
Abstract
We investigated the genetic causes of major mental disorders (MMDs) including schizophrenia, bipolar disorder I, major depressive disorder and attention deficit hyperactive disorder, in a large family pedigree from Alpujarras, South of Spain, a region with high prevalence of psychotic disorders. We applied a systematic genomic approach based on karyotyping (n = 4), genotyping by genome-wide SNP array (n = 34) and whole-genome sequencing (n = 12). We performed genome-wide linkage analysis, family-based association analysis and polygenic risk score estimates. Significant linkage was obtained at chromosome 9 (9q33.1–33.2, LOD score = 4.11), a suggestive region that contains five candidate genes ASTN2, BRINP1, C5, TLR4 and TRIM32, previously associated with MMDs. Comprehensive analysis associated the MMD phenotype with genes of the immune system with dual brain functions. Moreover, the psychotic phenotype was enriched for genes involved in synapsis. These results should be considered once studying the genetics of psychiatric disorders in other families, especially the ones from the same region, since founder effects may be related to the high prevalence.
Similar content being viewed by others

Introduction
Psychiatric disorders aggregate in families and their predisposition involve a complex, polygenic and pleiotropic genetic architecture1,2,3. Patterns of shared genetic material have shown across the five major mental disorders (MMD): autism spectrum disorder (ASD), schizophrenia (SCZ), bipolar disorder (BD), major depressive disorder (MDD) and alcoholism1,2,3,4,5. Genetic epidemiological studies have revealed that the risk of developing one of these disorders is proportional to the genomic material shared with an affected individual6. In fact, the heritability of MMDs has been estimated as being at least 80%6,7. Thanks to the application of whole-genome scan technologies, as genome-wide association studies (GWASs) and next generation sequencing, in the recent years we have observed a dramatic improvement in identifying genetic risk factors for these disorders8,9,10,11. Of those, common SNPs have shown to contribute to around 20% of the heritability, with individually weaker contributions (odds ratios, < 1.2)12. Meanwhile, copy number variants (CNVs) as well as rare de novo or recent single-nucleotide variants (SNVs) have evidenced higher impacts (odds ratios, 2–57)13,14. The challenge now resides in applying these technologies to establish personalized diagnoses. The Psychiatric Genomic Consortium (PGC) in his latest published agenda aims to study large pedigrees to search for genetic variants of large effect15. Pedigrees from genetic isolates with high degrees of consanguinity are of special interest. Several large pedigrees have been recently investigated, either looking for CNVs16,17,18, rare SNVs19,20,21,22,23,24 or common variant contributions25. But very few have followed the PGC suggestions15, aimed to analyze those pedigrees using comprehensive genomic assays26,27.
In this study we have applied a systemic genomic approach to uncover the genomic architecture of a large lineage, with 41 individuals affected of MMD in the last three generations, 27 of which have been diagnosed with psychotic disorders. This family is from a region of southern Spain, the Alpujarras, known to be a hotspot for psychiatric diseases, with a prevalence of 7.8%28, almost double of that from the rest of the country, suggestive of being due to founding genetic events.
Results
Pedigree description
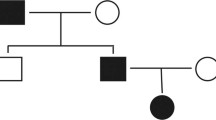
A large multigenerational family of Southern Spanish origin with high prevalence of mental disorder was recruited between the Psychiatry ward of the University Hospital Son Espases (HUSE) of the Balearic Islands and the Health Center of El Ejido. The full pedigree is shown in Fig. S1. Figure 1 shows the three subfamilies analyzed. Subjects 1–202 (subfamily 1), 4-211 (subfamily 2) and 3–208 (subfamily 3) are siblings. Out of the 41 individuals affected of MMD, 27 have been diagnosed with psychosis and 14 with a mental disease without psychosis. A clinical description of the family subjects is summarized in Table S1, showing the Global Assessment of Functioning (GAF) scale for all psychotic subjects studied and the Positive and Negative Syndrome Scale (PANSS) scores for all the schizophrenic patients analyzed (Table S1).

Pedigree structures of the three subfamilies analyzed. Subfamilies 1 (A), 3 (B) and 2 (C). DNA was available for all the subjects numbered from 1 till 35. Black indicates a diagnosis of psychosis, comprising SCZ, SCA and BD-I. Orange indicates a diagnosis of mental disorder without psychosis, comprising MDD and ADHD. Grey indicates undetermined diagnosis.
Phenotype definition
In order to perform the genomic analysis, two phenotypes were defined: the narrow phenotype was attributed only to patients with psychosis (n = 27), including: SCZ, (n = 17), schizoaffective disorder (SCA, n = 1), BD-I (n = 8) and acute psychotic episode F23 (n = 1). The wide phenotype of illness also included patients affected of mental disease, but who have not manifested any psychotic episode, as MDD (n = 14) and attention deficit hyperactive disorder (ADHD, n = 1). Within the narrow phenotype there were 10 females (24.3%) and 17 males (41.5%). By contrary, patients with a mental disease without psychosis included 12 females (29.3%) and only 3 males (7.3%). The mean age (± standard deviation) at participation was (30.5 ± 8) years for cases and (52 ± 6) years for controls.
Linkage analysis identified a locus at 9q33.1–33.2 associated with psychiatric disorders (wide phenotype)
The genome-wide results for nonparametric LOD (NPL) scores for the wide and narrow phenotypes are plotted in Fig. 2A and Table 1. A genomic region on chromosome 9 (113,117,183–124,200,417; 11 Mb) highlighted with significant LOD scores (LOD wide = 4.11; LOD narrow = 3.07) (Fig. 2). Moreover, eight other genomic regions identified in both phenotype analyses reached LOD scores above 1.5 for the wide phenotype suggestive of linkage and were considered for further analyses (Fig. 2A and Table 1). Within those regions it is worth to mention the one at chromosome 3 (169,411,792–183,303,037; 13.89 Mb) with a LOD narrow = 2.36, and LOD wide = 1.89 (Table 1). Once linkage analysis was performed only considering the narrow phenotype, there were no linkage regions that reached significance, although ten regions had suggestive LOD scores ≥ 1, highlighting two regions of chromosome 17 with suggestive LOD scores of 1.5 (Chr17: 51,166–6,296,217, 6.24 Mb and Chr17: 33,006,378–35,752,691, 2.74 Mb) (Table S2A). Regarding the linkage analysis considering only the wide phenotype, no significant regions were identified, although thirteen regions had LOD scores ≥ 1 (Table S2B). Two regions at chromosome 19 had suggestive LOD scores > 1.5 (Chr19: 301,639–3,030,118, 2.72 Mb and Chr19: 5,892,954–7,900,562, 7.3 Mb) (Table S2B).

Genome-Wide Linkage Analysis identified the 9q33.1–33.2 linkage region. (A) Genome-wide results for the NPL score analysis. In blue represented the LOD scores for the wide phenotype; in red the LOD scores for the narrow phenotype. (B) NPL score results for Chromosome 9. The − log10 (P value) of the family-based association test in regions with significant NPL scores are shown as dark green or light green dots for the wide and narrow phenotype, respectively. (C) Regional association plot for the 9q33.1–33.2 linked region. The dashed grey line represents the significance threshold for the associated SNPs. In red, genes previously associated with MMD.
To narrow down the linkage regions of chromosomes 9 and 3, family-based association analyses were performed. 114 SNPs were found to be nominally significant on chromosome 9. Four SNPs (rs117920810, rs10760030, rs1888737 and rs16908402), associated with the wide phenotype analyses, remained significant after adjusting for multiple testing (p-value = 0.042617) (Fig. 2C, Table 1). The associated SNPs map at the highly conserved ASTN2/BRINP1 locus at chr9q33.1–33.2, which contains five genes (ASTN2, BRINP1, TRIM32, TLR4 and C5) that have been previously associated with neurodevelopmental disorders (reviewed in29). To closely analyze the linkage and the association region, haplotype estimation was conducted using SHAPEIT430, phasing the entire chromosome 9 and carefully analyzing the linkage region (Figs. S2 and S3). Two different approaches were followed: we first used the SNP array genotyping to include all SNPs with MAF < 30% (Fig. S2). And second, haplotype phasing was also performed on those patients from whom we had whole genome sequencing data using SNPs with MAF < 0.5% (Fig. S3), searching for rare haplotypes that would segregate with the disease phenotypes. Only branch-specific haplotypes were identified. The subfamily 1 has three major haplotype blocks (H) shared by all affected subjects, but subject 1–2: H1 (chr9: 113,492,976–116,372,543, delimited by SNPs rs192009474–rs186260426, 2.87 Mb); H2 (chr9: 116,794,577–120,925,469, delimited by SNPs rs530450539–rs544077840, 4.1 Mb) and H3 (chr9: 121,694202–123,966,682, delimited by SNPs rs188485361–rs186909636, 2.2 Mb). It is important to emphasize that these three haplotypes are also shared by one healthy subject (1–21). The subfamily 3 has four haplotypes shared by wide affected subjects only: H1 (chr9: 116,857,705–117,615,594, delimited by SNPs rs34417627–rs142269627, 757 kb); H2 (chr9: 117,843,831–119,629,686, between SNPs rs944511–rs190965203, 1.78 Mb); H3 (chr9: 119,707,309–120,925,469, between SNPs rs118070509–rs544077840, 1.2 Mb); and H4 (chr9: 121,694,202–122,542,663, delimited by SNPs rs188485361–rs150164433, 848 kb), which is the haplotype located in the association region (Fig. S3).
On the other hand, on chromosome 3 a total number of 179 SNPs resulted to be significant, but none of them remained significant after correcting for multiple testing (Fig. S4). Family-based association analyses were also performed for all the suggestive linkage regions. Significant SNPs of common regions are summarized in Table 1, and those significant SNPs for the wide and narrow phenotype are summarized in Table S2.
Due to the high prevalence of psychiatric disorders in the pedigree, we hypothesized that related-affected subjects would share SNVs and structural variants (SVs) inherited from common ancestors within the linkage and the suggestive linkage regions identified.
Identification of different CNVs in psychiatric and neurodevelopmental-associated loci
We next search for SVs in the linkage regions, first performing clinical karyotyping of four individuals (subjects 1–18, 1–25, 3–30 and 3–31) to discard structural variants, as balanced translocations. All four patients had normal karyotypes (data not shown). Next, we performed SNP-array based copy-number variant (CNV) analysis. Nine CNVs were identified (Table S3), but none of them were in the linkage regions 9q33.1–33.2 and 3q26.32–26.33. Remarkably, two psychotic patients (mother 3–11 and her son 3–31) harbored a 450 Kb duplication in the 3q29-schizophrenia locus31,32. This duplication shares 74.5% overlap with the 3q29 duplication syndrome, which is characterized by delayed development (particularly speech delay) and intellectual disability or learning difficulties, although its manifestation varies widely (DECIPHER and31). Moreover, the same subjects (3–11 and 3–31) plus the brother (3–12) of 3–11 also harbored a 127 Kb duplication at 4q35.2, a genomic region also associated with behavioral disorders as autism and ADHD (DECIPHER). Another interesting CNV identified was a 198 Kb duplication at 22q11.23, right next to the major risk locus for SCZ33. Phenotypes associated with duplications of similar size comprise cognitive impairment, emotional/affect behavior, hyperactivity and intellectual disability (DECIPHER and34,35). The mother 1–1, affected of MDD, transmitted the DUP22q11.23 to her two psychosis-affected children (1–18 and 1–19). Three other MDD subjects (3–9, 3–10 and 4–15) and a healthy control (3–34) also harbor the DUP22q11.23. It is also worth mentioning the deletion DEL12q14.1, only identified in affected subjects, that encodes the leucine rich repeats and immunoglobulin likes domain 3 (LRIG3) gene. Siblings 1–24, 1–27 (MDD) and 1–25 (SCZ) inherited this deletion from their mother 1–6 (MDD). Phenotypes associated with similar deletions at 12q14.1 include intellectual disability and delayed speech and language development (DECIPHER and36).
SNVs and INDELs identified only in MMD subjects at 9q33.1–33.2
To search for rare (MAF < 0.01) coding SNVs and SVs below detection thresholds for SNP arrays, we conducted WGS (30× coverage) of 12 subjects: 8 affected of psychosis (1–2, 1–6, 1–18, 1–25, 3–11, 3–12, 3–30, 3–31), 2 MDDs (1–3, 3–13), and 2 healthy controls (1–21, 2–28). The genomic linkage region 9q33.1–33.2 and its surroundings were deeply analyzed. The coordinates used for variants identification were (chr9: 111,617,397–140,033,609). We first searched for rare SNVs with protein impact affecting conserved residues within 9q33.1–33.2. We did not identify any coding variant shared by all affected subjects within these coordinates. Six variants were identified in some affected subjects and were not present in any healthy control: two in ZNF618 gene (Zinc Finger Protein 618, rs762985449 and rs770522574), one in TNC (Tenascin C, rs61729478), one in CDK5RAP2 (CDK5 Regulatory Subunit Associated Protein 2, rs41296081), and two in C5 (Complement C5, rs139479771 and rs34552775) (Table 2). Many other rare intronic or intergenic variants were only identified in affected subjects in the linkage chromosome 9 region (Table 2). Some of these rare SNVs were branch-specific and defined the four rare-haplotype blocks identified in the linkage region of Subfamily 3 (Table S5). In the association region, four rare intergenic variants (rs191347609, rs181505483, rs191352043, and rs4837653) are located within the H4 haplotype block of Subfamily 3 at chr9: 121,694,202–122,542,663 (Fig. S2 and Table S5), and they were shared by all the wide-affected individuals of subfamily 3.
We next searched for small SVs that could not be detected by SNP-array, using different algorithms, HaplotypeCaller of GATK, CNVnator, Manta, BreakDancerMax and CREST. CNVnator identified 7 non-reported small INDELs in non-coding regions, only in MMD subjects, affecting the genes LPAR1, HSDL2, DELEC1, PAPPA, ASTN2 and ZNF618 (Table 2). Two of these INDELs, located at the H2 haplotype of subfamily 3 (Fig. S2 and Table S5), were shared by all the MMD subjects of the subfamily 3 and were not present in the healthy controls: a three base pair (AGG) deletion in an intronic region of DELEC1 gene (chr9: 118,113,219), predicted to affect a histone H3 lysine 4 trimethylation (H3K4me3) site in the brain frontal cortex37, and a two base pairs (TG) deletion at the long intergenic non-coding RNA 474 (LINC00474) (chr9: 118,667,077) (Table 2 and Table S5)). Moreover, CNVnator also identified a non-reported larger deletion of 3099 bp that overlapped with the expression of DELEC1 gene (Table 2). This deletion is only present in two psychotic siblings, 1–18 (SCZ) and 1–19 (BD-I). Manta identified four other non-reported deletions in intergenic regions at 9q33.1–33.2 (Table 2). All these INDELs were checked by PCR and Sanger sequencing and were not identified in any healthy family control.
The search for rare SNVs was extended to the rest of the genome. In Supplementary Table S4 are summarized the coding rare SNVs identified in susceptible linkage regions. Only the rs145032100 in the ARHGAP19 gene was shared by all affected subjects but was also carried by some healthy controls (Table S4A). This SNV is located at chr10q24.1, a suggestive region associated with the narrow phenotype (LOD score = 1.02).
Regions associated with the wide phenotype are enriched for genes involved in voltage-gated ion channels, microtubule organization and immune system
Functional enrichments were performed using GREAT38, searching for gene ontology (GO) terms associated with the significant SNPs of the linked region 9q33.1–33.2 plus the ones identified by both phenotype analyses (LOD > 1.5) (Table 1). The background used was composed of all the filtered SNPs, previously used to perform the association analysis. Regarding GO cellular component highlighted ontologies associated with voltage-gated ion channels and tubulin cytoskeleton (Fig. 3A). Interestingly, GO Biological Process terms were enriched for genes related to neuronal migration and differentiation and genes associated with the immune response (Fig. 3B). Mouse Genome Informatics (MGI) identified enriched expression in cerebral cortex (Fig. 3C), and within GO Disease Ontology, recurrent major depression appeared as the eight most significant enriched term (Fig. 3D).

MMD is enriched for genes associated with the immune system and the cytoskeleton of tubulin (A–D). Psychosis is enriched for genes involved in synaptic function (E–H). GO term enrichment analyses with GREAT38, including the significant SNPs of the suggestive linkage regions with LOD > 1.5 for the wide phenotype (A–D) and with LOD > 1 for the narrow phenotype (E–H). GO terms identified by: (A, E) Molecular Function; (B, F) Biological Process; (C, G) Cellular Component; and (D, H) Disease Ontology.
Regions associated with the narrow phenotype are enriched for genes involved in synaptic vesicle function
We also investigated whether the significant SNPs associated with the narrow phenotype from the suggestive narrow linkage regions (LOD > 1) (Table 1 and Table S2A) showed functional enrichments related to the disease etiology. Interestingly those regions appeared enriched for synaptic vesicle function, composition and transport (Fig. 3E–H).
Psychotic subjects have increased risk associated with common variants
We finally measured polygenic risk scores (PRS) to evaluate the contribution of common variants to the psychotic phenotype. PRS were calculated using GWAS data from9. We observed a clear gradient in the PRS results. All psychotic members of the pedigree scored positive PRS, either using SCZ as a base dataset to calculate the PRS or the combination of SCZ and BD (Table S6). By contrary, some subjects affected of MDD had negative PRS scores (subjects 1–24, 1–22, 4–15 and 1–20), suggestive of being protective. Interestingly, out of the 13 healthy controls analyzed, only one subject scored positive PRS (4–14, PRS = 0.36) (Table S6).
Discussion
The genetics paradigm of mental illness has changed substantially in recent years. Families with high prevalence, such as the one studied, are expected to encode variants of large effect. But since MMDs are polygenic, we obviously cannot search for a single cause of the disease and whole genome approaches need to be made.
In this pedigree we identified a susceptibility locus with a predominant involvement, the 9q33.1–33.2. Previous linkage analysis in families with mental disorders have reported the same region or very close coordinates, some of which could be considered partial linkage replications39,40,41. Badenhop et al. found suggestive evidence for linkage for 9q31–q33 when analyzing 13 families with high prevalence of BD-I39. Kaufmann et al. found suggestive evidence for linkage for 9q32–9q34 when analyzing 30 nuclear SCZ African–American families comprising 98 subjects (NPL Zmax = 2.17, p = 0.017)40. Interestingly, the highest evidence for linkage was found when considering individuals diagnosed with either BD I and II, SZA manic type, or depression as affected (NPL = 2.5 between D9S1690 and D9S1677 at 9q31–q33)40. Venken et al. found evidence of linkage at 9q31.1–q33 for affective disorder susceptibility analyzing nine multigenerational families from Northern Sweden41. Interestingly, some of these linkage reports have included depression and BD in their analyses; this is congruent with our report of linkage which is higher when including depressive subjects as affected (wide phenotype). Others have reported linkage peaks very close to the one identified in here42,43,44. Labbe et al. found suggestive evidence for linkage at 9q33 adopting a symptom dimension approach for delusional symptoms, in where most of the patients contributing to this signal were diagnosed as SCZ42. Liu et al. found suggestive evidence for linkage at 9q31 analyzing 373 individuals from 40 BD pedigrees43. Park et al. also found evidence for linkage at 9q31 analyzing psychotic BD in 40 extended pedigrees comprising 373 individuals44. Interestingly, the same genomic region 9q33.1–33.2 has also been associated with psychotic disorders through GWAS45.
The linked 9q33.1–33.2 region contain five candidate genes from the immune system that participate in synaptic processes and have been previously associated with neurodevelopmental disorders, ASTN2, BRINP1, C5, TLR4 and TRIM32. ASTN2 (Astrotactine 2) and BRINP1 (Bone Morphogenetic Protein/Retinoic Acid-inducible Neural specific Protein) encode proteins from the Membrane Attack Complex Perforin (MACPF) family, highly expressed in the developing brain (reviewed in29). Both genes have been associated with SCZ45,46, BD-I47, and other neurodevelopmental disorders48 and with structural abnormalities of the hippocampal volume49. ASTN2 facilitates glial-guided migration during brain development50, and regulates synaptic trafficking by modulating the composition of surface synaptic vesicle proteins51. BRINP1 function in neuronal development is less studied, although it has been implicated in neurogenesis52 and cell cycle regulation53. In fact, Brinp1 knock-out (KO) mice evidence altered hippocampal neurogenesis52 and exhibit altered behaviors that could model MMDs52,54.
C5, which encodes Complement 5 protein, is another interesting candidate gene in the linked region. Recent evidences has implicated the complement system as a promising immune mediator of SCZ (reviewed in55). GWAS studies have identified association of complement components as C4 and CSMD1 with SCZ8,56. In addition to the genetic findings, different studies have reported increased complement expression and overall activity in the plasma or serum of SCZ patients (reviewed in55). Recently, increased C5 levels have also been observed in cerebrospinal fluid of SCZ patients57.
TLR4 encodes the Toll-like Receptor 4, which plays a fundamental role in pathogen recognition and activation of innate immunity. TLRs express in the developing and adult CNS, in where have been involved in neurogenesis, axonal growth and structural plasticity (reviewed in58). Altered TLR4 counts have been observed in SCZ patients (reviewed in59), and interestingly antipsychotic treatment could normalize those counts60. Increased TLR4 expression has also reported in postmortem frontal cortex from SCZ patients and depressed suicide victims (reviewed in59). Other evidence supporting the role of TLR4 on psychiatric diseases come from animal models. TLR4 KO mice show improved spatial memory61, due to increased neuronal progenitor cell proliferation and neuronal differentiation in the hippocampus, suggesting that TLR4 may act to reduce hippocampal neurogenesis62.
The last candidate gene in the linked region is TRIM32, a small gene nested within an intron of ASTN2 and transcribed from the opposite strand. It encodes the Tripartite motif‐containing protein 32 (TRIM32). TRIM32 is a cell fate-determinant for a balanced embryonic development of the neocortex63 and the adult neurogenesis64. Recent reports have associated TRIM32 with psychiatric disorders, such as MDD, ASD, ADHD, anxiety and obsessive–compulsive disorder (reviewed in48). Interestingly, TRIM32 loss protects against the development of anxiety and depression induced by chronic stress65.
Although we did not find any coding variant in the linked region that segregates with all affected MMD subjects, we found several rare SNVs in those genes harbored only by MMD patients. Moreover, we cannot discard a regulatory role of the genomic region containing the two small INDELs identified at DELEC1 (deleted in esophageal cancer 1) gene and at LINC00474 in all affected subjects of subfamily 3. Further studies will have to shed light on the potential pathogenic roles of the INDELs and the rare SNVs identified in the linked region.
Furthermore, it is also interesting to highlight the only coding rare SNV identified in the family which segregates with all affected subjects, the g.99006061 G>A transition at ARHGAP19, associated with the psychotic phenotype. ARHGAP19 is another hematopoietic cell regulator, a specific Rho GTPase-activating protein (GAP) that plays an essential role in the division of T lymphocytes66.
Overall, our results reinforce the growing evidence linking immune system modulators with specific brain functions and MMDs. The susceptibility locus 9q33.1–33.2 should be taken into consideration in further genetic analysis, especially in those families that come from the same region.
Methods
Clinical assessments
Psychiatric assessments included semi-structured interviews, using the Spanish version of the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) Axis I (SCID-I)67, GAF68, PANSS69 and the Diagnostic Interview for Genetic Studies (DIGS)70.
Sample collection
A total of n = 34 subjects were recruited, DNA samples were obtained, being n = 9 psychotic patients, n = 11 non-psychotic mental disorder patients and n = 14 healthy controls. Genomic assays were done on n = 34 individuals, including SNP arrays (n = 34), WGS (n = 12), and karyotyping (n = 4).
Ethics
The experimental protocol was approved by the ethics committee of the Balearic Islands (CEI-IB) and was carried out in accordance with the ethical standards of the 2013 Declaration of Helsinki. All studied family members gave their written informed consent to take part in the study.
Genotype data
Genotyping and SNP array
Whole-genome genotype was generated for all samples in the Research Unit of Molecular Epidemiology, Institute of Epidemiology II, Helmholtz Zentrum München, German Research Center for Environmental Health using the Infinium Global Screening Array-24 v1.0 (GSA) from Illumina, which includes 642,824 SNPs. In addition, a pool of 57,254 SNPs (Multi-disease Drop-In Panel (MD)) previously related to neurological disorders was also genotyped. The genotype calling and CNV analysis were performed using the Genome Studio 2.0 (Illumina Inc. San Diego, California, USA).
Nonparametric linkage (NPL) analysis was carried using the NPL scoring function71, implemented in Merlin v1.1.272. Evidence for Linkage was assessed with the Kong and Cox exponential model73. Allele frequencies were calculated using the maximum likelihood method. Due to the complexity of the pedigree, it was split up in three different ~ 24 bit-sized sub-pedigrees (See Fig. 1). Before running linkage, data was exhaustively quality controlled. Graphical Representation of Relationship Errors (GRR)74 was used to identify errors in the structure of the pedigree. Whole Genome Association Analysis Toolset (PLINK 1.7)75 was used for the SNPs quality control. SNPs were excluded when Minor Allele Frequency (MAF) < 0.05, and if showing Mendelian inconsistencies. A total of 1198 Mendelian inconsistencies were found (0.17%). Unlikely double recombinants were analyzed using the “error detection” option from Merlin v1.1.2 and subsequently excluded using the “pedwipe” option. Linkage was carried using the most heterozygous SNPs per chromosome after being modeled for LD. Model for LD was performed calculating r2 using PLINK and removing one SNP of a pair each time r2 > 0.5. Out of the initial 700,008 SNPs genotyped, 8,078 SNPs were selected for the analysis.
Association analyses of suggestive linkage regions and haplotyping
Family-based association analyses were conducted using the Linkage and Association Modelling in Pedigrees Software (LAMP)76. We included all the SNPs from significant and suggestive linkage regions, using both definitions of the phenotype, wide and narrow. The p-values were corrected for multiple testing using the Benjamini–Hochberg correction. LAMP allows to accommodate different family structures.
Haplotype phase was estimated using SHAPEIT 4 (version 4.2)30 and haplotypes were visualized using inPHAP77 mapping SNPs shared by at least four affected subjects. To perform phasing two approaches were followed: (1) Genotyped SNPs from SNP array with low minor allele frequency (MAF ≤ 30%) were included. (2) Phasing was also performed using SNPs with MAF < 1% from VCF files of WGS. Allele frequencies were extracted from gnomAD. For both approaches, SNPs with high individual missingness rate (> 80%), and high genotyping missingness rate (> 80%) were excluded. SNPs that were not called in all the genotyped subjects were also excluded. Chromosomes 9 was entirely phased.
Whole genome sequencing (WGS)
12 samples were whole-genome sequenced (1–2, 1–3, 1–5, 1–18, 1–21, 1–25, 3–11, 3–12, 3–13, 3–30, 3–31, 2–28) using the BGISEQ-500 service (BGI Genomics Co., Ltd.). The workflow to obtain variant call format (VCF) files from raw data (FASTQ) provided by BGI was based on GATK Best Practices. FASTQ files, containing raw unmapped reads and Phred scores were quality controlled using FastQC tool. Low-quality sequences (phred score < 20) and adaptors were removed using cutadapt. QC sequences were aligned against the reference human genome (GRCh37/hg19) using BWA-MEM algorithm implemented in Burrows–Wheeler Alignment tool (BWA). Aligned data in SAM (Sequence Alignment/Map) format were then sorted and converted into BAM files using SAMtools. To generate new BAM files, PCR duplicates were removed using Picard Tools and realignment around INDELs and base recalibration was performed (BQRS) using Genome Analysis Toolkit (GATK). SNP and INDEL calling were carried from the cleaned BAM files using GATK producing unfiltered primary VCF files; which were then filtered using the variant call recalibration procedure (VSQR) to generate the definitive VCF files. VCF files were directly analyzed using ENLIS Genome Research V1.9 (Berkeley, CA, USA) which uses its own annotation pipeline. Shared variation among affected individuals was filtered for read depth > 10 and MAF < 0.01, using ENLIS Genome Research V1.9 (Berkeley, CA, USA). Alternatively, VCF files were annotated using SnepEff78 including the prediction of different protein impact and conservation algorithms and allele frequencies from 1000G (https://www.internationalgenome.org/) and gnomAD (gnomAD; https://gnomad.broadinstitute.org). The resulting txt files generated were analyzed for rare, shared variation among affected individuals using R.
CNV and SVs detection
CNVs were analyzed from WGS and from SNP array data.
CNVs and SVs detection from WGS data were performed taking advantage of the paired-end sequencing configuration of the samples, and using the following algorithms: (1) CNVnator79, a read-depth based algorithm which is useful for detecting large INDELs, insertions and deletions; (2) BreakDancerMax80, a paired-read based algorithm, that allows the detection of large SVs such as deletions, insertions, inversions, and intrachromosomal and interchromosomal translocations; (3) CREST81, an split-read based algorithm that also allows the detection of the same SVs as BreakDancerMax; (4) Manta82, which combines both split-read and read-pair methods and it is useful for detecting large SVs, medium-sized INDELs and large insertions; and (5) HaplotypeCaller of GATK (v3.3.0), which was used for small INDELs detection (< 50 bp). CNVnator was run in all WGS samples using standard settings and a bin size of 100 bp (optimized for 20–30× coverage). Manta was run as a joint diploid sample analysis. BreakDancerMax and CREST were used with default settings.
CNVs were also detected from SNP array data using GenomeStudio 2.0 (Illumina Inc. San Diego, California, USA), taking as a reference GRCh37/hg19. This algorithm is based on two parameters: the B allele frequency (BAF) and the Log R Ratio (LRR) which can be used to test the genotyping quality of the samples and to check the presence of CNVs across the genome. The BAF is a measure of allelic imbalance. In a normal well-genotyped sample, three genotypes are expected, homozygous AA, heterozygous AB, and homozygous BB. Once referred to the B allele, BAF is expected to have three discrete values, 0, 0.5, and 1 (representing AA, AB, and BB genotypes, respectively). R is defined as the sum of the probe intensities used to genotype the different markers. When it is normalized becomes the LRR which is a measure of relative intensity, the logarithm (base 2) of the observed value of R (observed probe intensity) divided by the expected value (expected probe intensity)83.
All variants identified using the different algorithms were checked on the bam files using the software IGV, designed to visualize genomic data. This allowed the detection of artifacts or variants called in low coverage regions. Non-previously reported INDELs located on the linkage chromosome 9q33.1–33.2 were validated in all the studied subjects of the family by PCR followed by electrophoresis and Sanger sequencing.
Analysis of rare SNP and CNV variants
SIFT (http://sift.jcvi.org/)84, Polyphen (http://genetics.bwh.harvard.edu/pph2/)85, VarSome (http://varsome.com)86 and UniProt (https://www.uniprot.org)87 were used to predict the levels of variant penetrance. DisGeNET (http://www.disgenet.org/web/DisGeNET/menu)88, VarElect (http://varelect.genecards.org/), and Schizophrenia Exome Sequencing Genebook89,90 were also used to characterize variations. DECIPHER (DatabasE of genomiC varIation and Phenotype in Humans using Ensembl Resources; https://www.deciphergenomics.org)91 and CNVxplorer (http://cnvxplorer.com)92 were used to study the pathogenicity and conservation of the identified CNVs. All genomic data for molecular variants in this study were compatible with Genome build GRCh37. Database of genomic variants (DGV; http://dgv.tcag.ca/dgv/app/home)93 and Integrative Genomics Viewer (IGV; http://software.broadinstitute.org/software/igv/)94 were used for SNP analysis.
Functional enrichment of biological pathways was investigated using the online tool GREAT (Genomic Regions Enrichment of Annotations Tool; http://great.stanford.edu/public/html/)38. The enrichment analyses were based on the comparison between significant SNPs associated with the phenotype and the rest of the SNPs of the SNP array.
Polygenic risk scores (PRS) were calculated for each family member (n = 34) using the PRSice-2 v2.1.11, with the publicly available PGC schizophrenia GWAS as a base dataset (33,426 SCZ cases, 54,065 controls), in addition to BD (20,129 BD cases, 21,524 controls). Before computing PRS, data was quality controlled for missingness per SNP and per subject (excluding sample with a rate of missingness higher than 10%), assigned sex inconsistencies, MAF < 0.05 in the dataset, deviances from Hardy–Weinberg equilibrium and Mendelian inconsistencies. After quality control, data from the target dataset was transformed to match the base dataset. This step is vital since any inconsistencies in the effective allele (A1) might have a profound impact on the results. PRS were calculated with default clumping settings and normalizing PRS scores.
Ethics approval
The present study was approved by the ethics committee of the Balearic Islands (CEI-IB), Spain.
Consent to participate
Written informed consent was obtained from all studied family members.
Data availability
The datasets generated for this study can be shared upon reasonable request and are publicly available in the European Genome-Phenome Archive (EGA) (https://ega-archive.org/studies/EGAS00001004592).
References
Gandal, M. J. et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697. https://doi.org/10.1126/science.aad6469 (2018).
Cross-Disorder Group of the Psychiatric Genomics, C. et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat. Genet. 45, 984–994. https://doi.org/10.1038/ng.2711 (2013).
Sullivan, P. F. & Geschwind, D. H. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell 177, 162–183. https://doi.org/10.1016/j.cell.2019.01.015 (2019).
Cardno, A. G. & Owen, M. J. Genetic relationships between schizophrenia, bipolar disorder, and schizoaffective disorder. Schizophr Bull 40, 504–515. https://doi.org/10.1093/schbul/sbu016 (2014).
Cross-Disorder Group of the Psychiatric Genomics, C. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. https://doi.org/10.1016/S0140-6736(12)62129-1 (2013).
Lichtenstein, P. et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet 373, 234–239. https://doi.org/10.1016/S0140-6736(09)60072-6 (2009).
Hilker, R. et al. Heritability of schizophrenia and schizophrenia spectrum based on the Nationwide Danish Twin Register. Biol. Psychiatry 83, 492–498. https://doi.org/10.1016/j.biopsych.2017.08.017 (2018).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. https://doi.org/10.1038/nature13595 (2014).
Bipolar, D. & Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address, d. r. v. e., Bipolar, D. & Schizophrenia Working Group of the Psychiatric Genomics, C. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173, 1705-1715.e1716. https://doi.org/10.1016/j.cell.2018.05.046 (2018).
Manolio, T. A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753. https://doi.org/10.1038/nature08494 (2009).
International Schizophrenia, C. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455, 237–241. https://doi.org/10.1038/nature07239 (2008).
Lee, S. H. et al. Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat. Genet 44, 247–250. https://doi.org/10.1038/ng.1108 (2012).
Rees, E. et al. Analysis of copy number variations at 15 schizophrenia-associated loci. Br. J. Psychiatry 204, 108–114. https://doi.org/10.1192/bjp.bp.113.131052 (2014).
Xu, B. et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 40, 880–885. https://doi.org/10.1038/ng.162 (2008).
Sullivan, P. F. et al. Psychiatric genomics: An update and an agenda. Am. J. Psychiatry 175, 15–27. https://doi.org/10.1176/appi.ajp.2017.17030283 (2018).
Khan, F. F. et al. Whole genome sequencing of 91 multiplex schizophrenia families reveals increased burden of rare, exonic copy number variation in schizophrenia probands and genetic heterogeneity. Schizophr. Res. 197, 337–345. https://doi.org/10.1016/j.schres.2018.02.034 (2018).
Georgieva, L. et al. De novo CNVs in bipolar affective disorder and schizophrenia. Hum. Mol. Genet. 23, 6677–6683. https://doi.org/10.1093/hmg/ddu379 (2014).
Van Den Bossche, M. J. et al. Identification of rare copy nuber variants in high burden schizophrenia families. Am. J. Med. Genet. B Neuropsychiatr. Genet. 162B, 273–282. https://doi.org/10.1002/ajmg.b.32146 (2013).
Steinberg, S. et al. Truncating mutations in RBM12 are associated with psychosis. Nat. Genet. 49, 1251–1254. https://doi.org/10.1038/ng.3894 (2017).
Maaser, A. et al. Exome sequencing in large, multiplex bipolar disorder families from Cuba. PLoS ONE 13, e0205895. https://doi.org/10.1371/journal.pone.0205895 (2018).
Goes, F. S. et al. Exome sequencing of familial bipolar disorder. JAMA Psychiat. 73, 590–597. https://doi.org/10.1001/jamapsychiatry.2016.0251 (2016).
Rao, A. R., Yourshaw, M., Christensen, B., Nelson, S. F. & Kerner, B. Rare deleterious mutations are associated with disease in bipolar disorder families. Mol. Psychiatry 22, 1009–1014. https://doi.org/10.1038/mp.2016.181 (2017).
Ganesh, S. et al. Exome sequencing in families with severe mental illness identifies novel and rare variants in genes implicated in Mendelian neuropsychiatric syndromes. Psychiatry Clin. Neurosci. 73, 11–19. https://doi.org/10.1111/pcn.12788 (2019).
Forstner, A. J. et al. Whole-exome sequencing of 81 individuals from 27 multiply affected bipolar disorder families. Transl. Psychiatry 10, 57. https://doi.org/10.1038/s41398-020-0732-y (2020).
Andlauer, T. F. M. et al. Bipolar multiplex families have an increased burden of common risk variants for psychiatric disorders. Mol. Psychiatry https://doi.org/10.1038/s41380-019-0558-2 (2019).
Toma, C. et al. An examination of multiple classes of rare variants in extended families with bipolar disorder. Transl. Psychiatry 8, 65. https://doi.org/10.1038/s41398-018-0113-y (2018).
Szatkiewicz, J. et al. The genomics of major psychiatric disorders in a large pedigree from Northern Sweden. Transl. Psychiatry 9, 60. https://doi.org/10.1038/s41398-019-0414-9 (2019).
Moreno-Kustner, B., Martin, C. & Pastor, L. Prevalence of psychotic disorders and its association with methodological issues. A systematic review and meta-analyses. PLoS ONE 13, e0195687. https://doi.org/10.1371/journal.pone.0195687 (2018).
Berkowicz, S. R., Giousoh, A. & Bird, P. I. Neurodevelopmental MACPFs: The vertebrate astrotactins and BRINPs. Semin. Cell Dev. Biol. 72, 171–181. https://doi.org/10.1016/j.semcdb.2017.05.005 (2017).
Delaneau, O., Zagury, J. F., Robinson, M. R., Marchini, J. L. & Dermitzakis, E. T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 10, 5436. https://doi.org/10.1038/s41467-019-13225-y (2019).
Goobie, S. et al. Molecular and clinical characterization of de novo and familial cases with microduplication 3q29: Guidelines for copy number variation case reporting. Cytogenet. Genome Res. 123, 65–78. https://doi.org/10.1159/000184693 (2008).
Murphy, M. M. et al. Study protocol for The Emory 3q29 Project: Evaluation of neurodevelopmental, psychiatric, and medical symptoms in 3q29 deletion syndrome. BMC Psychiatry 18, 183. https://doi.org/10.1186/s12888-018-1760-5 (2018).
Arinami, T. Analyses of the associations between the genes of 22q11 deletion syndrome and schizophrenia. J. Hum. Genet. 51, 1037–1045. https://doi.org/10.1007/s10038-006-0058-5 (2006).
Rodriguez-Santiago, B. et al. Association of common copy number variants at the glutathione S-transferase genes and rare novel genomic changes with schizophrenia. Mol. Psychiatry 15, 1023–1033. https://doi.org/10.1038/mp.2009.53 (2010).
Chang, J. et al. Pachygyria, seizures, hypotonia, and growth retardation in a patient with an atypical 1.33Mb inherited microduplication at 22q11.23. Gene 569, 46–50. https://doi.org/10.1016/j.gene.2015.04.090 (2015).
Choi, J. H. et al. Targeted knockout of a chemokine-like gene increases anxiety and fear responses. Proc. Natl. Acad. Sci. USA 115, E1041–E1050. https://doi.org/10.1073/pnas.1707663115 (2018).
Maunakea, A. K. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. https://doi.org/10.1038/nature09165 (2010).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. https://doi.org/10.1038/nbt.1630 (2010).
Badenhop, R. F. et al. A genome screen of 13 bipolar affective disorder pedigrees provides evidence for susceptibility loci on chromosome 3 as well as chromosomes 9, 13 and 19. Mol. Psychiatry 7, 851–859. https://doi.org/10.1038/sj.mp.4001114 (2002).
Kaufmann, C. A. et al. NIMH genetics initiative millenium schizophrenia consortium: Linkage analysis of African–American pedigrees. Am. J. Med. Genet. 81, 282–289 (1998).
Venken, T. et al. Genomewide scan for affective disorder susceptibility Loci in families of a northern Swedish isolated population. Am. J. Hum. Genet. 76, 237–248. https://doi.org/10.1086/427836 (2005).
Labbe, A. et al. Symptom dimensions as alternative phenotypes to address genetic heterogeneity in schizophrenia and bipolar disorder. Eur. J. Hum. Genet. 20, 1182–1188. https://doi.org/10.1038/ejhg.2012.67 (2012).
Liu, J. et al. Evidence for a putative bipolar disorder locus on 2p13-16 and other potential loci on 4q31, 7q34, 8q13, 9q31, 10q21-24, 13q32, 14q21 and 17q11-12. Mol. Psychiatry 8, 333–342. https://doi.org/10.1038/sj.mp.4001254 (2003).
Park, N. et al. Linkage analysis of psychosis in bipolar pedigrees suggests novel putative loci for bipolar disorder and shared susceptibility with schizophrenia. Mol. Psychiatry 9, 1091–1099. https://doi.org/10.1038/sj.mp.4001541 (2004).
Wang, K. S., Liu, X. F. & Aragam, N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 124, 192–199. https://doi.org/10.1016/j.schres.2010.09.002 (2010).
Vrijenhoek, T. et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 83, 504–510. https://doi.org/10.1016/j.ajhg.2008.09.011 (2008).
Stahl, E. A. et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803. https://doi.org/10.1038/s41588-019-0397-8 (2019).
Lionel, A. C. et al. Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum. Mol. Genet. 23, 2752–2768. https://doi.org/10.1093/hmg/ddt669 (2014).
Hibar, D. P. et al. Novel genetic loci associated with hippocampal volume. Nat. Commun. 8, 13624. https://doi.org/10.1038/ncomms13624 (2017).
Wilson, P. M., Fryer, R. H., Fang, Y. & Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J. Neurosci. 30, 8529–8540. https://doi.org/10.1523/JNEUROSCI.0032-10.2010 (2010).
Behesti, H. et al. ASTN2 modulates synaptic strength by trafficking and degradation of surface proteins. Proc. Natl. Acad. Sci. USA 115, E9717–E9726. https://doi.org/10.1073/pnas.1809382115 (2018).
Kobayashi, M. et al. Absence of BRINP1 in mice causes increase of hippocampal neurogenesis and behavioral alterations relevant to human psychiatric disorders. Mol. Brain 7, 12. https://doi.org/10.1186/1756-6606-7-12 (2014).
Kawano, H. et al. Identification and characterization of novel developmentally regulated neural-specific proteins, BRINP family. Brain Res. Mol. Brain Res. 125, 60–75. https://doi.org/10.1016/j.molbrainres.2004.04.001 (2004).
Berkowicz, S. R. et al. Brinp1(−/−) mice exhibit autism-like behaviour, altered memory, hyperactivity and increased parvalbumin-positive cortical interneuron density. Mol. Autism 7, 22. https://doi.org/10.1186/s13229-016-0079-7 (2016).
Woo, J. J., Pouget, J. G., Zai, C. C. & Kennedy, J. L. The complement system in schizophrenia: Where are we now and what’s next?. Mol. Psychiatry 25, 114–130. https://doi.org/10.1038/s41380-019-0479-0 (2020).
Schizophrenia Psychiatric Genome-Wide Association Study, C. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976. https://doi.org/10.1038/ng.940 (2011).
Ishii, T. et al. Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem. Biophys. Res. Commun. 497, 683–688. https://doi.org/10.1016/j.bbrc.2018.02.131 (2018).
Okun, E., Griffioen, K. J. & Mattson, M. P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 34, 269–281. https://doi.org/10.1016/j.tins.2011.02.005 (2011).
Garcia Bueno, B., Caso, J. R., Madrigal, J. L. & Leza, J. C. Innate immune receptor Toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci. Biobehav. Rev. 64, 134–147. https://doi.org/10.1016/j.neubiorev.2016.02.013 (2016).
Keri, S., Szabo, C. & Kelemen, O. Antipsychotics influence Toll-like receptor (TLR) expression and its relationship with cognitive functions in schizophrenia. Brain Behav. Immun. 62, 256–264. https://doi.org/10.1016/j.bbi.2016.12.011 (2017).
Potter, O. V., Giedraitis, M. E., Johnson, C. D., Cox, M. N. & Kohman, R. A. Young and aged TLR4 deficient mice show sex-dependent enhancements in spatial memory and alterations in interleukin-1 related genes. Brain Behav. Immun. 76, 37–47. https://doi.org/10.1016/j.bbi.2018.10.010 (2019).
Rolls, A. et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 9, 1081–1088. https://doi.org/10.1038/ncb1629 (2007).
Schwamborn, J. C., Berezikov, E. & Knoblich, J. A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136, 913–925. https://doi.org/10.1016/j.cell.2008.12.024 (2009).
Hillje, A. L. et al. TRIM32-dependent transcription in adult neural progenitor cells regulates neuronal differentiation. Cell Death Dis. 4, e976. https://doi.org/10.1038/cddis.2013.487 (2013).
Ruan, C. S. et al. Deletion of TRIM32 protects mice from anxiety- and depression-like behaviors under mild stress. Eur. J. Neurosci. 40, 2680–2690. https://doi.org/10.1111/ejn.12618 (2014).
David, M. D., Petit, D. & Bertoglio, J. The RhoGAP ARHGAP19 controls cytokinesis and chromosome segregation in T lymphocytes. J. Cell Sci. 127, 400–410. https://doi.org/10.1242/jcs.135079 (2014).
Grilo, C. M., Anez, L. M. & McGlashan, T. H. The Spanish-language version of the diagnostic interview for DSM-IV personality disorders: Development and initial psychometric evaluation of diagnoses and criteria. Compr. Psychiatry 44, 154–161. https://doi.org/10.1053/comp.2003.50006 (2003).
Pedersen, G., Urnes, O., Hummelen, B., Wilberg, T. & Kvarstein, E. H. Revised manual for the Global Assessment of Functioning scale. Eur. Psychiatry 51, 16–19. https://doi.org/10.1016/j.eurpsy.2017.12.028 (2018).
Kay, S. R., Fiszbein, A. & Opler, L. A. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr. Bull. 13, 261–276. https://doi.org/10.1093/schbul/13.2.261 (1987).
Roca, M. et al. Diagnostic Interview for Genetic Studies (DIGS): Inter-rater and test-retest reliability and validity in a Spanish population. Eur. Psychiatry 22, 44–48. https://doi.org/10.1016/j.eurpsy.2006.10.004 (2007).
Whittemore, A. S. & Halpern, J. A class of tests for linkage using affected pedigree members. Biometrics 50, 118–127 (1994).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101. https://doi.org/10.1038/ng786 (2002).
Kong, A. & Cox, N. J. Allele-sharing models: LOD scores and accurate linkage tests. Am. J. Hum. Genet. 61, 1179–1188. https://doi.org/10.1086/301592 (1997).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. GRR: Graphical representation of relationship errors. Bioinformatics 17, 742–743. https://doi.org/10.1093/bioinformatics/17.8.742 (2001).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. https://doi.org/10.1086/519795 (2007).
Li, M., Boehnke, M. & Abecasis, G. R. Joint modeling of linkage and association: Identifying SNPs responsible for a linkage signal. Am. J. Hum. Genet. 76, 934–949. https://doi.org/10.1086/430277 (2005).
Jager, G., Peltzer, A. & Nieselt, K. inPHAP: Interactive visualization of genotype and phased haplotype data. BMC Bioinform. 15, 200. https://doi.org/10.1186/1471-2105-15-200 (2014).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6, 80–92. https://doi.org/10.4161/fly.19695 (2012).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. https://doi.org/10.1101/gr.114876.110 (2011).
Chen, K. et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 6, 677–681. https://doi.org/10.1038/nmeth.1363 (2009).
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat. Methods 8, 652–654. https://doi.org/10.1038/nmeth.1628 (2011).
Chen, X. et al. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. https://doi.org/10.1093/bioinformatics/btv710 (2016).
Peiffer, D. A. et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 16, 1136–1148. https://doi.org/10.1101/gr.5402306 (2006).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081. https://doi.org/10.1038/nprot.2009.86 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. https://doi.org/10.1038/nmeth0410-248 (2010).
Kopanos, C. et al. VarSome: The human genomic variant search engine. Bioinformatics 35, 1978–1980. https://doi.org/10.1093/bioinformatics/bty897 (2019).
UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515. https://doi.org/10.1093/nar/gky1049 (2019).
Pinero, J. et al. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015, bav028. https://doi.org/10.1093/database/bav028 (2015).
Purcell, S. M. et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. https://doi.org/10.1038/nature12975 (2014).
Fromer, M. et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. https://doi.org/10.1038/nature12929 (2014).
Firth, H. V. et al. DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. Am. J. Hum. Genet. 84, 524–533. https://doi.org/10.1016/j.ajhg.2009.03.010 (2009).
Requena, F. et al. CNVxplorer: A web tool to assist clinical interpretation of CNVs in rare disease patients. medRxiv. https://doi.org/10.1101/2021.03.19.21253806 (2021).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992. https://doi.org/10.1093/nar/gkt958 (2014).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. https://doi.org/10.1038/nbt.1754 (2011).
Acknowledgements
We are extremely grateful to the family members that participated in this study. Thanks to Dr. Francisco J. Gázquez, psychiatrist at El Ejido, for providing valuable clinical information.
Funding
This work was supported by the Carlos III Institute of Health (ISCIII), Ministry of Economy and Competitiveness (Spain) (Grants PI15/00809 and PI18/00608), co-funded with European Union ERDF (European Regional Development Fund). JPF was funded by the FPU fellowship Granted by Ministry of Education, Culture and Sport (Spain) (FPU2014/03876). LRG, AMD and BBC were funded by the FPI fellowship granted by General Direction of Innovation and Research, Ministry of Innovation, Research and Tourism, Balearic Government (FPI/1800/2015, FPI/2059/2017, FPI/2111/2018), co-funded by European Union ERDF. KS and AF were funded by the Munich Center of Health Sciences (MC-Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. DHS is funded by ISCIII, Ministry of Economy and Competitiveness (Spain) (Grant PI1800847), co-funded with European Union ERDF. JHR is funded by a fellowship from IDISBA (Folium Program-INTRES Project).
Author information
Authors and Affiliations
Contributions
J.P.-F. wrote the paper and performed most of the analyses, including linkage, Whole Genome Sequencing and Copy Number Variant analyses; F.C. recruited the patients and performed their clinical characterization; L.R.-G., A.M.-D., B.B.C., B.O.-V. and J.L. performed DNA extraction, SNVs, CNVs and INDELs validations and segregation analysis by PCR and Sanger senquencing; K.S. and A.F. performed linkage analyses and family-based association analyses; J.L., G.O., D.H.-S. contributed to NGS data analyses and experimental design. J.H.-R. performed haplotype phasing. C.V.-B. conceived and designed the analysis, supervised the study, contributed to analyze the NGS data and gene ontology, and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pol-Fuster, J., Cañellas, F., Ruiz-Guerra, L. et al. The conserved ASTN2/BRINP1 locus at 9q33.1–33.2 is associated with major psychiatric disorders in a large pedigree from Southern Spain. Sci Rep 11, 14529 (2021). https://doi.org/10.1038/s41598-021-93555-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93555-4
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
