouse, P. E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 6, 288–295 (2006).
Goudet, J. Fstat (Version 1.2): A computer program to calculate F-Statistics. J. Hered. 86, 485–486 (1995).
De Meeûs, T., Guégan, J.-F. & Teriokhin, A. T. MultiTest V.1.2, a program to binomially combine independent tests and performance comparison with other related methods on proportional data. BMC Bioinform. 10, 443 (2009).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300 (1995).
Oosterhout, C. V., Weetman, D. & Hutchinson, W. F. Estimation and adjustment of microsatellite null alleles in nonequilibrium populations. Mol. Ecol. Notes 6, 255–256 (2006).
Petit, R. J., Mousadik, A. E. & Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 12, 844–855 (1998).
El Mousadik, A. & Petit, R. J. High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor. Appl. Genet. 92, 832–839 (1996).
Raymond, M. & Rousset, F. GENEPOP (Version 1.2): Population genetics Software for exact tests and ecumenicism. J. Hered. 86, 248–249 (1995).
Szulkin, M., Bierne, N. & David, P. Heterozygosity-fitness correlations: A time for reappraisal. Evolution 64, 1202–1217 (2010).
David, P., Pujol, B., Viard, F., Castella, V. & Goudet, J. Reliable selfing rate estimates from imperfect population genetic data. Mol. Ecol. 16, 2474–2487 (2007).
Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 19, 395–420 (1965).
Weir, B. S. & Cockerham, C. C. Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370 (1984).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 164, 1567–1587 (2003).
Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Jakobsson, M. & Rosenberg, N. A. Clumpp: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Goudet, J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186 (2005).
Séré, M., Thévenon, S., Belem, A. M. G. & De Meeûs, T. Comparison of different genetic distances to test isolation by distance between populations. Heredity 119, 55–63 (2017).
Rousset, F. & Raymond, M. Statistical analyses of population genetic data: New tools, old concepts. Trends Ecol. Evol. 12, 313–317 (1997).
Hijmans, R. J. Geosphere: Spherical Trigonometry. https://CRAN.R-project.org/package=geosphere. R package version 1.5–5. (2016).
Korotenko, K. A. Effects of mesoscale eddies on behavior of an oil spill resulting from an accidental deepwater blowout in the Black Sea: An assessment of the environmental impacts. PeerJ 6, e5448 (2018).
López-Márquez, V. et al. Seascape genetics and connectivity modelling for an endangered Mediterranean coral in the northern Ionian and Adriatic seas. Landsc. Ecol. 34, 2649–2668 (2019).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228 (1997).
Watts, P. C. et al. Compatible genetic and ecological estimates of dispersal rates in insect (Coenagrion mercuriale: Odonata: Zygoptera) populations: Analysis of ‘neighbourhood size’ using a more precise estimator. Mol. Ecol. 16, 737–751 (2007).
Hill, W. G. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 38, 209–216 (1981).
Waples, R. S. Seed banks, salmon, and sleeping genes: Effective population size in semelparous, age-structured species with fluctuating abundance. Am. Nat. 167, 118–135 (2006).
Waples, R. S. & Do, C. ldne: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 8, 753–756 (2008).
Do, C. et al. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214 (2014).
Cavalli-Sforza, L. L., & Edwards, A. W. F. Phylogenetic analysis: Model and estimation procedures. Am. J. Hum. Genet. 19, 233–257 (1967).
Acknowledgements
The work was funded by the CoCoNet (FP7 European project, ref. 287844), CONVOST (UCAJEDI Investments ANR-15-IDEX-01), MARFOR (Biodiversa, ERA-LEARN 2020 grant agreement No 645782, 31/12/2016) and AFRIMED (EASME/EMFF/2017/1.2.1.12) projects. Authors would like to thank all the participants of WP3 of CoCoNet project for the fruitful discussions and the sampling campaigns in both the Black Sea and the Adriatic Sea. Authors would also like to thank Emeline Deleury and Eric Lombaert for running the preliminary analysis with the STRUCTURE software.
Funding
This project, funded by the European FP7 CoCoNet project (Ocean.2011-4, grant agreement 773 #287844), follows ethic statement established by the EU. The collections were conducted with country partners and base on respective local authorizations and also by LabEx Corail (ANR-France).
Author information
Authors and Affiliations
Contributions
L.M. and L.B. conceived the project. L.M., S.P., S.F., N.M. and L.A. performed the sampling. L.B. performed the laboratory work. F.R. performed the analyses and drafted the manuscript with input from C. F., L.M. and L.B. All authors read and improved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Riquet, F., De Kuyper, CA., Fauvelot, C. et al. Highly restricted dispersal in habitat-forming seaweed may impede natural recovery of disturbed populations. Sci Rep 11, 16792 (2021). https://doi.org/10.1038/s41598-021-96027-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-96027-x
This article is cited by
-
Climate change and species facilitation affect the recruitment of macroalgal marine forests
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
