
Abstract
Cigarette smoking is a modifiable behaviour associated with mental health. We investigated the degree of genetic overlap between smoking behaviours and psychiatric traits and disorders, and whether genetic associations exist beyond genetic influences shared with confounding variables (cannabis and alcohol use, risk-taking and insomnia). Second, we investigated the presence of causal associations between smoking initiation and psychiatric traits and disorders. We found significant genetic correlations between smoking and psychiatric disorders and adult psychotic experiences. When genetic influences on known covariates were controlled for, genetic associations between most smoking behaviours and schizophrenia and depression endured (but not with bipolar disorder or most psychotic experiences). Mendelian randomization results supported a causal role of smoking initiation on psychiatric disorders and adolescent cognitive and negative psychotic experiences, although not consistently across all sensitivity analyses. In conclusion, smoking and psychiatric disorders share genetic influences that cannot be attributed to covariates such as risk-taking, insomnia or other substance use. As such, there may be some common genetic pathways underlying smoking and psychiatric disorders. In addition, smoking may play a causal role in vulnerability for mental illness.
Similar content being viewed by others


Introduction
Tobacco smoking is a modifiable risk factor and despite declining rates of smoking, 14–15% of adults in the US and UK and 28% across Europe still smoke regularly1,2,3. High co-occurrence between smoking behaviours and psychiatric disorders is well-established4,5,6. Smoking rates among individuals diagnosed with schizophrenia or bipolar disorder are five times greater and with depression two-fold greater compared to healthy controls7,8. Smoking behaviours also co-occur with psychotic experiences9,10,11, traits in the general population that typically resemble positive symptoms of psychotic disorders (e.g., paranoia, hallucinations and delusions). Smoking behaviours co-occur with schizotypy12, a construct that has parallels with psychotic experiences and which assesses personality characteristics thought to reflect schizophrenia vulnerability. Regular smoking during adolescence has been associated with psychotic experiences and negative symptom traits (PENS) such as anhedonia13.
In terms of underlying causes, shared genetic influences could explain the co-occurrence of smoking and psychiatric traits or disorders. A recent twin study found that regular smoking during adolescence shared genetic influences with paranoia and cognitive disorganization and familial influences with hallucinations13. A previous study found no evidence that adolescent PENS were predicted by polygenic liability to initiate smoking14 but used less well-powered genome-wide association study (GWAS) summary statistics than currently available. Schizophrenia and major depression share genome-wide genetic influences with smoking behaviours15,16, and polygenic liability to bipolar disorder has been associated with nicotine dependence17. Genetic variants for smoking being identified in GWAS for schizophrenia could also indicate possible causal associations between these traits18.
Several covariates could, at a genetic level, account for genetic overlap between psychiatric traits or disorders and smoking behaviour. Epidemiological studies have investigated cannabis and alcohol use, stressful or traumatic events, sociodemographic characteristics, novelty-seeking behaviour and sleep disturbances as covariates in the association between smoking and mental health problems9,10,11,13,19,20,21,22,23. Risk-taking has also been associated with smoking24,25 and psychiatric outcomes26. Many of these covariates, including cannabis and alcohol use, risk-taking and insomnia, are partly under genetic influence and have been genetically associated with smoking or with psychiatric traits or disorders27,28,29,30,31,32.
Smoking behaviour may be a causal risk factor for psychiatric disorders based on evidence from longitudinal and dose–response associations and Mendelian randomization studies19,21,23,33,34. Uncertainty remains over whether smoking may be causally linked to psychotic experiences prior to the onset of psychiatric conditions or in individuals who may not meet diagnostic thresholds: The association between psychotic experiences and smoking is not fully explained by known confounding factors9,35,36,37,38,39, (but see35) and a dose–response relationship has been reported9,40 although not consistently35,38. Furthermore, most longitudinal studies report an association between smoking and later reports of psychotic experiences10,41,42,43 while adolescents do not appear to start smoking to alleviate pre-existing psychotic experiences41. However, these observational studies cannot rule out the possibility that psychotic experiences were present prior to smoking initiation and vice-versa. Triangulation of longitudinal findings on the association between smoking and psychotic experiences with evidence from methods such as Mendelian randomization is needed.
In this study we employed a range of methods utilizing genetic data to systematically investigate the association between smoking behaviours and psychotic experiences across adolescence and adulthood as well as with psychiatric disorders. We assessed the degree to which smoking behaviours are genetically correlated with major psychiatric disorders (schizophrenia, major depression and bipolar disorder), with psychotic experiences during adolescence (paranoia and hallucinations, cognitive disorganization, anhedonia and negative symptoms), psychotic experiences in adults (auditory hallucinations, visual hallucinations, delusions of persecution and delusions of reference), and with schizotypy in adults (hypomania, perceptual aberrations, physical anhedonia and social anhedonia) after controlling for genetic overlap with cannabis and alcohol use, risk taking behaviour and sleep disturbances27,28,29,30,31. Furthermore, we aimed to assess causal associations between smoking initiation and psychotic experiences across development and confirm previous reports of causal associations with psychiatric disorders.
Results
Conditional genetic overlap between smoking behaviours and mental health
Bivariate genetic correlations between smoking behaviours and psychotic experiences and psychiatric disorders are summarised in Fig. 1.

Heat map showing genetic correlations between smoking behaviours, psychotic experiences and psychiatric disorders. PENS Psychotic experiences (PE) and negative symptom traits; NA indicates that genetic correlations could not be computed due to low SNP-heritability or sample size; *indicates statistically significant genetic correlations at p < .05; **indicates significance at FDR < .05 using Benjamini–Hochberg correction for 60 tests; Genetic correlations reported using unconstrained LD score regression intercept between phenotypes with sample overlap (for example, smoking behaviour and major depression GWASs contained participants from the UK Biobank, and smoking behaviours and adolescent PENS contained participants from ALSPAC). Note that age of smoking initiation was coded in the direction of lower scores reflecting younger ages of initiation. Note that current smoking cases are current smokers (compared to ex-smokers).
To investigate the degree to which these genetic correlations exist beyond genetic influences associated with covariates (lifetime cannabis use, alcohol consumption, insomnia and risk-taking behaviour), we specified genomic multiple regression models using Genomic Structural Equation Modelling (Genomic SEM)44. Models were run for phenotype pairs that shared at least nominally significant (p < 0.05) genetic overlap. Genetic correlations between smoking behaviours and the covariates are shown in Supplementary Figure S1.
Figure 2 shows the path diagram between schizophrenia and smoking initiation to illustrate the specified covariance paths in the models (see Supplementary Figures S2-5 for path diagrams of all models). Table 1 summarizes the conditional genetic correlations (bg) between smoking behaviours and psychiatric disorders/psychotic experiences obtained from these models.

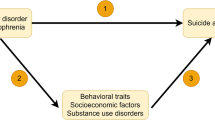
Path diagram illustrating genetic multiple regression models. SmkInitg = Genetic component of smoking initiation; CANg = Genetic component of cannabis use; ALCg = Genetic component of alcohol consumption; INSg = Genetic component of insomnia; SCZg = Genetic component of schizophrenia; u = residual genetic variance; bg = Conditional genetic correlation between the genetic components of smoking initiation and schizophrenia as summarized in Table 1; Double-headed arrow represents genetic covariance; Single-headed arrow represents regression paths; Path diagrams for models between all smoking behaviours and psychiatric phenotypes are provided in Supplementary Figures S2-5.
The genetic component of cigarettes per day, together with the genetic components of the four covariate predictors, accounted for 11–41% of genetic variation in psychiatric disorders and traits (calculated as one minus the residual variance shown in the figures). The equivalent values for smoking initiation, age of smoking initiation and current smoking status were 9–52%, 9–41%, and 11–41%, respectively.
Table 1 shows that after accounting for the genetic influences of the covariates, conditional genetic associations (bg) were significant between cigarettes per day and schizophrenia (unattenuated compared to rg estimates that did not account for covariates), depression (with bg accounting for 53% of rg; calculated as bg/rg × 100) and adolescent anhedonia (unattenuated) but not with bipolar disorder, auditory hallucinations, visual hallucinations and hypomania.
Significant conditional genetic associations were found between smoking initiation and depression, accounting for 42% of the rg estimate from LD score regression. The conditional genetic association between smoking initiation and bipolar disorder was negative. This suggests that overlapping genetic influences of the covariates drove the positive genetic correlation observed in LD score regression. No genetic association was found between smoking initiation and schizophrenia, visual hallucinations, delusions of persecution, hypomania and adolescent cognitive disorganization after accounting for the genetic influences of covariates.
No significant conditional genetic associations were found between age of smoking initiation with schizophrenia, depression, auditory hallucinations, visual hallucinations and hypomania. Negative conditional genetic association estimates indicate an association with a younger age of smoking initiation.
Significant genetic overlap not accounted for by other predictors in the model was found between current smoking and schizophrenia (with bg being higher than the rg estimate from LD score regression), depression (accounting for 79% of rg) and visual hallucinations (74% of rg) and not with auditory hallucinations. In summary, after controlling for genetic overlap with cannabis and alcohol use, insomnia and risk-taking behaviour, most smoking behaviours still showed significant genetic overlap with psychiatric disorders, but not with most measures of psychotic experiences.
Causal associations between smoking initiation and mental health
Table 2 presents the results from Mendelian randomization (MR; see also Supplementary Figures S6-13 and Supplementary Tables S2-4). All instrumental variables had a mean F > 10 indicating adequate strength of association with the exposures, and an I2 > 0.9 and therefore MR-Egger sensitivity analyses can be considered reliable. Generalized summary-based Mendelian randomization (GSMR)45 and inverse-variance weighted Mendelian randomization (MR-IVW)46 analyses yielded consistent results across analyses. These methods suggested a causal effect of smoking initiation on schizophrenia liability with this finding replicated in Weighted Median but not in MR-Egger or Weighted Mode sensitivity analyses, suggesting that results may have been biased due to violation of the instrumental variable assumptions47,48,49. A significant but smaller causal effect of schizophrenia liability on smoking initiation was also found, but the MR-Egger intercept was significantly different from zero (p = 0.043; Supplementary Table S2), indicating the presence of directional pleiotropy which could bias results from GSMR.
Evidence of a causal effect of smoking initiation on liability to major depression was found, and replicated in Weighted Median MR. However, this effect was not replicated in Weighted Mode MR and the GSMR effect size fell outside of the MR-Egger confidence intervals. Therefore, biased results due to violation of the instrumental variable assumptions cannot be ruled out. The MR-Egger intercept was significantly different from zero (p = 0.048), suggesting that results may have been affected by directional pleiotropy. No consistent evidence of a causal effect of depression liability on smoking initiation was detected.
Evidence of a causal effect of smoking initiation on liability for bipolar disorder was reported in GSMR and Weighted Median analyses with a similar effect size observed in MR-Egger, but not replicated in the Weighted Mode analysis. No evidence of reverse causation was found.
Evidence of a causal effect of smoking initiation on cognitive disorganisation was observed in GSMR and Weighted Median analyses. Effect sizes fell within the MR-Egger confidence intervals but was not replicated in Weighted Mode MR. We found a causal effect of smoking initiation on negative symptoms in the main MR analysis, but this effect was not replicated in MR sensitivity analyses. No evidence of reverse causation was observed.
Discussion
This study aimed to investigate systematically the degree of overlapping common genetic influences between smoking behaviours and psychiatric traits and disorders across adolescence and adulthood. We found evidence of overlapping genetic influences at a genome-wide level between smoking behaviours and psychiatric disorders and with some psychotic experiences and negative symptom traits reported by adolescents and adults in the community. For schizophrenia and depression, the overlap in common genetic influences shown with smoking frequency and current smoking status remained after controlling for genetic influences on known covariates, namely cannabis use, alcohol use, risk taking and insomnia. Genetic associations between smoking behaviours and most positive psychotic experiences were explained by genetic influences shared with the covariates. We found evidence of causal effects of smoking initiation on adolescent cognitive and (to some degree) negative symptoms as well as on schizophrenia, depression and bipolar disorder. These findings hint at plausible pathways by which smoking during adolescence could be involved in the development of psychiatric disorders. However, evidence of causation should be considered moderate to weak because not all sensitivity analyses confirmed findings.
Our GSEM findings support the hypothesis that schizophrenia and depression share genetic pathways with smoking frequency and persistent smoking. This shared genetic aetiology may point toward common biological pathways such as those involving nicotine, the principal pharmacologically active component of smoking that acts as an agonist on the nicotinic acetylcholine receptor (nAChR). Variants within the CHRNA5‐A3‐B4 gene cluster which encodes for subunits of nAChR are among the most robust associations with nicotine dependence50,51,52,53 and have also been implicated in schizophrenia18. Presynaptic activation of nAChR stimulates the release of several neurotransmitters including dopamine, serotonin and glutamate54,55,56,57. Dysregulation of dopaminergic and glutamatergic pathways could both explain why some people may be more susceptible to the positive reinforcing effects of smoking58 and have an increased vulnerability to develop schizophrenia59.
We found support for a causal role of smoking initiation on liability to develop schizophrenia, depression and bipolar disorder, corroborating recent findings33,34 whilst applying different Mendelian randomization methods. Compared to previous MR studies, we employed a method to remove likely pleiotropic variants from genetic instruments which aims to reduce confounding from biological pleiotropy. Despite this, not all sensitivity analyses yielded consistent findings in our study, and therefore biased causal effect estimates due to pleiotropy or a proportion of invalid instruments cannot be ruled out. Interestingly, our GSEM findings indicated that the covariates accounted for genetic overlap between schizophrenia and smoking initiation, raising the possibility that these traits may be involved in causal pathways of smoking initiation on schizophrenia or that the Mendelian randomization methods may not have adequately controlled for pleiotropy. Our results of a possible causal effect of smoking initiation on schizophrenia and depression (and a small causal effect of schizophrenia on smoking initiation) concurs with meta-analytic findings of longitudinal studies21,23. The action of nicotine on nAChR could underlie a mechanism by which smoking causally influences mental health. Chronic exposure to nicotine may result in long-lasting alterations of dopaminergic and cholinergic pathways, leading to an increase in risk of psychiatric disorders60,61,62. Beyond nicotine, other toxic compounds released during combustion of tobacco cause neuro-inflammation and oxidative stress63, factors that are associated with psychiatric disorders64,65,66.
To our knowledge, this is the first study to report that genome-wide genetic influences on bipolar disorder overlap with those on smoking frequency and initiation. This finding supports recent evidence of an association between polygenic scores for bipolar disorder and nicotine dependence17. Genetic overlap between bipolar disorder and smoking quantity was accounted for by genetic influences on the covariates. Interestingly, we found that genetic influences on smoking initiation was negatively associated with genetic influences on bipolar disorder after accounting for genetic overlap with the covariates, whereas the bivariate genetic correlation between these two phenotypes was positive. This negative genetic association was likely masked by overlapping genetic influences of the covariates. As such, currently identified common genetic variation shared between smoking and bipolar disorder is unlikely to explain causal pathways or may reflect complex pathways underlying multiple, related addiction, personality and psychiatric phenotypes.
Until recently, research into the genetic aetiology underlying the association between psychotic experiences in the community and smoking behaviours was lacking. Here, we report novel evidence that smoking behaviours share genetic influences with some types of psychotic experiences during adulthood (notably with visual and auditory hallucinations) and with hypomania. Interestingly, unlike our findings for schizophrenia and depression, the genetic overlap between most psychotic experiences and smoking was shared with the covariates. This suggest that overlapping genome-wide genetic influences (captured by current GWASs) that act on both psychotic experiences and smoking behaviours are likely shared with co-occurring traits such as other substance use, sleep problems and risk taking, or mechanisms underlying these traits.
Our findings suggest that smoking may increase a propensity to experience cognitive and negative psychotic experiences during adolescence, although the association with negative symptom traits was not confirmed by sensitivity analyses. Our findings that smoking may cause early cognitive and negative symptoms during adolescence could hint at developmental pathways by which smoking could increase the risk of developing psychiatric disorders. A recent preclinical study found that during adolescence, exposure to nicotine could lead to persistent alteration of neurotransmitter pathways61 likely relevant to psychotic experiences. Smoking initiation and positive psychotic experiences in adulthood did not appear to be causally related. This is somewhat surprising given the known phenotypic association between psychotic experiences and psychiatric disorders67,68,69,70,71,72 and could be explored further. Evidence suggests that psychotic experiences in adults have somewhat different underlying causal influences compared to psychotic experiences during adolescence73 which may explain that lack of causal associations between smoking and psychotic experiences in later life. We also note that the GWASs on positive psychotic experiences were based on a small number of cases, which could have resulted in low power to detect causal effects in our study.
This study had limitations to be considered. In some genomic multiple regression models, the estimated residual variances were imprecise due to small GWAS sample sizes, particularly for schizotypy and adolescent PENS. To mitigate this, we performed GSEM models only for traits that had significant bivariate genetic correlations. However, non-significant genetic correlations may have reflected smaller GWAS sample sizes rather than the absence of genetic overlap, and these analyses should be revisited once more powerful summary statistics become available. It is also possible that genetic influences on co-occurring factors other than those we considered (such as exposure to adverse life events or sociodemographic characteristics) could account for the genetic overlap between smoking and psychiatric disorders. Our Mendelian randomization analyses that used instruments from psychotic experiences summary statistics requires replication once better-powered GWASs for these psychiatric traits become available. We performed Mendelian randomization using instruments for smoking initiation, which has the advantage of being a trait measurable to the whole adolescent and adult population (unlike number of cigarettes per day, which only applies to smokers). Nevertheless, it is noted that smoking initiation may partly pick up on traits such as risk taking or impulsivity. Future studies should consider using instruments for objective measures of smoking on samples stratified by smoking status to confirm our results. However, such designs have their own limitations since gene-environment interactions are likely (genes affecting tobacco consumption will only do so in those who took up smoking) and in such cases Mendelian randomization on stratified samples may introduce collider bias74. Possible pleiotropic effects can be further investigated using multivariable Mendelian randomization, but GWASs with overlapping samples for exposures and confounders (as was the case here) require additional adjustments and individual-level phenotypic data. The GWAS on smoking initiation included a small number of ALSPAC participants and overlapping samples may have inflated Mendelian randomization estimates in analyses with adolescent PENS. Despite our approach to exclude possible pleiotropic instruments from Mendelian randomization analyses, the sensitivity analyses indicated that we cannot rule out bias due to violation of the instrumental variable assumptions. Finally, results based on GWASs from large cohort and biobank studies may be affected by ascertainment bias.
In conclusion, there is genetic overlap between smoking behaviours and schizophrenia and depression, and we show it exists beyond genetic influences shared with other risk factors. Furthermore, smoking appears to play a role in the development of traits relating to mental illness during adolescence and our findings strengthen the evidence base for smoking as a causal risk associated with psychiatric disorders. Our study indicates that the mental health risks of smoking during adolescence require further investigation. Exploration of the biological links underlying smoking and psychiatric illness, and smoking interventions as part of mental health prevention strategies, are well-justified based on current evidence.
Methods
Samples and measures
Summary statistics for participants of European descent used in the analyses are presented in Table 3. Smoking behaviours50 included smoking initiation, a binary phenotype with smokers defined as those who reported having ever smoked regularly. The average number of cigarettes per day was assessed in current and former smokers, with never-smokers excluded. Age of smoking initiation was defined as the age at which current or former smokers started smoking regularly. Current smoking was assessed among smokers and coded as either current smokers or former smokers.
Summary statistics for adolescent PENS were obtained from a mega-GWAS of four continuous scales of PENS, assessed when participants were 15 to 19 years old. Ethical approval for the original adolescent PENS GWAS75 was obtained for TEDS76 from the Institute of Psychiatry ethics committee (ref: 05/Q0706/228), for ALSPAC77,78 from the ALSPAC Ethics and Law Committee and the Local Research Ethics Committees, and for CATSS79 from the Karolinska Institute Ethical Review Board. All research participants and their parents granted informed consent.
Summary statistics for schizotypy in adulthood80,81 were available for four continuous schizotypy scales82,83,84. Positive psychotic experiences in adults were assessed using four dichotomous items on lifetime occurrence. The average age of onset of psychotic experiences was 31.6 (s.d. = 17.6) years. Psychiatric disorders were defined based on standard diagnostic criteria and assessed during clinical interviews or obtained from health records and self-report16,85,86.
For covariates in genomic multiple regression, we selected phenotypes considered to be likely confounders of the association between smoking and psychotic experiences/psychiatric disorders9,10,11,19,20,21,22,23 and for which GWAS summary statistics were available. Summary statistics were obtained for: cannabis use32, a binary phenotype for whether participants had ever used cannabis; alcohol consumption50, defined as average number of weekly drinks; risk taking was assessed with the item “Would you describe yourself as someone who takes risks?”; and insomnia defined as having trouble falling asleep at night or waking up in the middle of the night27.
Additional details on summary statistics are provided in Supplementary Table S1 and the Supplementary Methods. The Birkbeck Department of Psychological Sciences’ Ethics Committee approved this study and all research was performed in accordance with institutional guidelines.
Analyses
Prior to analyses, single nucleotide polymorphisms (SNPs) with incomplete association statistics and strand ambiguous and non-biallelic SNPs were excluded. Variants were matched and allele orders harmonized to the 1000 Genomes (phase 3) reference panel for European ancestry and restricted to HapMap 3 variants. Variants were excluded based on INFO scores < 0.9 and minor allele frequency (MAF) < 0.01. INFO scores were not provided in the summary statistics for smoking behaviours and drinks per week and were filtered on INFO < 0.3 by the study authors50.
LD score regression
Genetic correlations (rg) were estimated using linkage disequilibrium (LD) score regression87,88 on a liability scale based on population prevalences of 1% for schizophrenia, 15% for major depression and 2% for bipolar disorder89,90,91. Covariance intercepts were left unconstrained in analyses with overlapping samples (GWASs for smoking behaviours, depression and psychotic experiences in adults included UK Biobank participants; GWASs for smoking behaviours and adolescent PENS included ALSPAC participants). Correction for multiple testing (60 tests) was performed using Benjamini–Hochberg correction at a false discovery rate (FDR) of 0.05.
Genomic structural equation modelling
To investigate genetic overlap between psychiatric phenotypes and smoking behaviours after accounting for the genetic influences on confounds, Genomic Structural Equation Modelling (Genomic SEM)44 was used to model shared genetic architecture using genetic covariance structures.
Summary statistics were converted to include LD scores in LD score regression software using the parameters described above. Genomic covariance structures were computed, and genomic multiple regression models specified in the Genomic SEM package for R version 3.5.292 for phenotype pairs that had nominally significant genetic correlations (at p < 0.05). Models allowed for genetic covariation between smoking phenotypes and the covariates included as predictors and regressed onto psychiatric outcomes. Standardized estimates were reported thereby allowing the association between a given predictor and each outcome to be interpreted as the genetic correlation conditional on all other predictors. Conditional genetic correlations were considered statistically significant at p < 0.05.
Mendelian randomization
Mendelian randomization (MR)93 was performed to test for bi-directional causal associations between smoking initiation and psychiatric phenotypes using summary statistics (see also Supplementary Methods).
Generalised Summary-data-based Mendelian Randomisation (GSMR) was used as the main method due to its advantages of accounting for sampling variation in the exposure and outcome GWASs and for residual LD structure between variants used as instrumental variables (IVs)45. As a baseline to compare against sensitivity analyses, we also report MR-IVW46 results. MR-Egger regression, Weighted Median and Weighted Mode MR were conducted as sensitivity analyses as these methods make different assumptions to GSMR and MR-IVW by allowing for a proportion of invalid IVs in the analyses47,48,49. To assess whether MR-Egger will be an appropriate sensitivity analysis, we computed the I2 statistic, with I2 > 0.9 indicating that MR-Egger results can be considered reliable94. MR-Egger is typically less powerful to detect significant effects, but the MR-Egger confidence intervals are expected overlap with valid effect sizes from other methods. Directional pleiotropy was assessed using the MR-Egger intercept test. An intercept significantly different from zero indicates that MR-Egger causal estimates may be more robust compared to GSMR estimates. The strength of association between the IVs and exposures was assessed using the mean F-statistic, with F > 10 considered adequate instrument strength. Consistent findings across MR methods indicates that results from GSMR are less likely to be biased due to violations of IV assumptions.
Summary statistics for major depression excluded 23andMe participants. To ensure at least 20 IVs were used, genome-wide significant variants were however obtained from the publication that did include the 23andMe participants16. Major depression and adult psychotic experiences summary statistics included UK Biobank participants. To avoid overlapping samples in these MR analyses, we used a version of the smoking initiation summary statistics that excluded UK Biobank participants (N = 249,171) and selected IVs at a p value threshold of 5 × 10–7, resulting in 20 independent depression variants to use as IVs in these MR analyses. Only seven variants reached genome-wide significance in the summary statistics for bipolar disorder86. IVs for bipolar disorder were obtained from the publication of a recent meta-GWAS, also conducted on Psychiatric Genomics Consortium samples, but for which full summary statistics were not available95. For all other exposures, IVs were identified using the clumping algorithm in PLINK96 based on an r2 threshold = 0.05 within a 500 kb window. Recent GWASs on psychotic experiences have not yet been replicated using equivalent measures in independent samples or have been based on relatively small sample sizes. Therefore, IVs for psychotic experiences were selected at p < 5 × 10–5.
Analyses were performed in GSMR and MR Base R packages45,97. GCTA98 was used to calculate the LD structure between lead variants based on the 1000 Genomes (phase 3) reference panel for European ancestry. IVs excluded from GSMR analyses due to being Heidi-outliers and in residual LD at r2 > 0.1 were also removed from MR sensitivity analyses. MR was conducted for smoking initiation but not for smoking phenotypes assessed in smokers only. This is because variant associations among smokers may not explain smoking liability in samples that include non-smokers. Smoking initiation is also temporally relevant to the adolescent PENS since most smokers initiate smoking during adolescence99. Significance thresholds were set at p < 0.05.
Data and code availability
GWAS summary statistics used in the analyses for this paper are available at https://atlas.ctglab.nl/ and at https://www.med.unc.edu/pgc/download-results/. Summary data for adolescent psychotic experiences and for schizotypy were obtained from the authors with permission from the participating cohorts. Code to perform genomic structural equation modelling of GWAS summary statistics within Genomic SEM can be found at https://github.com/MichelNivard/GenomicSEM/wiki. Code to perform the Mendelian randomization analyses is available at https://cnsgenomics.com/software/gsmr/ and at https://www.mrbase.org/.
References
Office for National Statistics. Adult smoking habits in the UK: 2018. (2019). https://www.ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/healthandlifeexpectancies/bulletins/adultsmokinghabitsingreatbritain/2018. Accessed August 14, 2019.
Centers for Disease Control and Prevention. Current cigarette smoking among adults in the United States. (2019). https://www.cdc.gov/tobacco/data_statistics/fact_sheets/adult_data/cig_smoking/index.htm. Accessed August 14, 2019.
World Health Organization. European tobacco use: Trends report 2019. (2019). http://www.euro.who.int/__data/assets/pdf_file/0009/402777/Tobacco-Trends-Report-ENG-WEB.pdf?ua=1. Accessed September 6, 2019.
de Leon, J. & Diaz, F. J. A meta-analysis of worldwide studies demonstrates an association between schizophrenia and tobacco smoking behaviors. Schizophr. Res. 76, 135–157. https://doi.org/10.1016/j.schres.2005.02.010 (2005).
Lawrence, D., Mitrou, F. & Zubrick, S. R. Smoking and mental illness: results from population surveys in Australia and the United States. BMC Public Health 9, 285. https://doi.org/10.1186/1471-2458-9-285 (2009).
Thomson, D. et al. Tobacco use in bipolar disorder. Clin. Psychopharmacol. Neurosci. 13, 1–11. https://doi.org/10.9758/cpn.2015.13.1.1 (2015).
Prochaska, J. J., Das, S. & Young-Wolff, K. C. Smoking, mental illness, and public health. Annu. Rev. Public Health 38, 165–185. https://doi.org/10.1146/annurev-publhealth-031816-044618 (2017).
Hartz, S. M. et al. Comorbidity of severe psychotic disorders with measures of substance use. JAMA Psychiat. 71, 248–254. https://doi.org/10.1001/jamapsychiatry.2013.3726 (2014).
Bhavsar, V. et al. Tobacco smoking is associated with psychotic experiences in the general population of South London. Psychol. Med. 48, 123–131. https://doi.org/10.1017/s0033291717001556 (2018).
Gage, S. H. et al. Associations of cannabis and cigarette use with psychotic experiences at age 18: findings from the Avon Longitudinal Study of Parents and Children. Psychol. Med. 44, 3435–3444. https://doi.org/10.1017/s0033291714000531 (2014).
Jones, H. J. et al. Association of combined patterns of tobacco and cannabis use in adolescence with psychotic experiences. JAMA Psychiat. 75, 240–246. https://doi.org/10.1001/jamapsychiatry.2017.4271 (2018).
Rössler, W. et al. Subclinical psychosis syndromes in the general population: Results from a large-scale epidemiological survey among residents of the canton of Zurich, Switzerland. Epidemiol. Psychiatr. Sci. 24, 69–77. https://doi.org/10.1017/s2045796013000681 (2015).
Barkhuizen, W., Taylor, M. J., Freeman, D. & Ronald, A. A twin study on the association between psychotic experiences and tobacco use during adolescence. J. Am. Acad. Child Adolesc. Psychiatry 58, 267-276.e268. https://doi.org/10.1016/j.jaac.2018.06.037 (2019).
Krapohl, E. et al. Phenome-wide analysis of genome-wide polygenic scores. Mol. Psychiatry 21, 1188–1193. https://doi.org/10.1038/mp.2015.126 (2016).
Hartz, S. M. et al. Genetic correlation between smoking behaviors and schizophrenia. Schizophr. Res. 194, 86–90. https://doi.org/10.1016/j.schres.2017.02.022 (2018).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681. https://doi.org/10.1038/s41588-018-0090-3 (2018).
Reginsson, G. W. et al. Polygenic risk scores for schizophrenia and bipolar disorder associate with addiction. Addict. Biol. 23, 485–492. https://doi.org/10.1111/adb.12496 (2018).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. https://doi.org/10.1038/nature13595 (2014).
Mustonen, A. et al. Smokin’ hot: adolescent smoking and the risk of psychosis. Acta Psychiatr. Scand. 138, 5–14. https://doi.org/10.1111/acps.12863 (2018).
Fergusson, D. M., Goodwin, R. D. & Horwood, L. J. Major depression and cigarette smoking: Results of a 21-year longitudinal study. Psychol. Med. 33, 1357–1367. https://doi.org/10.1017/s0033291703008596 (2003).
Chaiton, M. O., Cohen, J. E., O’Loughlin, J. & Rehm, J. A systematic review of longitudinal studies on the association between depression and smoking in adolescents. BMC Public Health 9, 356. https://doi.org/10.1186/1471-2458-9-356 (2009).
Diaz, F. J. et al. Tobacco smoking behaviors in bipolar disorder: A comparison of the general population, schizophrenia, and major depression. Bipolar Disord. 11, 154–165. https://doi.org/10.1111/j.1399-5618.2009.00664.x (2009).
Gurillo, P., Jauhar, S., Murray, R. M. & MacCabe, J. H. Does tobacco use cause psychosis? Systematic review and meta-analysis. Lancet Psychiatry 2, 718–725. https://doi.org/10.1016/s2215-0366(15)00152-2 (2015).
Wood, A. P., Dawe, S. & Gullo, M. J. The role of personality, family influences, and prosocial risk-taking behavior on substance use in early adolescence. J. Adolesc. 36, 871–881. https://doi.org/10.1016/j.adolescence.2013.07.003 (2013).
Gullo, M. J. & Dawe, S. Impulsivity and adolescent substance use: Rashly dismissed as “all-bad”?. Neurosci. Biobehav. Rev. 32, 1507–1518. https://doi.org/10.1016/j.neubiorev.2008.06.003 (2008).
Reddy, L. F. et al. Impulsivity and risk taking in bipolar disorder and schizophrenia. Neuropsychopharmacology 39, 456–463. https://doi.org/10.1038/npp.2013.218 (2014).
Hammerschlag, A. R. et al. Genome-wide association analysis of insomnia complaints identifies risk genes and genetic overlap with psychiatric and metabolic traits. Nat. Genet. 49, 1584–1592. https://doi.org/10.1038/ng.3888 (2017).
Clifton, E. A. D. et al. Genome-wide association study for risk taking propensity indicates shared pathways with body mass index. Commun. Biol. 1, 36. https://doi.org/10.1038/s42003-018-0042-6 (2018).
Nivard, M. G. et al. Connecting the dots, genome-wide association studies in substance use. Mol. Psychiatry 21, 733. https://doi.org/10.1038/mp.2016.14 (2016).
Walters, R. K. et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci. 21, 1656–1669. https://doi.org/10.1038/s41593-018-0275-1 (2018).
Taylor, M. J., Gregory, A. M., Freeman, D. & Ronald, A. Do sleep disturbances and psychotic-like experiences in adolescence share genetic and environmental influences?. J. Abnorm. Psychol. 124, 674–684. https://doi.org/10.1037/abn0000057 (2015).
Pasman, J. A. et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat. Neurosci. 21, 1161–1170. https://doi.org/10.1038/s41593-018-0206-1 (2018).
Vermeulen, J. et al. Smoking and the risk for bipolar disorder: evidence from a bidirectional Mendelian randomisation study. Br. J. Psychiatry 218, 88–94. https://doi.org/10.1192/bjp.2019.202 (2021).
Wootton, R. E. et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: A Mendelian randomisation study. Psychol. Med. 50, 2435–2443. https://doi.org/10.1017/S0033291719002678 (2020).
Wolfe, R. M., Reeves, L. E., Gibson, L. E., Cooper, S. & Ellman, L. M. Attenuated positive psychotic symptoms in relation to cigarette smoking in a nonclinical population. Nicotine Tob. Res. 19, 124–128. https://doi.org/10.1093/ntr/ntw240 (2017).
Mallet, J., Mazer, N., Dubertret, C. & Le Strat, Y. Tobacco smoking and psychotic-like experiences in a general population sample. J. Clin. Psychiatry https://doi.org/10.4088/JCP.17m11994 (2018).
van Gastel, W. A. et al. Cigarette smoking and cannabis use are equally strongly associated with psychotic-like experiences: a cross-sectional study in 1929 young adults. Psychol. Med. 43, 2393–2401. https://doi.org/10.1017/S0033291713000202 (2013).
Saha, S. et al. The association between delusional-like experiences, and tobacco, alcohol or cannabis use: A nationwide population-based survey. BMC Psychiatry 11, 202. https://doi.org/10.1186/1471-244x-11-202 (2011).
McGrath, J. J. et al. Age at first tobacco use and risk of subsequent psychosis-related outcomes: A bir th cohort study. Aust. N. Z. J. Psychiatry 50, 577–583. https://doi.org/10.1177/0004867415587341 (2016).
Koyanagi, A., Stickley, A. & Haro, J. M. Psychotic symptoms and smoking in 44 countries. Acta Psychiatr. Scand. 133, 497–505. https://doi.org/10.1111/acps.12566 (2016).
Davies, J., Sullivan, S. & Zammit, S. Adverse life outcomes associated with adolescent psychotic experiences and depressive symptoms. Soc. Psychiatry Psychiatr. Epidemiol. 53, 497–507. https://doi.org/10.1007/s00127-018-1496-z (2018).
Ferdinand, R. F., van der Ende, J. & Verhulst, F. C. Associations between visual and auditory hallucinations in children and adolescents, and tobacco use in adulthood. Soc. Psychiatry Psychiatr. Epidemiol. 39, 514–520. https://doi.org/10.1007/s00127-004-0777-x (2004).
Vermeulen, J. et al. Smoking, symptoms, and quality of life in patients with psychosis, siblings, and healthy controls: A prospective, longitudinal cohort study. Lancet Psychiatry 6, 25–34. https://doi.org/10.1016/s2215-0366(18)30424-3 (2019).
Grotzinger, A. D. et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav. 3, 513–525. https://doi.org/10.1038/s41562-019-0566-x (2019).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224. https://doi.org/10.1038/s41467-017-02317-2 (2018).
Burgess, S., Butterworth, A. S. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665. https://doi.org/10.1002/gepi.21758 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314. https://doi.org/10.1002/gepi.21965 (2016).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998. https://doi.org/10.1093/ije/dyx102 (2017).
Liu, M. et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat. Genet. 51, 237–244. https://doi.org/10.1038/s41588-018-0307-5 (2019).
Thorgeirsson, T. E. et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452, 638–642. https://doi.org/10.1038/nature06846 (2008).
Erzurumluoglu, A. M. et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol. Psychiatry 25, 2392–2409. https://doi.org/10.1038/s41380-018-0313-0 (2020).
Brazel, D. M. et al. Exome chip meta-analysis fine maps causal variants and elucidates the genetic architecture of rare coding variants in smoking and alcohol use. Biol. Psychiatry 85, 946–955. https://doi.org/10.1016/j.biopsych.2018.11.024 (2019).
McKinney, D. L. & Vansickel, A. R. in Neuropathology of drug addictions and substance misuse (ed Victor R. Preedy) 93–103 (Academic Press, 2016).
Levin, E. D. in Advances in neurotoxicology Vol. 2 (eds Michael Aschner & Lucio G. Costa) 189–196 (Academic Press, 2018).
McCallum, S. E., Cowe, M. A., Lewis, S. W. & Glick, S. D. alpha3beta4 nicotinic acetylcholine receptors in the medial habenula modulate the mesolimbic dopaminergic response to acute nicotine in vivo. Neuropharmacology 63, 434–440. https://doi.org/10.1016/j.neuropharm.2012.04.015 (2012).
Brody, A. L. et al. Smoking-induced ventral striatum dopamine release. Am. J. Psychiatry 161, 1211–1218. https://doi.org/10.1176/appi.ajp.161.7.1211 (2004).
Ashok, A. H., Mizuno, Y. & Howes, O. D. Tobacco smoking and dopaminergic function in humans: A meta-analysis of molecular imaging studies. Psychopharmacology 236, 1119–1129. https://doi.org/10.1007/s00213-019-05196-1 (2019).
Coyle, J. T. Glutamate and schizophrenia: Beyond the dopamine hypothesis. Cell. Mol. Neurobiol. 26, 365–384. https://doi.org/10.1007/s10571-006-9062-8 (2006).
Mineur, Y. S. & Picciotto, M. R. Biological basis for the co-morbidity between smoking and mood disorders. J. Dual Diagn. 5, 122–130. https://doi.org/10.1080/15504260902869964 (2009).
Jobson, C. L. M. et al. Adolescent nicotine exposure induces dysregulation of mesocorticolimbic activity states and depressive and anxiety-like prefrontal cortical molecular phenotypes persisting into adulthood. Cereb. Cortex 29, 3140–3153. https://doi.org/10.1093/cercor/bhy179 (2019).
Quigley, H. & MacCabe, J. H. The relationship between nicotine and psychosis. Ther. Adv. Psychopharmacol. 9, 2045125319859969. https://doi.org/10.1177/2045125319859969 (2019).
Goncalves, R. B. et al. Impact of smoking on inflammation: Overview of molecular mechanisms. Inflamm. Res. 60, 409–424. https://doi.org/10.1007/s00011-011-0308-7 (2011).
Berk, M. et al. Pathways underlying neuroprogression in bipolar disorder: Focus on inflammation, oxidative stress and neurotrophic factors. Neurosci. Biobehav. Rev. 35, 804–817. https://doi.org/10.1016/j.neubiorev.2010.10.001 (2011).
Howes, O. D. & McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptualization. Transl. Psychiatry 7, e1024. https://doi.org/10.1038/tp.2016.278 (2017).
Miller, A. H., Maletic, V. & Raison, C. L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741. https://doi.org/10.1016/j.biopsych.2008.11.029 (2009).
Poulton, R. et al. Children’s self-reported psychotic symptoms and adult schizophreniform disorder: A 15-year longitudinal study. Arch. Gen. Psychiatry 57, 1053–1058 (2000).
Welham, J. et al. Emotional and behavioural antecedents of young adults who screen positive for non-affective psychosis: A 21-year birth cohort study. Psychol. Med. 39, 625–634. https://doi.org/10.1017/S0033291708003760 (2009).
Hanssen, M., Bak, M., Bijl, R., Vollebergh, W. & van Os, J. The incidence and outcome of subclinical psychotic experiences in the general population. Br. J. Clin. Psychol. 44, 181–191. https://doi.org/10.1348/014466505x29611 (2005).
Werbeloff, N. et al. Self-reported attenuated psychotic symptoms as forerunners of severe mental disorders later in life. Arch. Gen. Psychiatry 69, 467–475. https://doi.org/10.1001/archgenpsychiatry.2011.1580 (2012).
Zammit, S. et al. Psychotic experiences and psychotic disorders at age 18 in relation to psychotic experiences at age 12 in a longitudinal population-based cohort study. Am. J. Psychiatry 170, 742–750. https://doi.org/10.1176/appi.ajp.2013.12060768 (2013).
Dominguez, M. D., Wichers, M., Lieb, R., Wittchen, H. U. & van Os, J. Evidence that onset of clinical psychosis is an outcome of progressively more persistent subclinical psychotic experiences: An 8-year cohort study. Schizophr. Bull. 37, 84–93. https://doi.org/10.1093/schbul/sbp022 (2011).
Barkhuizen, W., Pain, O., Dudbridge, F. & Ronald, A. Genetic overlap between psychotic experiences in the community across age and with psychiatric disorders. Transl. Psychiatry 10, 86. https://doi.org/10.1038/s41398-020-0765-2 (2020).
Dudbridge, F. Commentary: Tobacco consumption and body weight: Mendelian randomization across a range of exposure. Int. J. Epidemiol. 45, e1–e3. https://doi.org/10.1093/ije/dyv033 (2016).
Pain, O. et al. Genome-wide analysis of adolescent psychotic-like experiences shows genetic overlap with psychiatric disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 177, 416–425. https://doi.org/10.1002/ajmg.b.32630 (2018).
Haworth, C. M., Davis, O. S. & Plomin, R. Twins Early Development Study (TEDS): A genetically sensitive investigation of cognitive and behavioral development from childhood to young adulthood. Twin Res. Hum. Genet. 16, 117–125. https://doi.org/10.1017/thg.2012.91 (2013).
Boyd, A. et al. Cohort Profile: The ’children of the 90s’—The index offspring of the Avon Longitudinal Study of Parents and Children. Int. J. Epidemiol. 42, 111–127. https://doi.org/10.1093/ije/dys064 (2013).
Fraser, A. et al. Cohort Profile: The Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol. 42, 97–110. https://doi.org/10.1093/ije/dys066 (2013).
Anckarsater, H. et al. The Child and Adolescent Twin Study in Sweden (CATSS). Twin Res. Hum. Genet. 14, 495–508 (2011).
Haapea, M. et al. Non-participation in a field survey with respect to psychiatric disorders. Scand. J. Public Health 36, 728–736. https://doi.org/10.1177/1403494808092250 (2008).
Ortega-Alonso, A. et al. Genome-wide association study of psychosis proneness in the Finnish population. Schizophr. Bull. 43, 1304–1314. https://doi.org/10.1093/schbul/sbx006 (2017).
Chapman, L. J., Chapman, J. P. & Raulin, M. L. Body-image aberration in Schizophrenia. J. Abnorm. Psychol. 87, 399–407 (1978).
Eckblad, M. & Chapman, L. J. Development and validation of a scale for hypomanic personality. J. Abnorm. Psychol. 95, 214–222 (1986).
Chapman, L. J., Chapman, J. P. & Raulin, M. L. Scales for physical and social anhedonia. J. Abnorm. Psychol. 85, 374–382 (1976).
Pardinas, A. F. et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 50, 381–389. https://doi.org/10.1038/s41588-018-0059-2 (2018).
Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic dissection of Bipolar Disorder and Schizophrenia, including 28 subphenotypes. Cell 173, 1705–1715. https://doi.org/10.1016/j.cell.2018.05.046 (2018).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241. https://doi.org/10.1038/ng.3406 (2015).
Bulik-Sullivan, B. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295. https://doi.org/10.1038/ng.3211 (2015).
Merikangas, K. R. et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch. Gen. Psychiatry 68, 241–251. https://doi.org/10.1001/archgenpsychiatry.2011.12 (2011).
Moreno-Kustner, B., Martin, C. & Pastor, L. Prevalence of psychotic disorders and its association with methodological issues. A systematic review and meta-analyses. PLoS ONE 13, e0195687. https://doi.org/10.1371/journal.pone.0195687 (2018).
Lim, G. Y. et al. Prevalence of depression in the community from 30 countries between 1994 and 2014. Sci. Rep. 8, 2861. https://doi.org/10.1038/s41598-018-21243-x (2018).
Core R Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2018).
Davey Smith, G. & Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease?. Int. J. Epidemiol. 32, 1–22 (2003).
Bowden, J. et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974. https://doi.org/10.1093/ije/dyw220 (2016).
Stahl, E. A. et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803. https://doi.org/10.1038/s41588-019-0397-8 (2019).
Chang, C. C. et al. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7. https://doi.org/10.1186/s13742-015-0047-8 (2015).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife https://doi.org/10.7554/eLife.34408 (2018).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82. https://doi.org/10.1016/j.ajhg.2010.11.011 (2011).
Marcon, A. et al. Trends in smoking initiation in Europe over 40 years: A retrospective cohort study. PLoS ONE 13, e0201881. https://doi.org/10.1371/journal.pone.0201881 (2018).
Acknowledgements
This work was supported by the UK Medical Research Council (G1100559 to AR) and a Wellcome Trust ISSF Grant (204770/Z/16/Z) and the Camara-Rijvers David Studentship to WB. This research was funded in whole by Wellcome [102215/2/13/2 and 204770/Z/16/Z]. For the purpose of open access, the author has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising from this submission. The authors gratefully acknowledge the ongoing contribution of the participants and their families in the Twins Early Development Study (TEDS), the Avon Longitudinal Study of Children and Parents (ALSPAC), the Child and Adolescent Twin Study in Sweden (CATSS) and participants in the North Finland Birth Cohort (NFBC) and the UK Biobank. We thank the funding bodies and research teams which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists and nurses. The data from TEDS was supported by a program grant to Robert Plomin from the UK Medical Research Council (MR/M021475/1) and UK Medical Research Council grant G1100559 to AR. The UK Medical Research Council and Wellcome (Grant Ref: 102215/2/13/2) and the University of Bristol provide core support for ALSPAC. This publication is the work of the authors and WB and AR will serve as guarantors for its contents. ALSPAC GWAS data was generated by Sample Logistics and Genotyping Facilities at Wellcome Sanger Institute and LabCorp (Laboratory Corporation of America) using support from 23andMe. We thank the Neale Lab and the Psychiatric Genomics Consortium (PGC) for providing genetic summary results and Dr. W. Hennah and A. Ortega-Alonso for preparing and sharing the schizotypy summary statistics. The authors are very grateful to Andrew Grotzinger for his advice on the implementation and interpretation of genomic multiple regression models performed within the Genomic Structural Equation Modelling software. WB had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Author information
Authors and Affiliations
Contributions
W.B. and A.R. designed the study. W.B. analysed the data. All authors interpreted the data. W.B. wrote the first draft. All authors contributed to the final draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Barkhuizen, W., Dudbridge, F. & Ronald, A. Genetic overlap and causal associations between smoking behaviours and mental health. Sci Rep 11, 14871 (2021). https://doi.org/10.1038/s41598-021-93962-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93962-7
This article is cited by
-
Deep sequencing of candidate genes identified 14 variants associated with smoking abstinence in an ethnically diverse sample
Scientific Reports (2024)
-
Cigarette Smoking and Psychiatric Illness Among Individuals with COPD: a Systematic Review
Current Addiction Reports (2024)
-
Mendelian randomization studies of depression: evidence, opportunities, and challenges
Annals of General Psychiatry (2023)
-
Evaluation of the causal relationship between smoking and schizophrenia in East Asia
Schizophrenia (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
