
Abstract
Hyperandrogenic women with PCOS show disrupted decidualization (DE) and placentation. Dihydrotestosterone (DHT) is reported to enhance DE in non-PCOS endometrial stromal cells (eSCCtrl); however, this has not been assessed in PCOS cells (eSCPCOS). Therefore, we studied the transcriptome profile of non-decidualized (non-DE) and DE eSCs from women with PCOS and Ctrl in response to short-term estradiol (E2) and/or progesterone (P4) exposure with/without (±) DHT. The non-DE eSCs were subjected to E2 ± DHT treatment, whereas the DE (0.5 mM 8-Br-cAMP, 96 h) eSCs were post-treated with E2 and P4 ± DHT, and RNA-sequenced. Validation was performed by immunofluorescence and immunohistochemistry. The results showed that, regardless of treatment, the PCOS and Ctrl samples clustered separately. The comparison of DE vs. non-DE eSCPCOS without DHT revealed PCOS-specific differentially expressed genes (DEGs) involved in mitochondrial function and progesterone signaling. When further adding DHT, we detected altered responses for lysophosphatidic acid (LPA), inflammation, and androgen signaling. Overall, the results highlight an underlying defect in decidualized eSCPCOS, present with or without DHT exposure, and possibly linked to the altered pregnancy outcomes. We also report novel factors which elucidate the mechanisms of endometrial dysfunction in PCOS.
Similar content being viewed by others


Introduction
Polycystic ovary syndrome (PCOS) is the most common cause for anovulatory infertility. It involves endometrial alterations that are thought to contribute to reproductive performance1. The majority (60%) of women with PCOS exhibit hyperandrogenism, which is thought to be responsible for the pathogenesis of the syndrome2. This hyperandrogenism may also contribute to the increased risk for adverse pregnancy outcomes and placental aberrations3. Clinical findings have indicated that restoring ovulations with medication does not result in an endometrial environment comparable to that of ovulatory non-PCOS women, suggesting an underlying pathology4.
Endometrial decidualization involves dramatic morphological and functional changes coordinated by ovarian steroid hormones estrogen (E2) and progesterone (P4). Cyclic adenosine monophosphate (cAMP) activation provides synergistic enhancement of decidualization by inducing the transcriptome of common decidualization genes5. Supporting the crucial role of androgens in decidualization and endometrial receptivity, androgen receptor (AR)-knockout mice are reported to be sub-fertile, whereas upregulation of the AR in the endometrium results in impaired endometrial receptivity6. Progesterone has also been shown to down-regulate AR, modulating androgen effects in the endometrium7. Imbalance of this synergy may result in a disadvantageous environment for the implanting embryo, and subsequently a dysregulated placentation.
The PCOS endometrium has previously been shown to display altered expression of sex hormone receptors and their co-receptors, suggesting an increased E2 effect and a decreased P4 effect8,9. Additionally, studies suggest disrupted decidualization and placentation in hyperandrogenic mouse models of PCOS but also in hyperandrogenic women with PCOS10,11. While dihydrotestosterone (DHT; a more potent metabolite of testosterone) was reported to enhance decidualization in non-PCOS endometrial stromal cells (eSCCtrl)12, this effect has not been observed in PCOS eSCs (eSCPCOS)13.
Based on these previous findings, the present study aims to evaluate the global gene expression profile of non-decidualized (non-DE) and decidualized (DE) eSCs in response to short-term steroid hormone exposure of PCOS compared to non-PCOS women. DHT was chosen over testosterone for the present study to bypass 5α-reductase dependence and to investigate isolated androgen effect with minimal testosterone to estradiol conversion14,15. The objective was to assess whether DHT differentially modulates non-DE eSCs when exposed to E2 (the model of non-decidualized/proliferative phase), compared to DE eSCs exposed to both E2 and P4 (the model of decidualized/mid-secretory phase) in PCOS and non-PCOS samples.
Results
Quantitative transcriptome profile after steroid hormone post-treatment in PCOS and Ctrl eSCs
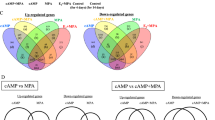
Regardless of the treatment, the eSCs from PCOS and Ctrl women clustered separately in the t-SNE (T-distributed stochastic neighbor embedding) plot (Fig. 1A). In both non-DE and DE eSCs, as well as with and without DHT, there were 20–36 DEGs between PCOS vs. Ctrl groups. Most of the differences were observed in the DE-eSCs after E2P4 exposure (E2P4) (Fig. 1B), regardless of the DHT treatment. Based on SOM (Self-organizing map) analyses for each treatment, the PCOS and Ctrl groups also clustered separately (Fig. 2A–D). The non-DE eSCPCOS post-treated with E2 without DHT featured differentially expressed genes (DEGs) related to cell signaling and metabolism (GALNT4, TOM1L1, PASK, MTRR, ZNF711, CIP2A), whereas E2 + DHT exposure triggered DEGs related to androgen action (CDADC1, FOXO1, PDGFRL, KLHDC1, IFI44L). Following E2P4 post-treatment, DE-eSCPCOS presented with DEGs related to cell cycle, proliferation, differentiation, and inflammation (E2P4: CDKN3, CD58, SNCA, CCNA2, PIFB1, MICB; E2P4DHT: ILF2, NDUFAF4, GABRE, IFIT3, IL24, PTX3, NEK3). In the presence of DHT (E2P4DHT vs. E2DHT), there was no major change in the number of DEGs in the eSCCtrl, whereas in eSCPCOS, there was an increase in the number of DEGs (p = 0.029, Fig. 1C). A Venn diagram depicting the DEGs for comparison of E2P4 ± DHT vs. E2 ± DHT, is presented in Fig. 1D. A detailed listing of the DEGs can be found in Supplemental Table S1A–C.

Quantitative transcriptome differences following steroid hormone exposure. (A) T-distributed stochastic neighbor embedding (t-SNE) plot of differentially expressed genes (DEGs) with FDR (False discovery rate) < 0.05 and absolute log2 fold change (LFC) > 2. Blue and red dots represent respectively endometrial stromal cells (eSCs) from Ctrl and PCOS. (B) Counts of up-regulated (red) and down-regulated (pink) DEGs in response to hormone treatment in PCOS group compared to Ctrl for non-decidualized (non-DE) eSCs post-treated with E2 with or without (±) DHT, and for decidualized (DE) eSCs post-treated with E2P4 ± DHT. (C) Counts of up- and down-regulated DEGs in Ctrl (blue) and PCOS (red) women in non-DE eSCs with E2 ± DHT and in DE eSCs with E2P4 ± DHT. (D) Venn diagram of group-wise comparisons illustrating the counts for the common vs. uniquely expressed DEGs for E2P4 vs. E2 and E2P4DHT vs. E2DHT in Ctrl (blue) and PCOS (red), respectively. E2, estrogen; P4, progesterone; DHT, dihydrotestosterone. The DEG counts, shown as ratios, illustrate the number of DEGs before and after applying Independent Hypothesis Weighting Bonferroni (IHW-BON) corrections for filtering the most significant hits. Validation targets were chosen from the DEG groups marked with a red circle.

Self-organizing map (SOM) clustering analysis. Gene expression levels for Ctrl (cyan, n = 6) and PCOS (pink, n = 6) eSCs treated with (A) estrogen; E2 and (B) E2 with dihydrotestosterone; DHT (E2DHT) in non-decidualized (non-DE) eSCs; (C) E2 and progesterone; P4 without DHT (E2P4) and (D) E2 and P4 with DHT (E2P4DHT) treatment in decidualized (DE) eSCs. Red indicates high and blue indicates low expression level. Differentially expressed genes (DEGs) with false discovery rate (FDR < 0.05) were taken into consideration in the analyses.
Common DEGs for decidualization with steroid post-treatment
In the Venn diagram (Fig. 1D), 65 genes were common for decidualization in all four group-wise comparisons regardless of PCOS status or hormone treatment. These genes were thus considered conserved decidualization genes of eSCs in vitro. An additional 19 genes were identified to be specific for E2P4 vs. E2 comparison, both in Ctrl and PCOS. The 65 conserved genes included commonly known decidualization genes such as SST, REN, MMP3, POSTN, PSAT1, and COL1A2, whereas the 19 DEGs included mostly genes related to cell cycle regulation: SPDYE16, FLT3, HDKC1, ACTG2, N4BP2, IFIT2, SFRP4 and ZWINT. A total of 18 DEGs detected comparing E2P4DHT vs. E2DHT were common for both PCOS and Ctrl, including genes involved in cytoskeleton and/or cell migratory properties (PLPPR4, LPAR1), and cell differentiation or proliferation (STC2, EZH2, HJURP, ANG, CENPN). A detailed list of these DEGs is shown in the Supplemental Table S2 A–C. Lysophosphatidic Acid Receptor 1 (LPAR1), which showed reduced expression in DE eSCs after DHT exposure in both Ctrl and PCOS, was chosen for validation.
Unique DEGs associated with steroid hormone post-treatment in decidualized vs. non-decidualized eSCs
The PCOS vs. Ctrl DEGs that were characteristic of the DE vs. non-DE comparison were assessed after steroid hormone post-treatment. This comparison revealed 140 unique group-specific DEGs for eSCCtrl and 251 for eSCPCOS without DHT exposure, and revealed 136 DEGs for eSCCtrl and 314 for eSCPCOS with DHT exposure (Fig. 1D, Supplemental Table S3A–D). After correction for multiple testing (IHW-BON), the remaining DEG count was 13/140 in Ctrl and 19/251 in PCOS for the E2P4 vs. E2 comparison in the absence of DHT (Supplemental Table S4A, B). The 13 unique genes for eSCCtrl included genes involved in cellular function (SKA3, GATA6, SAXO2), immune response (GPANK1, STK17B), and ion channel function (KCNK2, KCNB1). The 19 unique DEGs for the eSCPCOS were mostly related to fatty acid and mitochondrial energy metabolism (RARRES1, EGR2, FABP5, SLC2A3, CKMT1B, ABCB1/MDR1) and progesterone signaling (HHIP). The IHW-BON corrected DEGs (13/136) for E2P4DHT vs. E2DHT in eSCCtrl included genes predominantly related to immune response (CXCL8/IL-8, ATG4C, IL-24, MMP10, TNFSF4). The same comparison in eSCPCOS (17/314) included DEGs related to cellular calcium phosphate homeostasis and intracellular trafficking (PHEX, PGAP1, RAB3C, SNX1, C2CD6), inflammation (GBP1, RCN3, BBX), and androgen action related cell stemness (KDM7A, IFI27, ALDH1A1, C2CD6) (Supplemental Table S4C, D). From these DEGs, Aldehyde Dehydrogenase 1 Family Member A1 (ALDH1A1), which was downregulated in DE eSCPCOS after DHT exposure, was chosen as a validation target.
Functional enrichment analysis of DEGs
According to the Reactome and GO annotation assignment, the pathways related to the 65 common DEGs for decidualization (E2P4 ± DHT) were significantly (FDR < 0.05) enriched for processes such as cell proliferation, collagen formation, extracellular matrix modulation, and the cell cycle. The only significant pathway for the 18 common DEGs in the E2P4DHT vs. E2DHT comparison was related to the lysosphingolipid and the lysophosphatidic acid (LPA) receptor. For the 314 PCOS group-specific DEGs arising from the E2P4DHT vs. E2DHT comparison, pathways related to the cell cycle, interferon signaling, endosomal pathway, nucleosome assembly, and immunoregulatory interactions were found to be enriched. Detailed data are shown in Supplemental Table S5A–G.
In vitro and in vivo LPAR1 and ALDH1A1 protein validation
For validation of the RNA-seq data, two candidate genes were chosen representing two distinct groups of DEGs in the Venn diagram (Fig. 1D; red circles): LPAR1 (a DE-down-regulated gene in E2P4DHT vs. E2DHT that was common to both groups) and ALDH1A1 (a DE-down-regulated gene E2P4DHT vs. E2DHT that was unique to PCOS). The IF data for the in vitro decidualized eSCs post-treated with steroid hormones is presented in Fig. 3. According to the logarithmic fold change (LFC) ratio based on E2 and E2P4 treatments ± DHT, down-regulation of both LPAR1 and ALDH1A1 could be observed in DE eSCPCOS and eSCCtrl post-treated with DHT. Consistent with the RNA-seq data, ALDH1A1 showed more pronounced downregulation in eSCPCOS compared to eSCCtrl (Fig. 3A, B). The IHC analysis of all endometrium biopsy samples showed significant downregulation of LPAR1 and ALDH1A1 in the stromal compartment in SE vs. PE comparison only in the Ctrl group (p < 0.001), whereas no significant change was detected in PCOS samples (Fig. 4A–D).

Validation of in vitro protein expression by immunofluorescence (IF). (A) Radar plot showing in vitro effect of DHT on expression of LPAR1 (blue) or ALDH1A1 (orange) in non-DE eSCs with E2 ± DHT and in DE-eSCs with E2P4 ± DHT in both groups (Ctrl, PCOS; both n = 3). The y-axis shows log fold change (LFC) values calculated for ratios with and without DHT; LFC 0 line (no change in expression) is shown in dotted black. (B) IF panel for LPAR1 and ALDH1A1 protein expression in non-DE-eSCs with E2 or E2DHT post-treatment and DE-eSCs with E2P4 or E2P4DHT post-treatments in eSCCtrl and eSCPCOS; nuclei are stained with DAPI. Scale bar: 100 µm.

Validation of in vivo protein expression by immunohistochemistry (IHC). (A, B) In vivo protein expression showing LPAR1 and ALDH1A1 expression in the endometrial stroma in Ctrl and PCOS; n = 6, proliferative endometrium (PE): cycle day (6–9); Ctrl, PCOS; n = 5, secretory endometrium (SE): collected 9–10 days after the luteinizing hormone surge. (C, D) Whole biopsy samples from PE and SE in Ctrl and PCOS were used for IHC staining to identify LPAR1 and ALDH1A1 proteins. For every patient, 3 different areas (700 × 700 µm) on each slide were considered and stroma was separated from the glandular epithelium manually for staining quantification. Scale bar 100 µm; NC; negative control.
Discussion
This is the first study assessing a global gene expression profile after a short-term steroid hormone exposure in vitro in eSCPCOS compared to eSCCtrl. Our specific interest was to investigate the effect of DHT in the presence of E2 (modeling PE) or E2P4 (modeling SE) in non-DE and DE eSCs, respectively. We hypothesized that the hormonal challenges may reveal an underlying defect in the eSCPCOS, which could be linked to the altered placentation and adverse pregnancy outcomes reported in vivo.
The clear clustering of the RNA-seq study samples in PCOS and Ctrl group both in the t-SNE plot and the SOM suggests a distinct transcriptome profile for the eSCPCOS, which includes many previously unreported genes, especially for the PCOS endometrium (Figs. 1A, B, 2). It is known that the PCOS endometrium is burdened with an adverse metabolic environment arising from factors such as visceral obesity, insulin resistance, and hyperinsulinemia, all common for the syndrome4. In line with this, our study was also able to identify up-regulation of genes in the non-DE eSCPCOS with E2 post-treatment, which have been previously reported to be involved in lipid and glucose metabolism (GALNT4, GALNT8, PASK)16,17. On the other hand, the E2DHT post-treatment in eSCPCOS revealed several DEGs related to androgen action (CDADC1, FOXO1, PDGFRL), supporting the previous findings that estrogen promotes androgen action in the non-PCOS and PCOS endometrium18,19,20. Even though the eSCPCOS showed comparable decidualization capacity with the eSCCtrl by classical decidualization markers PRL and IGFBP-1, the global gene expression profile after short-term hormonal exposure in DE-eSCPCOS was different from the one in DE-eSCCtrl. Several of the increased (SNCA, CDKN3, CD58) or decreased (PIBF1, MICB) DEGs have been previously described in endometriosis, endometrial cancer, and other gynecological pathologies as being specifically involved in delayed decidualization and inflammation21,22,23,24. Moreover, the addition of DHT to E2P4 in DE eSCs resulted in a complete change in gene expression in eSCPCOS, including modulation of many progesterone receptor (PR) target genes such as IFIT3, IL24, PTX3, and NEK3, all of which are involved in cell proliferation, decidualization, and inflammation. Interestingly, these genes have also been reported to be associated with endometriosis and implantation failure25,26,27,28. Collectively, these findings support the impaired action of P4 in the PCOS endometrium in a hyperandrogenic milieu, in line with previous studies4.
Decidualization, a well-coordinated transformation process of eSCs from an E2-dominant proliferative state to an E2P4-driven differentiated state, is imperative for successful embryo attachment, implantation, and healthy pregnancy. The central role of decidualization also necessitates identifying the set of robust conservative genes that drive this important process. Accordingly, we identified 65 DEGs (Fig. 1D) common for decidualization, regardless of the study groups and DHT exposure. Of these 65 DE-consensus genes, 10 were identical and 6 were homologous to genes that have been included in the endometrial receptivity array (ERA)29. There were also 19 DEGs that were common for decidualization in the absence of DHT (E2P4 vs. E2) and were found in both groups; these are involved in the cell cycle and metabolic processes (HKDC1, ACTG2, ZWINT, IFIT2) and are crucial for ensuring the normal endometrial milieu30,31. In addition, 19 unique DEGs (Supplemental Table S4B) were identified for the E2P4 vs. E2 comparison in eSCPCOS without added DHT, underlining the distinct transcriptome profile for DE-eSCPCOS. Among the upregulated DEGs in this PCOS/DE-group, RARRES1 is involved in androgen signaling via retinoic acid synthesis32, and EGR2 and FABP5 are both involved in fatty acid uptake33,34. In the same DEG group, the downregulated transporter genes are associated with mitochondrial function (CKMT1B, ABCB1/MDR1)35,36. In addition, a well-known mediator of the progesterone signaling cascade HHIP was found to be downregulated, implying an impaired decidualization process for eSCPCOS37,38. This is consistent with our previously reported data, which indicated a robust impairment of decidualization in more severe PCOS cases39. The milder phenotype of the PCOS cases included in the present study may explain why the underlined impairment was not seen in classical decidualization markers but in the larger gene panel after steroid hormone exposure. The concept of progesterone resistance and mitochondrial dysfunction in the PCOS endometrium is also supported by a recent work in PCOS-mimicking animals, where DHT exposure during pregnancy resulted in impairment of the endometrial progesterone effect, decidualization, and mitochondrial function40,41.
Previous studies have reported that normal levels of androgens modulate the P4 effect, thereby facilitating decidualization and normal endometrial function42. The comparison of E2P4 vs. E2 in the presence of DHT (E2P4DHT vs. E2DHT) yielded 18 DEGs (Fig. 1D) common for both study groups, representing DHT-dependent decidualization genes. As expected, these DEGs were involved in androgen receptor (AR) regulation (PLPPR4, STC2, ANG, LPAR1), suggesting the interference of DHT with lysophosphatidic acid (LPA) signaling, which is involved in a variety of cellular processes such as proliferation, differentiation, adhesion, and tissue morphology43,44,45. Recently, the molecular influence of the LPA receptor on implantation was discovered, as its targeted deletion in mice resulted in significantly reduced litter size, which could be attributed to delayed implantation and altered embryo development46. However, the suppression of LPAR1 is apparent in the secretory DE stromal cells in control/fertile women, as seen in our study (Figs. 3, 4). These changes were not as uniform in PCOS women and may thus contribute to the endometrial dysfunction in PCOS. Thus, our study offers a unique and intriguing link between DHT effects on eSCs decidualization and altered phospholipid signaling. Future studies will be required to clarify whether this connection has a role in PCOS-related endometrial dysfunction.
In a PCOS mouse model, the DHT exposure has also been shown to induce impaired decidualization and implantation that could be linked to poor vasculature, angiogenesis, and placental formation in these animals47. In our study, the 17 DEGs unique to PCOS in the E2P4DHT vs. E2DHT comparison (Supplemental Table S4D), have been previously reported to be upregulated in cases with thin endometrium and implantation failure for example (PHEX, RNASE7, SNX1)48. Among the down-regulated DEGs in the same group, there were several genes connected to androgen-related cellular signaling (KDM7A, ALDH1A1, C2CD6)49,50, as well as genes important for implantation (GBP1, RCN3, BBX, IFI27)51,52,53,54. Of these, ALDH1A1 was chosen for validation as it has been shown to be specific for androgen action and plays a role in several physiological processes such as lipid and glucose metabolism55. In DE eSCPCOS, ALDH1A1 was downregulated after DHT exposure in RNA-seq analysis, suggesting a desynchronized androgen effect in PCOS women compared to non-PCOS Ctrl. In vitro validation by IF for this protein was in line with our RNA-seq data. Although in vivo IHC analysis confirmed down-regulation of ALDH1A1 during decidualization in non-PCOS Ctrl women, the trend was not evident in tissue samples derived from PCOS women in IHC studies. This apparent contradiction can be easily explained by the fact that intact tissue samples from the natural cycle do not explicitly mirror the RNA-seq results performed on isolated chemically induced DE eSCs in vitro.
Unlike eSCPCOS, the 13 DEGs in eSCCtrl (E2P4DHT vs. E2DHT comparison, Supplemental Table S4C) featured several inflammatory cascade-related genes suggesting that the eSCCtrl are highly stressed during androgen exposure. Considering all the differences found between the eSCs from fertile/Ctrl and PCOS women, we suggest that the eSCs from non-PCOS women should not be used to model PCOS, but rather primary cells from PCOS subjects should be used due to an inherently altered steroid hormone response in the PCOS endometrium. This change was also evident in the absence of DHT, and especially in the case of decidualized eSCs in vitro, as they presented with progesterone resistance and mitochondrial dysfunction. DHT was able to cause other changes unique to eSCPCOS. This likely implies altered epigenetic regulation in eSCPCOS56, although yet requires further research.
The strengths of the study include a well-defined sample set and experimental design including exposure with three central steroid hormones in different combinations and comparisons between cases and controls. Regarding limitations, the in vitro decidualization model and limited sample size is not well comparable with the in vivo system. In addition, the 24 h hormone exposure after decidualization can be considered short, although short-term exposure was already able to reveal unique differences between study groups. Further replication of the findings and functional validation in a larger sample set should be pursued in future studies.
Conclusion
The data herein provides, for the first time, insight into decidualized eSCPCOS and their steroid hormone response using global gene expression profiling. The results highlight an underlying defect in decidualized eSCPCOS that is present with or without DHT exposure. This may suggest the presence of cellular memory, likely involving the epigenetic mechanisms. These changes may provide functional insights into endometrial dysfunction in women with PCOS and pave the way for future studies on biological themes such as progesterone resistance, LPA signaling, and mitochondrial dysfunction. The results of the present study may also give insight into the altered endometrium milieu for embryo implantation and adverse pregnancy outcomes in PCOS-affected women. Further studies are also warranted to establish whether women with different PCOS phenotypes have different risks and mechanisms for endometrial dysfunction.
Materials and methods
Patient characteristics and sample collection
Endometrial biopsies were collected from women that were diagnosed with PCOS at the Oulu University Hospital (According to the Rotterdam criteria and the new PCOS guideline57,58) and from non-PCOS women (Ctrl). Exclusion criteria included pregnancy or breastfeeding within 6 months of collection and hormonal or anti-diabetic medication within 3 months of collection. The dating of the collected endometrial tissue samples utilized in all the experiments was evaluated by an experienced pathologist by examining the tissue morphology with hematoxylin and eosin (H/E) staining, and corresponded to the respective cycle phase. The study was approved by the Ethical Committee of Northern Ostrobothnia Hospital District. Informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
Isolation of endometrial stromal cells (eSCs), in vitro decidualization, and hormone exposure
Endometrial biopsy samples were collected at the proliferative phase (PE) of the menstrual cycle (cycle days [cd] 6–10) from Ctrl (mean age/body mass index (BMI) ± standard error of the mean (SEM); age 35 ± 2.3 years, BMI 27 ± 0.5 kg/m2) and from women with PCOS (age 35 ± 1.0 years, BMI 27 ± 2.6 kg/m2). Endometrial stromal cells were isolated from these biopsies as previously described39. There were no significant differences between the groups in age or BMI.
For decidualization (DE), eSCs from Ctrl and PCOS (eSCCtrl, eSCPCOS, ntotal = 12) were treated with 8-Br-cAMP (0.5 mM, Sigma-Aldrich, Germany) in duplicate for 96 h in a low serum medium5, while non-decidualized (non-DE) cells were cultured for 96 h using only DMSO (vehicle; Sigma-Aldrich, Germany). Cells were than washed and starved for 24 h in low serum media. Next, eSCs were post-treated for 24 h with hormones in low serum medium: non-DE eSCs were treated with E2 (10 nM, Sigma-Aldrich, Germany) with or without (±) DHT (100 nM, Sigma-Aldrich, Germany)59; while the DE eSCs were post-treated with E2 (10 nM, Sigma-Aldrich, Germany) in combination with P4 (1 µM, Sigma-Aldrich, Germany) (E2P4) ± DHT. Decidualization was confirmed by analyzing the increased expression of prolactin (PRL) and insulin-like growth factor-1 (IGFBP-1) both in individual Ctrl and PCOS. There were no significant differences between the groups (Supplemental Figure 1). The detailed study protocol is presented in Fig. 5.

Schematic diagram of workflow. Proliferative phase (PE, cycle day 6–9) endometrial stromal cells (eSCs) from women with PCOS and non-PCOS controls (both n = 6) were decidualized (DE; 0.5 mM 8-Br-cAMP; stock solution prepared in DMSO) and non-decidualized (non-DE; 0.1% DMSO, vehicle) for 96 h. Cells were washed and starved in low serum media for 24 h. Non-DE eSCs were subjected to post-treatment with estrogen alone (E2, 10 nM) or with dihydrotestosterone (DHT, 100 nM; E2 + DHT) for 24 h. The DE-eSCs were subjected to post-treatment with E2 and progesterone (P4, 1 µM; E2 + P4) or E2P4 with DHT (100 nM; E2 + P4 + DHT) for 24 h. All four-hormone combination treated cells were harvested, and RNA sequencing (NextSeq500, Illumina) was performed from all different treatments’ arms (n = 48).
RNA sequencing, data processing, and bioinformatic analysis
Forty-eight samples in duplicate (total number of 96 samples) from four different hormone combination treatments as well as both groups were used for RNA isolation and cDNA library preparation. Total RNA was extracted using RNeasy Mini Kits (Qiagen, Valencia, USA) and RNA sequencing (RNA-seq) libraries were prepared using the SMART-seq2 protocol with modifications60. Initial data retrieval and processing is outlined in the Supplemental Methods Part 1. The RNA-seq data are available in the GEO database (accession: GSE171507). The differential expression and subsequent bioinformatics analysis are presented in Supplemental Methods Part 2.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
For technical validation of sequencing data, RT-qPCR was performed according to the protocol described in Supplemental Methods Part 3. The results are shown in Fig. 6. The oligonucleotide primers are presented in Supplemental Table S6.

Technical validation of RNA-seq by reverse transcription quantitative polymerase chain reaction (RT-qPCR). Differentially expressed genes (DEGs) with false discovery rate, FDR < 0.05 were chosen from E2P4DHT vs. E2DHT comparison (eSCCtrl, n = 3). The log2 fold change (LFC) for RT-qPCR is shown on the y-axis as the blue bar. The numerical LFC value retrieved from RNA-seq data is specific for the treatment corresponding gene shown inside each bar. Data are presented as mean ± standard error of the mean (SEM). TBP and GAPDH were used as reference genes.
In vitro studies and immunofluorescence (IF) for LPAR1 and ALDH1A1 proteins
Proliferative phase (cd 6–10) eSCs from controls (n = 3, age 35 ± 2.1 years, BMI 28 ± 3.5 kg/m2) and women with PCOS (n = 3, age 31 ± 2.0 years, BMI 26 ± 2.8 kg/m2) were treated with 8-Br-cAMP or 0.1% DMSO (DE vs. non-DE) for 96 h, then treated with hormones for 24 h as described earlier. The cells were fixed with methanol, and then stained with primary antibodies (anti-LPAR1 and anti-ALDH1A1, 1:100) and a nuclear stain (DAPI). The detailed protocol is described in Supplemental Methods Part 4.
In vivo studies and immunohistochemistry (IHC) for LPAR1 and ALDH1A1 proteins
The PE phase endometrial biopsies were obtained in (cd 6–10); (Ctrl, n = 6, age 35 ± 2.3 years, BMI 27 ± 0.5 kg/m2; PCOS, n = 6, age 35 ± 1.0 years, BMI 27 ± 2.6 kg/m2). The SE phase endometrial biopsies were also obtained 9–10 days after a surge in luteinizing hormone (LH) confirmed by the Clearblue Advanced Digital Ovulation Test kit and by a pathologist based on endometrial histology (Ctrl, n = 5, age 28 ± 3.3 years, BMI 24 ± 2.2 kg/m2; PCOS, n = 5, age 33 ± 2.4 years, BMI 25 ± 0.6 kg/m2). Formalin-fixed paraffin-embedded sections were used for IHC analysis using primary antibodies (anti-LPAR1, 1:400 and anti-ALDH1A1, 1:1000). The detailed protocol is described in Supplemental Methods Part 5.
References
Balen, A. H. et al. The management of anovulatory infertility in women with polycystic ovary syndrome: An analysis of the evidence to support the development of global WHO guidance. Hum. Reprod. Update 22, 687–708 (2016).
Rodriguez Paris, V. & Bertoldo, M. J. The mechanism of androgen actions in PCOS etiology. Med. Sci. (Basel) https://doi.org/10.3390/medsci7090089 (2019).
Bahri Khomami, M. et al. Increased maternal pregnancy complications in polycystic ovary syndrome appear to be independent of obesity—A systematic review, meta-analysis, and meta-regression. Obes. Rev. 20, 659–674 (2019).
Palomba, S., Piltonen, T. T. & Giudice, L. C. Endometrial function in women with polycystic ovary syndrome: A comprehensive review. Hum. Reprod. Update 27, 584–618 (2021).
Gellersen, B. & Brosens, J. Cyclic AMP and progesterone receptor cross-talk in human endometrium: A decidualizing affair. J. Endocrinol. 178, 357–372 (2003).
Kajihara, T. et al. Androgen signaling in decidualizing human endometrial stromal cells enhances resistance to oxidative stress. Fertil. Steril. 97, 185–191 (2012).
Apparao, K. B., Lovely, L. P., Gui, Y., Lininger, R. A. & Lessey, B. A. Elevated endometrial androgen receptor expression in women with polycystic ovarian syndrome. Biol. Reprod. 66, 297–304 (2002).
Hulchiy, M., Nybacka, A., Sahlin, L. & Hirschberg, A. L. Endometrial expression of estrogen receptors and the androgen receptor in women with polycystic ovary syndrome: A lifestyle intervention study. J. Clin. Endocrinol. Metab. 101, 561–571 (2016).
Quezada, S. et al. Evaluation of steroid receptors, coregulators, and molecules associated with uterine receptivity in secretory endometria from untreated women with polycystic ovary syndrome. Fertil. Steril. 85, 1017–1026 (2006).
Palomba, S. et al. Early trophoblast invasion and placentation in women with different PCOS phenotypes. Reprod. Biomed. Online 29, 370–381 (2014).
Sun, M. et al. Elevated maternal androgen is associated with dysfunctional placenta and lipid disorder in newborns of mothers with polycystic ovary syndrome. Fertil. Steril. 113, 1275-1285.e2 (2020).
Kajihara, T. et al. Androgens modulate the morphological characteristics of human endometrial stromal cells decidualized in vitro. Reprod. Sci. 21, 372–380 (2014).
Younas, K. et al. Delayed endometrial decidualisation in polycystic ovary syndrome; the role of AR-MAGEA11. J. Mol. Med. (Berl) 97, 1315–1327 (2019).
Munzker, J. et al. Testosterone to dihydrotestosterone ratio as a new biomarker for an adverse metabolic phenotype in the polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 100, 653–660 (2015).
Torchen, L. C. et al. Evidence for increased 5alpha-reductase activity during early childhood in daughters of women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 101, 2069–2075 (2016).
Bennett, E. P. et al. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 22, 736–756 (2012).
Wu, X. et al. PAS kinase drives lipogenesis through SREBP-1 maturation. Cell Rep. 8, 242–255 (2014).
Li, N. et al. Upregulation of FoxO 1 signaling mediates the proinflammatory cytokine upregulation in the macrophage from polycystic ovary syndrome patients. Clin. Lab. 63, 301–311 (2017).
Marshall, E. et al. In silico analysis identifies a novel role for androgens in the regulation of human endometrial apoptosis. J. Clin. Endocrinol. Metab. 96, E1746–E1755 (2011).
Lee, M. H. et al. Hyperandrogenic milieu dysregulates the expression of insulin signaling factors and glucose transporters in the endometrium of patients with polycystic ovary syndrome. Reprod. Sci. https://doi.org/10.1177/1933719119833487 (2019).
Aghajanova, L. et al. The protein kinase A pathway-regulated transcriptome of endometrial stromal fibroblasts reveals compromised differentiation and persistent proliferative potential in endometriosis. Endocrinology 151, 1341–1355 (2010).
Basu, S., Pioli, P. A., Conejo-Garcia, J., Wira, C. R. & Sentman, C. L. Estradiol regulates MICA expression in human endometrial cells. Clin. Immunol. 129, 325–332 (2008).
Kolbe, D. L. et al. Differential analysis of ovarian and endometrial cancers identifies a methylator phenotype. PLoS ONE 7, e32941 (2012).
Zhou, M. et al. Decreased PIBF1/IL6/p-STAT3 during the mid-secretory phase inhibits human endometrial stromal cell proliferation and decidualization. J. Adv. Res. 30, 15–25 (2020).
Shao, J. et al. Macrophages promote the growth and invasion of endometrial stromal cells by downregulating IL-24 in endometriosis. Reproduction 152, 673–682 (2016).
Popovici, R. M. et al. The long pentraxin PTX3 in human endometrium: Regulation by steroids and trophoblast products. Endocrinology 149, 1136–1143 (2008).
Miller, S. L., Antico, G., Raghunath, P. N., Tomaszewski, J. E. & Clevenger, C. V. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene 26, 4668–4678 (2007).
Guo, Y. et al. Differential gene expression in bovine endometrial epithelial cells after challenge with LPS; specific implications for genes involved in embryo maternal interactions. PLoS ONE 14, e0222081 (2019).
Diaz-Gimeno, P. et al. A genomic diagnostic tool for human endometrial receptivity based on the transcriptomic signature. Fertil. Steril. 95(50–60), 60.e1–15 (2011).
Kenigsberg, S. et al. Gene expression microarray profiles of cumulus cells in lean and overweight-obese polycystic ovary syndrome patients. Mol. Hum. Reprod. 15, 89–103 (2009).
Ramly, B., Afiqah-Aleng, N. & Mohamed-Hussein, Z. A. Protein–protein interaction network analysis reveals several diseases highly associated with polycystic ovarian syndrome. Int. J. Mol. Sci. https://doi.org/10.3390/ijms20122959 (2019).
Wickenheisser, J. K. et al. Retinoids and retinol differentially regulate steroid biosynthesis in ovarian theca cells isolated from normal cycling women and women with polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 90, 4858–4865 (2005).
Zhou, X. J. et al. FCGR2B and FCRLB gene polymorphisms associated with IgA nephropathy. PLoS One 8, e61208 (2013).
Tsai, I. T. et al. FABP1 and FABP2 as markers of diabetic nephropathy. Int. J. Med. Sci. 17, 2338–2345 (2020).
Clark, H. et al. Dysfunctional MDR-1 disrupts mitochondrial homeostasis in the oocyte and ovary. Sci. Rep. 9, 9616 (2019).
Zhang, Y. et al. Sensorineural deafness and male infertility: A contiguous gene deletion syndrome. BMJ Case Rep. https://doi.org/10.1136/bcr.08.2008.0645 (2009).
Lee, K. et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat. Genet. 38, 1204–1209 (2006).
Hu, M. et al. Endometrial progesterone receptor isoforms in women with polycystic ovary syndrome. Am. J. Transl. Res. 10, 2696–2705 (2018).
Piltonen, T. T. et al. Endometrial stromal fibroblasts from women with polycystic ovary syndrome have impaired progesterone-mediated decidualization, aberrant cytokine profiles and promote enhanced immune cell migration in vitro. Hum. Reprod. 30, 1203–1215 (2015).
Risal, S. et al. Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat. Med. 25, 1894–1904 (2019).
Hu, M. et al. Hyperandrogenism and insulin resistance induce gravid uterine defects in association with mitochondrial dysfunction and aberrant reactive oxygen species production. Am. J. Physiol. Endocrinol. Metab. 316, E794–E809 (2019).
Gibson, D. A., Simitsidellis, I., Cousins, F. L., Critchley, H. O. & Saunders, P. T. Intracrine androgens enhance decidualization and modulate expression of human endometrial receptivity genes. Sci. Rep. 6, 19970 (2016).
Shin, J. & Sohn, Y. C. Identification of Ran-binding protein M as a stanniocalcin 2 interacting protein and implications for androgen receptor activity. BMB Rep. 47, 643–648 (2014).
Krikun, G. et al. Expression of angiopoietin-2 by human endometrial endothelial cells: Regulation by hypoxia and inflammation. Biochem. Biophys. Res. Commun. 275, 159–163 (2000).
Sheng, X., Yung, Y. C., Chen, A. & Chun, J. Lysophosphatidic acid signalling in development. Development 142, 1390–1395 (2015).
Ye, X. et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 435, 104–108 (2005).
Stener-Victorin, E. et al. Animal models to understand the etiology and pathophysiology of polycystic ovary syndrome. Endocr. Rev. https://doi.org/10.1210/endrev/bnaa010 (2020).
Maekawa, R. et al. Thin endometrium transcriptome analysis reveals a potential mechanism of implantation failure. Reprod. Med. Biol. 16, 206–227 (2017).
Lee, K. H. et al. Histone demethylase KDM7A controls androgen receptor activity and tumor growth in prostate cancer. Int. J. Cancer 143, 2849–2861 (2018).
Vazquez-Martinez, E. R. et al. DNA methylation in the pathogenesis of polycystic ovary syndrome. Reproduction 158, R27–R40 (2019).
Kumar, S. et al. Messenger ribonucleic acid encoding interferon-inducible guanylate binding protein 1 is induced in human endometrium within the putative window of implantation. J. Clin. Endocrinol. Metab. 86, 2420–2427 (2001).
Garcia-Herrero, S. et al. The transcriptome of spermatozoa used in homologous intrauterine insemination varies considerably between samples that achieve pregnancy and those that do not. Fertil. Steril. 94, 1360–1373 (2010).
Ponnampalam, A. P., Weston, G. C., Trajstman, A. C., Susil, B. & Rogers, P. A. Molecular classification of human endometrial cycle stages by transcriptional profiling. Mol. Hum. Reprod. 10, 879–893 (2004).
Gray, C. A. et al. Identification of endometrial genes regulated by early pregnancy, progesterone, and interferon tau in the ovine uterus. Biol. Reprod. 74, 383–394 (2006).
Petrosino, J. M., Disilvestro, D. & Ziouzenkova, O. Aldehyde dehydrogenase 1A1: Friend or foe to female metabolism?. Nutrients 6, 950–973 (2014).
Mimouni, N. E. H. et al. Polycystic ovary syndrome is transmitted via a transgenerational epigenetic process. Cell. Metab. 33, 513-530.e8 (2021).
Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil. Steril. 81, 19–25 (2004).
Teede, H. J. et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum. Reprod. 33, 1602–1618 (2018).
Li, X. et al. Regulation of androgen receptor expression alters AMPK phosphorylation in the endometrium: In vivo and in vitro studies in women with polycystic ovary syndrome. Int. J. Biol. Sci. 11, 1376–1389 (2015).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Funding
This work was financially supported by a grant from the Academy of Finland (grant no. 315921, 321763), the Finnish Medical Foundation, The Sigrid Juselius Foundation, Estonian Research Council (grant PRG1076), Horizon 2020 innovation (ERIN, grant no. EU952516) of the European Commission and Enterprise Estonia (grant EU48695).
Author information
Authors and Affiliations
Contributions
T.T.P., A.S., A.R. were involved in study conception, design and critical review of the manuscript; K.K., K.J., M.L. conducted SMART-seq2 and RT-qPCR, H.R.R. was involved in sample collection; R.K.A. performed critical revision of the manuscript; F.L. performed gene enrichment analysis; K.K. conducted IHC experiments; D.L. performed IF experiments and critical revision of the manuscript, A.M. performed bioinformatics analyses; M.K. was responsible for study design, sample collection, cell culture, decidualization,RT-qPCR, data interpretation and manuscript preparation. All authors have actively participated in the study design and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khatun, M., Meltsov, A., Lavogina, D. et al. Decidualized endometrial stromal cells present with altered androgen response in PCOS. Sci Rep 11, 16287 (2021). https://doi.org/10.1038/s41598-021-95705-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-95705-0
This article is cited by
-
Hyperinsulinemia impairs decidualization via AKT-NR4A1 signaling: new insight into polycystic ovary syndrome (PCOS)-related infertility
Journal of Ovarian Research (2024)
-
Polycystic ovary syndrome
Nature Reviews Disease Primers (2024)
-
Induced Pluripotent Stem Cells as a Possible Approach for Exploring the Pathophysiology of Polycystic Ovary Syndrome (PCOS)
Stem Cell Reviews and Reports (2024)
-
Targeted gene expression profiling for accurate endometrial receptivity testing
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.

{kind=link}