
Abstract
Resistance to drugs targeting the androgen receptor (AR) signaling axis remains an important challenge in the treatment of prostate cancer patients. Activation of alternative growth pathways is one mechanism used by cancer cells to proliferate despite treatment, conferring drug resistance. Through a kinome-centered CRISPR-Cas9 screen in CWR-R1 prostate cancer cells, we identified activated BRAF signaling as a determinant for enzalutamide resistance. Combined pharmaceutical targeting of AR and MAPK signaling resulted in strong synergistic inhibition of cell proliferation. The association between BRAF activation and enzalutamide resistance was confirmed in two metastatic prostate cancer patients harboring activating mutations in the BRAF gene, as both patients were unresponsive to enzalutamide. Our findings suggest that co-targeting of the MAPK and AR pathways may be effective in patients with an activated MAPK pathway, particularly in patients harboring oncogenic BRAF mutations. These results warrant further investigation of the response to AR inhibitors in BRAF-mutated prostate tumors in clinical settings.
Similar content being viewed by others


Introduction
Prostate cancer is the second most common cancer diagnosed in men, accounting for over 350,000 cancer-related deaths worldwide each year1. The androgen receptor (AR) pathway is a key driver in prostate tumorigenesis, regulating genes that drive prostate cancer cell proliferation2. In recent years, new compounds have been introduced clinically that target the AR signaling axis resulting in tumor regression. These include drugs such as abiraterone, which blocks biosynthesis of androgen precursor molecules, and enzalutamide, which functions through antagonistic binding of AR. Even though these AR-directed drugs have shown to be clinically effective3,4, evasion of AR blockade through adaptation inevitably leads to disease progression and eventually death5,6,7. Acquired resistance to enzalutamide has been the focus of intense research, and several mechanisms have been described. These resistance mechanisms include activation of other signaling pathways such as the PI3K pathway8, NF-κB signaling9 and glucocorticoid receptor (GR) overexpression10,11.
Primary resistance is commonly defined by unresponsiveness to treatment, characterized by clinical progression within the first 3 months after commencing systemic therapy5. Primary resistance to enzalutamide, even though relatively under-examined, occurs in about 10% to 20% of prostate cancer patients3,12. A better mechanistic understanding of primary resistance will allow for better patient stratification and improved therapeutic avenues.
Functional genetic screens using CRISPR-Cas9 are a powerful tool for the unbiased identification of genes that have a central role in a wide range of biological processes in various genetic and pharmacological backgrounds, including cancer11,13,14. For example, through a genome-wide CRISPR-Cas9 screen, TLE3 was identified as a novel modulator of enzalutamide sensitivity which, together with AR, regulates GR expression and drug response in prostate cancer11. Using a similar approach, we set out to identify kinases whose inhibition could potentiate enzalutamide efficacy in prostate cancer cells, with the aim to discover biomarkers for resistance and potential drug combinations that are able to overcome enzalutamide resistance. We found that inhibition of BRAF, or downstream MAPK components MEK and ERK, enhanced enzalutamide sensitivity in prostate cancer cells harboring a mutation in the activating kinase domain of the BRAF gene. Our findings suggest therapeutic potential for co-inhibition of the MAPK and AR pathways in BRAF-mutated prostate cancers.
Results
A kinome-centered dropout screen identifies BRAF as a modulator of enzalutamide sensitivity
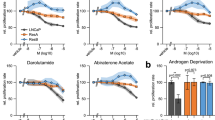
The AR inhibitor enzalutamide is successfully used for the treatment of prostate cancer. However, primary resistance is observed in a significant proportion of patients3,12, illustrating the need for improved therapeutic approaches for this subset of patients. To address this unmet clinical need, we performed a kinome-centered CRISPR-Cas9 screen to identify kinases whose inhibition synergize with AR inhibition in cells that show a poor response to enzalutamide. The cell line CWR-R1 is a prostate cancer cell line that shows moderate sensitivity to the AR inhibitor enzalutamide, as compared to the sensitive LNCaP cells and resistant 22rv1 cells (Fig. 1A,B). This moderate sensitivity makes the CWR-R1 cell line a suitable model system to screen for kinases whose inhibition may synergize with enzalutamide to enhance anti-tumor effects in vitro.

A kinome-centered CRISPR-Cas9 screen identifies BRAF as a modulator of enzalutamide sensitivity in CWR-R1 cells. (A). Enzalutamide sensitivity of prostate cancer cell lines LNCaP, CWR-R1 and 22rv1 in a long-term growth assay. (B) Quantified data of the results shown in (A). (C) Schematic representation of the kinome-centered CRISPR-Cas9 screen. (D) Representation of the relative abundance of the sgRNA barcode sequences of the screen. The y-axis shows the enrichment (relative abundance of enzalutamide treated/untreated) and the x-axis shows the average sequence reads of the untreated samples. (E) Long-term growth assay showing the enzalutamide response of CWR-R1 cells harboring sgRNAs targeting BRAF. Cells harboring a non-targeting sgRNA were used as a control. (F) Quantified growth data of the results shown in (E). (G) Western blot showing the protein expression levels of BRAF in indicated cell lines which were used in the assays shown in (E,F). Original western blots are presented in Supplemental Fig. S1D. For the bar graphs in (B,F), showing the quantified data of the growth assays, the bars represent the average data from at least three independent experiments ± SEM. p-values are indicated with ***p < 0.001, **p < 0.01 and *p < 0.05 (two-tailed t-test).
CWR-R1 cells were infected with the NKI Human Kinome CRISPR pooled sgRNA library targeting 578 human kinases. Infected cells were seeded at low density and treated with 10 μM enzalutamide or vehicle for 2 weeks to allow selection. The single gRNA (sgRNA) cassettes were recovered from the genomic DNA by PCR and their relative abundance was determined through massively parallel sequencing (Fig. 1C). The sgRNA abundance of the enzalutamide-treated and vehicle-treated populations were compared, and depleted sgRNAs were identified using DESeq215 and MAGeCK16 analyses (Supplemental Tables S1, S2). We found that all 10 sgRNAs targeting BRAF were under-represented in enzalutamide-treated cells when compared to vehicle-treated cells (Fig. 1D).
We validated the results of the CRISPR-Cas9 screen using sgRNAs targeting BRAF in CWR-R1 cells, using polyclonal knockout cell populations. Knockout of BRAF using two independent sgRNAs (sgBRAF-9 and sgBRAF10) yielded viable cells with growth kinetics mirroring those of control cells harboring a non-targeting sgRNA (sgNT) (Fig. 1E,F). CRISPR-mediated loss of BRAF protein expression in BRAFKO cells was confirmed by western blot (Fig. 1G). Importantly, BRAFKO cells showed increased sensitivity to enzalutamide in long-term growth assays when compared to control cells (Fig. 1E,F), concordant with the results from the screen. In contrast to our findings in CWR-R1 cells, knockout of BRAF using CRISPR-Cas9 in LNCaP cells did not result in increased sensitivity to enzalutamide (Supplemental Fig. S1A–C).
MAPK inhibition potentiates enzalutamide sensitivity in AR-driven prostate cancer cells harboring a BRAF mutation
Given the increased sensitivity to enzalutamide upon knockout of BRAF, we sequenced the BRAF kinase domain in CWR-R1 cells using a clinically validated, NGS-based, targeted sequencing assay. Through this approach, we identified a p.L597R mutation in the activating kinase domain of the BRAF gene. Sequencing of LNCaP cells using the same assay, revealed no BRAF alterations, consistent with previous reports for this cell line2,17.
Next, we assessed whether the increased sensitivity of BRAFKO CWR-R1 cells to enzalutamide could be confirmed by pharmacological inhibition of the MAPK pathway in combination with enzalutamide. Short-term and long-term growth assays showed that CWR-R1 cells were unresponsive to the RAF inhibitor LY3009120 (Fig. 2A–C). However, the combination of LY3009120 and enzalutamide resulted in strong inhibition of cell proliferation when compared to monotherapy treatment using these two inhibitors (Fig. 2A–C). The combination of enzalutamide and dabrafenib was also tested and found to be more effective than single drug treatments (Supplemental Fig. S2A,B), but to a lesser extent when compared to the combination of enzalutamide and LY3009120. These findings are consistent with the reduced efficacy of BRAF V600E-targeting drugs, such as dabrafenib, in non-V600E BRAF mutant cancer cells18.

Pharmacological validation of screen hit BRAF in CWR-R1 prostate cancer cells. (A) Short-term growth assay for CWR-R1 cells treated with enzalutamide and MAPK pathway inhibitors. The percentage of growth relative to the untreated control is shown, with the standard error of the mean (SEM) for n = 3 experimental replicates. (B,C) Long-term growth assay of CWR-R1 cells treated with AR inhibitor enzalutamide and RAF inhibitor LY3009120 as monotherapy or in combination. (D,E) Long-term growth assay of CWR-R1 cells cultured in the presence of enzalutamide and MEK inhibitor trametinib as indicated. (F,G) Long-term growth assay of CWR-R1 cells treated with enzalutamide in the presence of ERK inhibitor sch772984 at indicated concentrations. (H) Western blot showing the expression levels and phosphorylation status of MAPK pathway components in CWR-R1 cells treated with inhibitors as indicated. Vinculin was used as a loading control. Original western blots are shown in Supplemental Fig. S6A. For the bar graphs showing the quantified data of the growth assays, the bars represent the average data from at least three independent experiments ± SEM. p-values are indicated with ***p < 0.001, **p < 0.01 and *p < 0.05 (two-tailed t-test).
Pharmacological inhibition of the MAPK pathway downstream of BRAF was performed using trametinib, a MEK inhibitor, and sch772984, an ERK inhibitor. Trametinib monotherapy did not affect cell proliferation in short-term and long-term growth assays with concentrations up to 500 nM. However, when combined with enzalutamide, a strong inhibitory effect on growth was observed (Fig. 2A,D,E). Similar results were obtained with the ERK inhibitor sch772984, which showed a strong synergistic effect only when used in combination with enzalutamide (Fig. 2A,F,G). Growth assays using low concentrations of enzalutamide showed that co-treatment with MAPK inhibitors enhanced enzalutamide efficacy, making the drug effective in the nanomolar (nM) range, though the effect was strongest at 0.5 μM (Supplemental Fig. S2C–H).
Although LNCaP cells do not contain an activating BRAF mutation, other oncogenic alterations may cause MAPK pathway activation in these cells. However, inhibition of AR in combination with MEK or ERK inhibitors in this cell line did not result in increased sensitivity to enzalutamide (Supplemental Fig. S3A–D). These result are concordant with the absence of activating MAPK alterations2,17. LNCaP cells are PTEN-deficient and were shown to be more dependent on PI3K signaling upon enzalutamide-mediated AR inhibition, through reciprocal feedback regulation of the AR and PI3K pathway. It was shown that co-targeting of the PI3K and AR pathway in LNCaP cells resulted in strong anti-tumor effects when compared to single drug treatment8.
We also tested the combination of AR and RAF/MAPK inhibitors in two enzalutamide-resistant cell lines: AR-negative PC3 cells and the 22rv1 cell line. In 22rv1 cells, enzalutamide resistance was shown to be mediated by the AR-V7 splice variant19. Both cell lines did not respond to enzalutamide, as expected. Addition of inhibitors targeting MAPK pathway components RAF, MEK and ERK did not affect the enzalutamide response of PC3 cells (Supplemental Fig. S4A–C), excluding off-target effects of the used inhibitors. We found that 22rv1 cells showed modest sensitivity to MAPK pathway inhibitors (Supplemental Fig. S5A–C). MAPK inhibitor treatment had no effect on the enzalutamide response (Supplemental Fig. S5A–C), and knockout of BRAF did not confer enzalutamide sensitivity in these cells (Supplemental Fig. S5D–F). The lack of synergy between AR and MAPK inhibitors is most likely caused by the fact that enzalutamide is incapable of targeting the AR-V7 splice variant that drives resistance in 22rv1 cells, as AR-V7 lacks the ligand-binding domain.
Next, we biochemically investigated MAPK pathway activation in response to AR and MAPK inhibition as monotherapy and when used in combination in CWR-R1 cells. We found that upon enzalutamide treatment, MAPK signaling is upregulated, as shown by increased p-ERK and p-RSK levels (Fig. 2H)20. Inhibition of the MAPK pathway by LY3009120 or trametinib significantly downregulated p-ERK and p-RSK levels. When cells were treated with enzalutamide in addition to LY3009120, MAPK signaling was slightly increased, but still significantly lower when compared to enzalutamide monotherapy (Fig. 2H). This upregulation as a result of enzalutamide treatment was not observed when cells were treated with the combination of enzalutamide and trametinib (Fig. 2H). Together, these data suggest that MAPK inhibition in combination with enzalutamide may be effective in AR-driven prostate cancer cells having an activated MAPK pathway, through an activating BRAF mutation.
Clinical response to enzalutamide in BRAF-mutant CRPC patients
Although infrequent, prostate cancers harbor BRAF mutations in around 2% of cases (Supplemental Fig. S7A)21,22, mostly involving hotspot mutations p.K601E and p.G469A (Supplemental Fig. S7B). To explore the role of BRAF mutations in primary resistance to enzalutamide in CRPC patients, we analyzed sequencing data from a biopsy study (CPCT-02) at our center to identify BRAF-mutated patients. Biopsies were collected prior to enzalutamide or abiraterone treatment, and sequenced by either exome or whole genome sequencing23. We identified two patients harboring a BRAF p.K601E mutation. Both of these patients showed early clinical progression after commencing enzalutamide treatment (Fig. 3A,B). Out of the 30 similarly treated patients in the cohort, having clinical and PSA data available, 77% (n = 23) showed a decline of ≥ 50% in PSA levels in the first 3 months of enzalutamide or abiraterone treatment. After 6 months of treatment, 83% (n = 25) of patients had a lower PSA level compared to baseline (Supplemental Fig. S7C). These numbers are concordant with previously reported rates of primary resistance in enzalutamide-treated patients3,12. Our findings suggest that activating mutations in BRAF are associated with primary resistance to enzalutamide, though confirmation in a larger cohort of BRAF-mutated prostate cancer patients is needed.

Enzalutamide response in two patients with BRAF mutant tumors. (A) Computed tomography (CT) imaging for two patients with tumors harboring the BRAF p.K601E mutation prior to starting treatment (left) and 3 months on treatment (right). The yellow arrow indicates a mediastinal lymph node in CRPC-1 and liver metastases in CRPC-2. (B) On-treatment PSA levels for the two patients shown in (A).
Together, we find that co-inhibition of the AR and MAPK pathway activity is synergistic in prostate cancer cells carrying a BRAF mutation. Furthermore, the poor clinical response to enzalutamide in two CRPC patients harboring BRAF mutations indicates that oncogenic mutations in the kinase domain of BRAF may result in primary resistance to therapy, which could be addressed by co-treatment with MAPK inhibitors. These findings warrant further investigation of BRAF alterations in the context of the enzalutamide response in larger cohorts, to further elucidate the clinical relevance of activating BRAF mutations in AR inhibitor-treated patients.
Discussion
AR antagonists, such as enzalutamide, are effective in the treatment of AR-driven prostate cancer3. Still, resistance to AR inhibitors commonly arises during therapy, and primary resistance occurs in 10–20% of patients3,12. To improve treatment outcome, more insight into the primary resistance mechanisms is essential and may lead to the development of new treatment avenues.
In our study, we employed a kinome-centered CRISPR-Cas9 screen to identify genes that can be targeted to improve sensitivity to AR inhibition. We found that genetic knockout of the BRAF gene resulted in increased sensitivity to enzalutamide in CWR-R1 cells. These findings were confirmed through pharmacological inhibition of BRAF, or downstream components of the MAPK pathway. Through genetic profiling of CWR-R1 cells, the BRAF p.L597R mutation was identified, potentially conferring specific vulnerability to BRAF inhibition in CWR-R1 cells. The clinical significance of BRAF p.L597R has been shown in melanoma patients, where expression of this mutant was associated with sensitivity to MEK inhibitors24,25. Moreover, knockdown experiments comparing WT BRAF to several mutant forms of BRAF, including p.L597R, demonstrated the oncogenic function of this BRAF mutant in non-small cell lung cancer (NSCLC) cells26. In CWR-R1 cells, increased BRAF activity as a result of this mutation may be responsible for the moderate sensitivity of these cells to enzalutamide, and their sensitivity to MAPK inhibition in combination with AR blockade. The fact that no mutations affecting BRAF are present in LNCaP2,17, may explain the lack of synergy of combined AR/MAPK inhibition in this cell line.
Driver mutations in BRAF are found in a variety of cancers and are characterized by activating hotspot mutations in the kinase domain of the gene, most notably the p.V600E mutation27,28. Cancers harboring BRAF mutations are often sensitive to BRAF inhibitors, such as dabrafenib or vemurafenib28. Sensitivity can be increased by combination with MEK inhibitors, such as trametinib29. BRAF mutations in prostate cancer are rare but do occur in around 2% of patients21,22, predominantly involving hotspot mutations in the activating kinase domain (p.K601E and p.G469A; Supplemental Fig. S7A,B)21,22,30. In our study, we describe two mCRPC patients with tumors harboring a p.K601E BRAF mutation with early disease progression after commencing enzalutamide treatment, indicating potential relevance of these mutations in driving prostate cancer cell growth and enzalutamide resistance. Validation of these findings in larger cohorts is needed to confirm whether presence of these mutations correlates with primary resistance in patients. Our findings suggest that co-inhibition of AR and BRAF in BRAF-mutant prostate cancer patients could be particularly effective.
Alterations in the MAPK pathway are observed in about 40% of primary and 90% of metastatic prostate cancer cases2. Amplification of MAPK components is frequent, while mutations in members of this pathway are less common in prostate cancer2,31. Whereas the clinical significance of BRAF mutations has been demonstrated in various disease settings, it is unclear what proportion of the MAPK alterations found in prostate cancer lead to meaningful activation of the MAPK pathway. Emerging evidence suggests that targeting the MAPK pathway may represent a viable treatment approach for advanced prostate cancer cells fully resistant to enzalutamide20,31.
In conclusion, the findings by our group and others warrant further investigation of combined inhibition of the MAPK and AR pathway at an early stage of systemic treatment of AR-driven prostate cancer to overcome primary or acquired resistance. However, investigation of the clinical progression and longitudinal biochemical responses of enzalutamide-treated patients in larger cohorts is needed to further validate our findings in a clinical setting. The increase in availability of genetic profiling for cancer mutations in clinical settings32 may aid further exploration of the potential for combined BRAF/AR inhibition in BRAF-mutant prostate cancer.
Materials and methods
Cell culture and generation of knockout cells
The human prostate cancer cell lines LNCaP, CWR-R1, 22rv1 and PC3 were a kind gift from Prof. W. Zwart (Netherlands Cancer Institute). All prostate cancer cell lines were maintained in RPMI. HEK293T cells were obtained from ATCC and were cultured in DMEM. Medium was supplemented with 10% FBS (Serana) and 1% penicillin/streptomycin. Cells were maintained at 37 °C in 5% CO2. All cell lines were STR profiled. Control and BRAFKO cells were created by infecting target cells with lentiviral particles containing LentiCRISPR v2.0 harboring non-targeting or BRAF-targeting gRNAs, which were cloned into the vector using Gibson Assembly (NEB cat#: E2611S) utilizing BsmBI restriction sites. For gRNA sequences see Supplemental Table S3. For virus production, HEK293T cells were co-transfected with lentiviral CRISPR constructs, using PEI. Target cells were seeded 1 day prior to infection. Lentiviral supernatant was added to the medium along with 5 μg/ml polybrene. Infected cells were selected with 2 μg/ml puromycin.
Kinome-centered CRISPR-Cas9 dropout screen
CWR-R1 cells were infected with lentiviral particles containing the NKI Human Kinome CRISPR Knockout library at low M.O.I. (~ 0.2) for single viral integration, at a ~ 500-fold coverage, and cultured in the presence of vehicle or 10 μM enzalutamide for ~ 2 weeks. Barcodes were recovered and sequenced as described11. For sequence depth normalization a relative total size factor was calculated for each sample, by dividing the total counts of each sample by the geometric mean of all totals. After normalization, a differential test between the treated and untreated condition for each sgRNA was performed using DESeq215. The output from the DESeq2 analysis contains the DESeq2 test statistic. Positive DESeq2 test statistic indicate positive log2FoldChange value, negative DESeq2 test statistic indicate negative log2FoldChange value. We sorted the output of DESeq2 on the test statistic in increasing order, putting the most significant depleted sgRNA at the top. We then used the MAGeCK16 Robust Rank Algorithm to determine for each gene if its sgRNAs are enriched towards the top of the result list. The resulting enrichment p-values were corrected for multiple testing using the Benjamini–Hochberg correction, resulting in a FDR value. As hits we considered the genes with a FDR rounded on two decimals ≤ 0.1.
Proliferation assays
Colony formation assays were performed as previously described11. Enzalutamide, LY3009120, Dabrafenib, Trametinib, sch772984 were obtained from Medkoo Biosciences, all drugs were dissolved in DMSO and stored at − 20 °C. Used seeding densities were 20,000 (LNCaP) or 10,000 (22rv1, CWR-R1) cells/well in 6-well plates, and drugs were added as indicated the next day. For 12-well assays, the used seeding densities were 10,000 (LNCaP), or 5000 (CWR-R1, 22rv1, PC3). The growth medium, containing vehicle or drugs, was refreshed every 36–48 h. After 12–14 days of growth in presence of the drugs, when the control cells reached ~ 90% confluency, all cells were fixed in 2% formaldehyde and stained with 0.1% crystal violet.
For quantification of the growth assays, crystal violet was extracted by incubating the stained plates with 5% acetic acid for 1 h at room temperature. The solution, containing the crystal violet, was transferred to a 96-well plate and measured using the Envision 2104 Multilabel Reader (PerkinElmer). Growth assays were performed at least three times for each experiment. Therefore, when quantified data is shown, bars represent the average data from at least three independent experiments ± SEM. P-values are indicated with ***p < 0.001, **p < 0.01 and *p < 0.05 (two-tailed t-test).
Protein lysate preparation and western blot
Typically, CWR-R1 cells were plated at a density of 200,000 cells per well in 6-well plates and cultured in the presence of drugs as indicated for 5 days before harvesting. Samples were prepared and western blot was performed as described previously11, using the spectra Multicolor Broad Range Protein Ladder (ThermoFisher Scientific). Antibodies directed against BRAF (14814), GAPDH (5174), t-ERK (9102), p-ERK (4377), t-RSK (8408) were purchased from Cell Signaling Technology; antibody against p-RSK (04-419) was purchased from Millipore; antibody targeting Vinculin (V9131) was purchased from Sigma.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68(6), 394–424 (2018).
Taylor, B. S. et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18(1), 11–22 (2010).
Beer, T. M. et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 371(5), 424–433 (2014).
Ryan, C. J. et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 368(2), 138–148 (2013).
Buttigliero, C. et al. Understanding the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 41(10), 884–892 (2015).
Prekovic, S. et al. Molecular underpinings of enzalutamide resistance. Endocr. Relat. Cancer 25(11), R545–R557 (2018).
Bungaro, M., Buttigliero, C. & Tucci, M. Overcoming the mechanisms of primary and acquired resistance to new generation hormonal therapies in advanced prostate cancer: Focus on androgen receptor independent pathways. Cancer Drug Resist. https://doi.org/10.20517/cdr.2020.42 (2020).
Carver, B. S. et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19(5), 575–586 (2011).
Nadiminity, N. et al. NF-κB/p52 induces resistance to enzalutamide in prostate cancer: Role of androgen receptor and its variants. Mol. Cancer Ther. 12(8), 1629–1637 (2013).
Arora, V. K. et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 155(6), 1309–1322 (2013).
Palit, S. A. L. et al. TLE3 loss confers AR inhibitor resistance by facilitating GR-mediated human prostate cancer cell growth. Elife 8, e47430. https://doi.org/10.7554/eLife.47430 (2019).
Scher, H. I. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 367(13), 1187–1197 (2012).
Prahallad, A. et al. Unresponsiveness of colon cancer to BRAF (V600E) inhibition through feedback activation of EGFR. Nature 483(7387), 100–103 (2012).
Evers, B. et al. CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat. Biotechnol. 34(6), 631–633 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 550 (2014).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scare CRISPR/Cas9 knockout screens. Genome Biol. 15(12), 554 (2014).
Tate, J. G. et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47(D1), D941–D947 (2019).
Yao, Z. et al. BRAF mutants evade ERK-dependent feedback by different mechanisms that determine their sensitivity to pharmacological inhibition. Cancer Cell 28(3), 370–383 (2015).
Li, Y. et al. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res. 73(2), 483 (2013).
Li, S. et al. Activation of MAPK signaling by CXCR7 leads to enzalutamide resistance in prostate cancer. Cancer Res. 79(10), 2580–2592 (2019).
Cerami, E. et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2(5), 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioportal. Sci. Signal 6(269), pl1 (2013).
Van Dessel, L. F. et al. The genomic landscape of metastatic castration-resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat. Commun. 10(1), 5251 (2019).
Dahlman, K. B. et al. BRAF (L597) mutations in melanoma are associated with sensitivity to MEK inhibitors. Cancer Discov. 2(9), 791–797 (2012).
Bahadoran, P. et al. Major clinical response to BRAF inhibitor in a patient with a BRAF L597R-mutated melanoma. J. Clin. Oncol. 31(19), e324–e326 (2013).
Okimoto, R. A. et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 13456–13461 (2016).
Davies, H. et al. Mutations of the BRAF gene in human cancer. Nature 417(6892), 949–954 (2002).
Zaman, A., Wu, W. & Bivona, T. G. Targeting oncogenic BRAF: Past, present, and future. Cancers (Basel) 11(8), 1197 (2019).
Flaherty, K. T. et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367(18), 1694–1703 (2012).
Quigley, D. A. et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174(3), 758–769 (2018).
Nickols, N. G. et al. MEK-ERK signaling is a therapeutic target in metastatic castration resistant prostate cancer. Prostate Cancer Prostatic Dis. 22(4), 531–538 (2019).
Malone, E. R., Oliva, M., Sabatini, P. J. B., Stickley, T. L. & Siu, L. L. Molecular profiling for precision cancer therapies. Genome Med. 12(1), 8 (2020).
Acknowledgements
This work was funded by a KWF-Alpe d’HuZes Grant (NKI 2014–7080). W Zwart is supported by a KWF-Alpe d’HuZes Grant (10084). RB is supported by a KWF national infrastructure grant ScreeninC. The authors thank the NKI Genomics Core Facility for bioinformatics support. This publication and the underlying study have been made possible partly on the basis of the data that Hartwig Medical Foundation and the Center of Personalized Cancer Treatment (CPCT) have made available to the study.
Author information
Authors and Affiliations
Contributions
M.v.H. supervised the project and acquired funding. S.P. and M.v.H. conceptualized the project, methodology, interpreted the results and edited the manuscript. S.P. designed the experiments, wrote the initial draft and edited the manuscript. S.P. and J.v.D. carried out the experiments, and J.v.D. edited the manuscript. R.B., D.V. and C.L. performed computational analyses, interpreted the data and provided bioinformatics support. R.B., A.M.B., S.L. and W.Z. provided critical feedback on the results throughout the project and edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Palit, S.A.L., van Dorp, J., Vis, D. et al. A kinome-centered CRISPR-Cas9 screen identifies activated BRAF to modulate enzalutamide resistance with potential therapeutic implications in BRAF-mutated prostate cancer. Sci Rep 11, 13683 (2021). https://doi.org/10.1038/s41598-021-93107-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93107-w
This article is cited by
-
Recent Advances and Therapeutic Strategies Using CRISPR Genome Editing Technique for the Treatment of Cancer
Molecular Biotechnology (2023)
-
Strategies to overcome the main challenges of the use of CRISPR/Cas9 as a replacement for cancer therapy
Molecular Cancer (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
