
Abstract
Human induced pluripotent stem (iPS) cell technologies coupled with genetic engineering now facilitate the study of the molecular underpinnings of disease in relevant human cell types. Application of CRISPR/Cas9-based approaches for genome-scale functional screening in iPS-derived cells, however, has been limited by technical constraints, including inefficient transduction in pooled format, loss of library representation, and poor cellular differentiation. Herein, we present optimized approaches for whole-genome CRISPR/Cas9 based screening in human iPS derived cardiomyocytes with near genome-wide representation at both the iPS and differentiated cell stages. As proof-of-concept, we perform a screen to investigate mechanisms underlying doxorubicin mediated cell death in iPS derived cardiomyocytes. We identified two poorly characterized, human-specific transporters (SLCO1A2, SLCO1B3) whose loss of function protects against doxorubicin-cardiotoxicity, but does not affect cell death in cancer cells. This study provides a technical framework for genome-wide functional screening in iPS derived cells and identifies new targets to mitigate doxorubicin-cardiotoxicity in humans.
Similar content being viewed by others
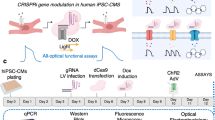


Introduction
The discovery of human cellular plasticity and the reprogramming of adult somatic cells to induced pluripotent stem (iPS) cells have ushered in new tools and approaches for interrogating human biology1,2. iPS cells may be readily differentiated into a number of human cell types3,4,5, including terminally differentiated cells not typically amenable to isolation and culture, such as specialized neurons or cardiomyocytes5,6,7,8. iPS derived cells have provided invaluable insights into biological processes and mechanisms underlying patient specific characteristics9,10, cell type specific differentiation3,4,5, human disease states11,12,13, and drug toxicity14,15,16.
Facile genetic manipulation particularly at genome-wide scale is crucial to leveraging the discovery potential of iPS cells. Traditionally, however, human iPS cells have proven resistant to conventional targeting methods relative to transformed human cells or even mouse iPS cells17,18,19,20. Recently, application of clustered regularly interspersed palindromic repeat (CRISPR)/Cas9 approaches has greatly improved the efficiency with which single functional mutations may be introduced in a wide range of organisms and cell types, including human iPS lines21,22,23,24,25,26. Coupled with large scale, pooled libraries of CRISPR/Cas9 reagents for gene disruption iPS cells now, theoretically, allow for the genome-scale functional study in relevant primary human cell types27,28. To date, efforts to apply genome-wide CRISPR/Cas9 to forward genetic screens in iPS derived cells have been limited. A key challenge for these approaches has been the efficient infection of iPS cells with virus encoding CRISPR/Cas9 reagents29. At too high an infection rate, iPS cells are likely to be infected with multiple CRISPR/Cas9 reagents, resulting in simultaneous disruption of multiple genes, thereby complicating deconvolution of results29. A lower than optimal infection rate, however, precludes interrogation of the whole genome29. In addition, it remains to be determined whether infection with pooled lentiviral reagents required for genome-wide CRISPR sgRNA expression would alter either iPS cell stemness or differentiation capacity. Finally, it is unclear whether these approaches may be applied to reveal novel human-specific mechanisms underlying cardiomyocyte phenotypes.
Herein, we find that reverse transduction greatly enhances lentiviral infection of human iPS cells. Using this method, we optimize approaches for CRISPR/Cas9 forward genetic screening in human iPS derived cells and achieve near genome-wide representation of CRISPR sgRNAs in both stem cells and differentiated cardiomyocytes, without altering makers of stemness or differentiation, respectively. Finally, in a proof of concept genome-wide CRISPR/Cas9 screen, we uncover cell-autonomous, human-specific mediators of doxorubicin induced cardiotoxicity in iPS derived cardiomyocytes, further highlighting the discovery potential of these approaches. This study presents an accessible strategy for CRISPR/Cas9 mediated screening in human iPS derived cells at genome scale. Application of these techniques will facilitate discovery of novel to human biology and disease mechanisms in relevant differentiated cell types.
Results
Lentiviral targeting of human iPS cells
Application of genome-wide CRISPR/Cas9 approaches in pooled human iPS cells necessitates efficient transduction at the iPS stage, followed by selection, expansion, and differentiation into relevant human cell types for study (Fig. 1a). Chief among these steps is optimized infection of iPS cells with lentivirus encoding CRISPR/Cas9 reagents at an infection rate of ~ 30%, to allow for genome-wide representation while statistically limiting the likelihood of incorporation of multiple sgRNAs into a single cell29. Using a lentiviral mCherry reporter system, we found that human iPS cells were resistant to viral infection using standardized infection protocols previously utilized in immortalized and/or transformed non-iPS cell lines30,31,32, with less than 10% infection rate (Fig. 1b). Application of methods that have been reported to enhance viral infection, including exposure to increased concentration of bovine serum albumin (BSA)33, or low-density culture conditions32,34, did not significantly improve lentiviral infection in human iPS cells, nor did removal of ROCK inhibitor thiazovivin (Fig. 1b). Prior work has suggested that viral infection of cells during plating, also known as ‘reverse transduction’ may enhance infection rates35,36. Indeed, we found that reverse transduction greatly increased infection efficiency relative to control methods (Fig. 1b, Supplementary Fig. 1).

Optimization of CRISPR/Cas9 transfer in human iPS cells. (a) Schematic for forward genetic CRISPR/Cas9 screening in induced pluripotent stem (iPS) derived cardiomyocytes. (b) Optimization of infection efficiency with mCherry expressing virus. Cells were treated with no virus (blank), standard infection protocols (control), addition of 1% BSA during standard infection (+ BSA), removal of the ROCK inhibitor thiazovivin during plating (− Thiaz), plating at half density prior to standard infection (low density) or use of reverse transduction (n = 2 per condition, student’s T-test, **: p < 0.01). (c) Infection efficiency in iPS cells using standard (control) or reverse transduction approaches (n = 2 per condition). (d) Bright-field image of iPS cells during plating (top) and 24 h after plating (bottom).
We next determined whether the enhanced viral infectivity of iPS cells observed with reverse transduction translated to viral infection with a CRISPR/Cas9 lentiviral system. Using a well described lentiviral system expressing Cas9 and sgRNA in single vector28, human iPS cells were infected with increasing virus volume. Reverse transduction resulted in a marked increase in viral infection of greater than sixfold relative to standard infection protocols (Fig. 1c). Enhanced lentiviral infection was also noted in human embryonic stem (H1) cells (Supplementary Fig. 1). Notably, reverse transduction results in greater cellular surface area exposure during viral infection versus traditional adherent cell culture, particularly as iPS cells tend to grow in colonies with cell–cell contact occupying a significant portion of surface area (Fig. 1d), potentially accounting for the improved infection efficiency with reverse transduction.
Infection of human iPS cells with genome-wide CRISPR/Cas9 sgRNA library
We next determined whether viral infection or expression of a CRISPR/Cas9 library in human iPS cells alters cellular state. Infection of human iPS cells with lentivirus expressing Cas9-sgRNA followed by puromycin selection for guide expression did not alter expression of key transcriptional markers of pluripotency, POU5F1, Nanog or Sox2 (Fig. 2a, Supplementary Fig. 2) or iPS cell proliferation rate (Fig. 2b, Supplementary Fig. 2) relative to uninfected cells. Following differentiation into cardiomyocytes, CRISPR/Cas9 infected iPS cells exhibited robust expression of the cardiac-specific markers cardiac troponin T (cTNT) (Fig. 2c) and myosin light-chain 2 (MLC2) (Supplementary Fig. 2), similar to uninfected cells and clear rhythmic contraction (Supplementary Video 1) consistent with normal cardiomyocyte physiology. These data suggest that gross markers of cellular physiology are unchanged at the iPS cell or differentiated cell stage with introduction of whole-genome CRISPR/Cas9 reagents. Additionally, we performed Karyotype analysis on uninfected iPS cells (Supplementary Fig. 3) and confirmed that there were no clonal or structural abnormalities that would introduce systemic bias into a whole-genome CRISPR/Cas9 screen.

Phenotypic resilience in pooled CRISPR/Cas9 library infected iPS cells. Uninfected iPS cells (control) or those transduced with CRISPR/Cas9 library (virus) were assessed for (a) gene expression of pluripotency markers (n = 3 per condition), (b) proliferative potential (n = 3 per condition), and (c) cardiac troponin T (cTNT) protein expression after differentiation to cardiomyocytes. (d) Percent of CRISPR/Cas9 library sgRNAs present in iPS cells at 10 read-counts or higher. (e) Number of sgRNAs per gene present in plasmid, iPS cells, and iPS derived cardiomyocytes.
We next sought to determine whether human iPS cells infected with a genome-wide CRISPR/Cas9 library retain full coverage sgRNA expression. Human iPS cells (9 × 107 cells) were infected with lentivirus encoding a well-described genome-wide CRISPR/Cas9 library containing ~ 75,000 total sgRNAs targeting ~ 19,000 human genes28. Following puromycin selection and 10 × expansion, human iPS cells retained robust expression of the sgRNA library, with loss of only 8% of sgRNAs relative to the original plasmid library (Fig. 2d). Lost sgRNAs were found to target genes essential to cellular survival and proliferation, including ribosomal, mRNA processing and cell cycle genes (Supplementary Fig. 4). iPS cells were subsequently differentiated into cardiomyocytes and the sgRNA library again sequenced. At the cardiomyocyte stage, sgRNAs targeting only a small fraction of genes were lost relative to the iPS stage, with continued expression of at least 3 sgRNAs targeting greater than 13,000 genes (Fig. 2e, Supplementary Fig. 4), enabling near genome-wide representation required for comprehensive forward genetic screening.
Genome-wide CRISPR/Cas9 screening for modulators of cardiotoxicity
Cardiotoxicity remains among the most significant impediments for new drug approval, particularly among many anti-cancer agents37. While a number of potential contributors may lead to drug associated cardiotoxicity, the fundamental mechanisms underlying chemotherapy related cardiotoxicity in humans still remain unclear37,38. Recently, iPS derived cardiomyocytes have been demonstrated to be a particularly useful system for probing mechanisms of cardiotoxicity associated with doxorubicin (DOX), a common first line chemotherapy used in breast cancer14,39. As proof of concept, we applied our whole-genome CRISPR/Cas9 knockout screening approach to identify new cell-autonomous mechanisms of DOX induced cardiotoxicity. Our rationale was that cardiomyocytes containing knockout genes that facilitate DOX-toxicity would have higher resistance to the chemotherapeutic, and thus would enrich over time due to prolonged survival. Exposure of iPS derived cardiomyocytes to DOX resulted in significant cell death at reported circulating concentrations (Fig. 3a)14,40,41. We next evaluated the impact of CRISPR/Cas9 library infection on DOX-toxicity in iPS derived cardiomyocytes (Supplementary Fig. 2) and found a small, but significant increase in survival in library infected cells. This increase in survival likely represents to the protective effect of sgRNAs targeting genes involved in DOX-toxicity, we thus selected representative concentration of 3 μM for genetic screening.

Genome-scale CRISPR/Cas9 screen in iPS derived cardiomyocytes for mediators of doxorubicin-toxicity. (a) Dose response curve for doxorubicin-toxicity in iPS derived cardiomyocytes (circulating concentration range highlighted in red, n = 8 per condition). (b) Distribution of fold change scores in doxorubicin (dox) versus control (ctrl) treated cardiomyocytes. SLCO1A2 and SLCO1B3 indicated by blue and red arrows, respectively. (c), and (d) Enrichment scores for individual sgRNAs targeting SLCO1A2 and SLCO1B3. (e) Western-blot for protein expression of SLCO1A2 and SLCO1B3 in human iPS derived cardiomyocytes. Full-length blots are included in Supplementary Figure 10a–b. (f) Western-blot for protein expression of SLCO1A2 and SLCO1B3 in human heart tissue. Full-length blots are included in Supplementary Figure 10c–d.
iPS cells were transduced with a whole-genome CRISPR/Cas9 library, selected, expanded, and differentiated into highly pure cardiomyocytes and exposed to DOX for 72 h. gDNA was then collected for PCR amplification of sgRNA guides and sequencing. Gene ontology analysis revealed DOX-toxicity affected processes related to iron metabolism and mitochondrial function (Supplementary Fig. 5), both key biological processes previously implicated in DOX-toxicity42. Of note, we found significant enrichment for membrane protein transporters (Supplementary Fig. 5), particularly among SLC solute carrier family, whose silencing protected against DOX-cardiotoxicity (Fig. 3b). Examination of individual sgRNAs found multiple sequence-independent guides targeting SLCO1A2 (solute carrier organic anion transporter family member 1A2, also known as OATP1; p = 0.017) and SLCO1B3 (solute carrier organic anion transporter family member 1B3, also known as OATP8 and LST2; p = 0.0012), two related organic anion transporters (Fig. 3c, d)43. Both SLCO1A2 and SLCO1B3 protein were found to be expressed in in human iPS derived cardiomyocytes (Fig. 3e) and adult human hearts (Fig. 3f). We therefore hypothesized that these transporters may be essential for DOX-cardiotoxicity.
SLCO1A2 is essential for doxorubicin cardiotoxicity
After leveraging CRISPR/Cas9 based screening to uncover SLCO1 family transporters as novel disease mediators in iPS derived cardiomyocytes, as a proof of concept we further determined the role of SLCO1A2 in doxorubicin induced cardiotoxicity. We selected two sequence-independent shRNAs to reduce the expression of SLCO1A2 in iPS derived cardiomyocytes (Supplementary Fig. 6), utilizing an approach orthogonal to the CRISPR/Cas9 based method where SLCO1A2 initially highlighted as a mediator of DOX-cardiotoxicity. Loss of SLCO1A2 protected against cell death within the reported circulating range of DOX (0.1–3 μM) but not at higher concentrations (Fig. 4a), suggesting that other low-affinity transporters may also be capable of DOX uptake at elevated concentration. We further confirmed that SLCO1A2 knockdown also impacted DOX mediated cell death in hES derived cardiomyocytes at the lower end of the reported circulating range (0.1–0.3 μM) (Supplementary Fig. 7). We next evaluated whether target cancer cells similarly utilize SLCO1A2 for transport of DOX. Silencing of SLCO1A2 did not affect DOX mediated cell death in BT549 breast cancer cells at reported circulating (Fig. 4a) or even at lower (Supplementary Fig. 8) concentrations, suggesting that other key transporters may mediate DOX-related cell death in BT549 breast cancer cells44. Pharmacologic inhibition of SLCO1A2 with the small molecule naringin phenocopied genetic silencing, and similarly protected against DOX-cardiotoxicity in cardiomyocytes, but not breast cancer cells (Fig. 4b)45. To further validate the role of specific human transporters in DOX toxicity, SLCO1A2 was overexpressed (Supplementary Fig. 9). Overexpression of SLCO1A2 enhanced DOX mediated cell death (Fig. 4c) and directly mediated uptake of DOX (Fig. 4d), suggesting a direct role for SLCO1A2 mediated transport in DOX-toxicity.

SLCO1A2 mediates doxorubicin-toxicity in iPS derived cardiomyocytes. (a) Dose response for doxorubicin toxicity in cells expressing control Scramble shRNA, or two sequence independent shRNAs targeting SLCO1A2 in iPS derived cardiomyocytes or BT549 breast cancer cells (**: p < 0.01 vs scramble shRNA). (b) Doxorubicin-toxicity in cells treated with the SLCO1A2 inhibitor naringin in iPS derived cardiomyocytes or BT549 cancer cells (**: p < 0.01). (c) Doxorubicin toxicity (n = 8 per condition) and (d) uptake (n = 3 per condition) in cells over-expressing SLCO1A2 (**: p < 0.01). (a–d) p from student’s T-test.
Discussion
In the present study we establish and apply approaches for genome-scale CRISPR/Cas9 genetic screening in iPS derived cells. We identify and optimize conditions for efficient lentiviral infection of cells at the iPS state, which allows for infection with genome-wide sgRNA libraries. We further demonstrate that CRISPR/Cas9 library infected iPS cells maintain their pluripotent state and retain their characteristics of proliferation and differentiation. Library infected cells retained cell-type specific physiology upon differentiation, including cell-type specific protein expression and function, such as contraction/relaxation in differentiated cardiomyocytes, despite expression of sgRNAs targeting > 91% of the genome. Collectively, these advances enable pooled, genome-scale, forward, loss of function screening in human cells. As a proof of concept, we apply these described approaches to uncover previously unrecognized modulators of drug related cardiotoxicity and identify SLCO1A2 as a key transport mechanism underlying DOX toxicity specifically in human iPS derived cardiomyocytes.
The differentiation of iPS cells affords a unique opportunity to study development and disease in the relevant human cell types and has proven especially useful for difficult to isolate cell types such as cardiomyocytes or neurons17,46,47. Until recently, insights into cardiomyocyte- or neuron-specific biology were largely limited to rodent and other small animal models10. While certainly valuable as model systems, lower vertebrates including mice and rats have well described and key genetic, molecular and physiological discrepancies relative to humans potentially limiting discovery and slowing the translation of findings from rodents to man48,49,50,51. This limitation is underscored in our study: the transporter proteins SLCO1A2 and SLCO1B3, identified here as mediators of DOX-cardiotoxicity, do not have direct one-to-one orthologs in commonly used small model organisms43. As such, the importance of these transporters in DOX-cardiotoxicity would not have been uncovered using mouse or rat models, highlighting the importance of functional genomics approaches specifically in human cells. Moreover, given that these transporters are found to mediate cellular toxicity uniquely in cardiomyocytes rather than in typically studied transformed cell lines, the importance of iPS derived cells is further noted. Importantly, the approaches and advantages of unbiased CRISPR/Cas9 based discovery in iPS derived cells outlined here are readily transferrable to other iPS based systems and cell types of interest.
To date, CRISPR/Cas9 approaches have been largely applied to iPS derived cells to examine a subset of human genes, such as long non-coding RNAs or largely focused upon iPS cell biology and cell-type specific differentiation52,53,54, rather than unbiased analysis of the whole genome in differentiated cell types, which necessitates optimized infection rates. Other studies, have similarly centered on CRISPR/Cas9 screening in selected iPS cells or specialized iPS derived cells, such as neural progenitor cells, that are proliferative and typically more amenable to viral infection55,56,57. Our optimized methods in human iPS cells indicate that ‘reverse-transduction’ approaches greater improve lentiviral infection efficiency for a pooled CRISPR/Cas9 library. iPS cells typically grow in densely packed colonies with minimal surface area exposed thereby limiting viral contact. Reverse transduction allows for interaction of viral particles with dissociated cells in suspension, during which time the exposed surface area is significantly increased. This reverse transduction approach closely resembles the suspension infection techniques first used with lentiviral infection of human embryonic stem cells58,59, and similar techniques have been previously employed to increase adenoviral infection efficiency in human cells35,36.
In applying the described CRISPR/Cas9 approaches in iPS derived cells, we have also uncovered and validated a role for the cell surface transporters SLCO1A2 and SLCO1B3 in doxorubicin induced cardiotoxicity. Whereas the transporters required for efflux of doxorubicin and related chemotherapies have been described60, to date, the transport systems that promote uptake of doxorubicin under physiologic concentrations into human cardiomyocytes have remained unclear. While SLCO1A2 and SLCO1B3 likely mediate doxorubicin uptake and clearance61 at reported circulating concentrations14,40,41, it is notable that cardiomyocytes and cancer cells exhibit distinct uptake kinetics62 and we find that other transport systems likely mediate doxorubicin uptake into breast cancer cells. Given this selectivity for doxorubicin uptake via SLCO1A2 in cardiomyocytes, this transporter represents an intriguing drug target to mitigate doxorubicin induced cardiotoxicity, while preserving the drug’s chemotherapeutic effect. Taken together our results demonstrate the utility of an optimized functional genomics approach in iPS derived cells for novel human discovery.
Methods
Detailed methods are included in the Supplementary Information appendix, Materials and Methods.
Cell culture and reagents
BT549 breast cancer cells were obtained from ATCC. HEK293T cells were obtained from Sonia Sharma(LJI), H1ESC line was obtained from Aitor Aguirre(UCSD), and IPSL1 was a obtained from Eric Adler(UCSD). Cells were maintained as described in the Supplementary Methods and cardiomyocyte differentiation was performed was described previously8. Antibodies were obtained according to the following: Anti-cTnT primary antibody (ThermoFisher MA5-12960), Anti-SLCO1A2 (abcam ab221804), Anti-SLCO1B3 (abcam ab224064), Anti-βActin primary antibody (Cell Signaling 3700S), Ant- Myosin Light Chain (abcam ab89594), HRP Conjugated Anti Rabbit (Enzo ADI-SAB-300J), HRP Conjugated Anti Mouse (Cell Signaling 7076P2), and Alexa Fluor 594 anti-mouse secondary antibody (ThermoFisher A-21203). Doxorubicin (Sigma D1515) and Narignin (71162) were obtained from Sigma Aldrich.
Heart tissue
Heart tissue protein lysate was obtained from explanted human cardiac tissue. All research was conducted in accordance with relevant guidelines and regulations. All human subjects work was conducted under approval of the UCSD Institutional Review Board (IRB) and Human Research Protection Program and informed consent was obtained from all subjects.
CRISPR/Cas9 screening
The Brunello LentiCRISPR plasmid library28 was obtained from Addgene (Cat # 73178), amplified according to established Broad Institute GPP Protocol, and virus was subsequently produced using HEK293T cells as described in the Supplementary Methods. The multiplicity of infection for each cell type was determined empirically as previously described27. For the genome wide screening, 87.5 million iPS cells were infected with the Brunello library at an efficiency rate of 30% and selected for lentiviral incorporation using puromycin. CRISPR/Cas9 library iPS cells were then differentiated to cardiomyocytes as described previously8. For the doxorubicin (DOX) treatment, iPS cells were treated with 3 μM doxorubicin for 72 h. DNA was collected for sgRNA sequencing from iPS cell, differentiated cardiomyocyte and doxorubicin treated conditions. Data was processed using Mageck analysis software and the RIGER algorithm to analyze sgRNA and gene level enrichment in doxorubicin treated versus no-doxorubicin control63,64.
SLCO1A2 validation
Plasmid encoding scramble or SLCO1A2 shRNA were purchased from VectorBuilder and lentivirus was generated as in the Supplementary Methods. iPS cells, ES cells and BT549 breast cancer cells were infected in media containing polybrene during plating. Cells expressing the shRNA cassette were selected using puromycin. For transient over-expression, HEK cells were transfected with plasmid encoding for SLCO1A2 or were mock transfected without plasmid for control. For stable over-expression HEK cells were infected with virus encoding SLCO1A2 in media containing polybrene during plating. iPS derived cardiomyocytes and Bt549 breast cancer cells were treated with SLCO1A2 inhibitor Naringin at 50 μM or vehicle control. Doxorubicin toxicity was assessed using nuclei counting, as described in Supplementary Methods. Doxorubicin uptake was detected using mass-spectrometry based measures of cell lysate, as outlined in the Supplementary Methods.
Statistical analysis
Statistical significance was determined using students T-test or linear regression using R statistical analysis software. Data is illustrated as mean +/− SEM. GraphPad PRISM software was used to fit a dose–response curve to doxorubicin toxicity data using the bell-shape model.
References
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. https://doi.org/10.1016/j.cell.2007.11.019 (2007).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. https://doi.org/10.1016/j.cell.2006.07.024 (2006).
Yoshida, Y. & Yamanaka, S. Recent stem cell advances: Induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation 122, 80–87. https://doi.org/10.1161/CIRCULATIONAHA.109.881433 (2010).
Song, Z. et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 19, 1233–1242. https://doi.org/10.1038/cr.2009.107 (2009).
Zhang, J. et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ. Res. 104, e30-41. https://doi.org/10.1161/CIRCRESAHA.108.192237 (2009).
Boyer, L. F., Campbell, B., Larkin, S., Mu, Y. & Gage, F. H. Dopaminergic differentiation of human pluripotent cells. Curr. Protoc. Stem Cell Biol. Chapter 1, Unit1H 6, https://doi.org/10.1002/9780470151808.sc01h06s22 (2012).
Shi, Y., Kirwan, P., Smith, J., Robinson, H. P. & Livesey, F. J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–486. https://doi.org/10.1038/nn.3041 (2012).
Lian, X. et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. U. S. A. 109, E1848–E1857. https://doi.org/10.1073/pnas.1200250109 (2012).
Karakikes, I., Ameen, M., Termglinchan, V. & Wu, J. C. Human induced pluripotent stem cell-derived cardiomyocytes: Insights into molecular, cellular, and functional phenotypes. Circ. Res. 117, 80–88. https://doi.org/10.1161/CIRCRESAHA.117.305365 (2015).
Bellin, M., Marchetto, M. C., Gage, F. H. & Mummery, C. L. Induced pluripotent stem cells: The new patient?. Nat. Rev. Mol. Cell Biol. 13, 713–726. https://doi.org/10.1038/nrm3448 (2012).
Moretti, A. et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409. https://doi.org/10.1056/NEJMoa0908679 (2010).
Carvajal-Vergara, X. et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808–812. https://doi.org/10.1038/nature09005 (2010).
Kurian, L. et al. Conversion of human fibroblasts to angioblast-like progenitor cells. Nat. Methods 10, 77–83. https://doi.org/10.1038/nmeth.2255 (2013).
Burridge, P. W. et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 22, 547–556. https://doi.org/10.1038/nm.4087 (2016).
Mordwinkin, N. M., Burridge, P. W. & Wu, J. C. A review of human pluripotent stem cell-derived cardiomyocytes for high-throughput drug discovery, cardiotoxicity screening, and publication standards. J. Cardiovasc. Transl. Res. 6, 22–30. https://doi.org/10.1007/s12265-012-9423-2 (2013).
Takayama, K. et al. Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 111, 16772–16777. https://doi.org/10.1073/pnas.1413481111 (2014).
Hockemeyer, D. & Jaenisch, R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18, 573–586. https://doi.org/10.1016/j.stem.2016.04.013 (2016).
Rapti, K. et al. Effectiveness of gene delivery systems for pluripotent and differentiated cells. Mol. Ther. Methods Clin. Dev. 2, 14067. https://doi.org/10.1038/mtm.2014.67 (2015).
Schinzel, R. T. et al. Efficient culturing and genetic manipulation of human pluripotent stem cells. PLoS ONE 6, e27495. https://doi.org/10.1371/journal.pone.0027495 (2011).
Xia, X., Zhang, Y., Zieth, C. R. & Zhang, S. C. Transgenes delivered by lentiviral vector are suppressed in human embryonic stem cells in a promoter-dependent manner. Stem Cells Dev. 16, 167–176. https://doi.org/10.1089/scd.2006.0057 (2007).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. https://doi.org/10.1126/science.1225829 (2012).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. https://doi.org/10.1126/science.1231143 (2013).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat. Biotechnol. 31, 227–229. https://doi.org/10.1038/nbt.2501 (2013).
Long, C. et al. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science 345, 1184–1188. https://doi.org/10.1126/science.1254445 (2014).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826. https://doi.org/10.1126/science.1232033 (2013).
Ding, Q. et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12, 393–394. https://doi.org/10.1016/j.stem.2013.03.006 (2013).
Shalem, O. et al. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343, 84–87. https://doi.org/10.1126/science.1247005 (2014).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR–Cas9. Nat. Biotechnol. 34, 184–191. https://doi.org/10.1038/nbt.3437 (2016).
Doench, J. G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 19, 67–80. https://doi.org/10.1038/nrg.2017.97 (2018).
Elegheert, J. et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 13, 2991–3017. https://doi.org/10.1038/s41596-018-0075-9 (2018).
Denning, W. et al. Optimization of the transductional efficiency of lentiviral vectors: Effect of sera and polycations. Mol. Biotechnol. 53, 308–314. https://doi.org/10.1007/s12033-012-9528-5 (2013).
Zhang, B. et al. The significance of controlled conditions in lentiviral vector titration and in the use of multiplicity of infection (MOI) for predicting gene transfer events. Genet. Vaccines Ther. 2, 6. https://doi.org/10.1186/1479-0556-2-6 (2004).
Mekkaoui, L. et al. Lentiviral vector purification using genetically encoded biotin mimic in packaging cell. Mol. Ther. Methods Clin. Dev. 11, 155–165. https://doi.org/10.1016/j.omtm.2018.10.008 (2018).
Braam, S. R. et al. Improved genetic manipulation of human embryonic stem cells. Nat. Methods 5, 389–392. https://doi.org/10.1038/nmeth.1200 (2008).
Oehmig, A. et al. A novel reverse transduction adenoviral array for the functional analysis of shRNA libraries. BMC Genom. 9, 441. https://doi.org/10.1186/1471-2164-9-441 (2008).
Lee, E. J., Robinson, T. M., Tabor, J. J., Mikos, A. G. & Suh, J. Reverse transduction can improve efficiency of AAV vectors in transduction-resistant cells. Biotechnol. Bioeng. 115, 3042–3049. https://doi.org/10.1002/bit.26830 (2018).
Ferri, N. et al. Drug attrition during pre-clinical and clinical development: Understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 138, 470–484. https://doi.org/10.1016/j.pharmthera.2013.03.005 (2013).
Bowes, J. et al. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 11, 909–922. https://doi.org/10.1038/nrd3845 (2012).
Maillet, A. et al. Modeling doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci. Rep. 6, 25333. https://doi.org/10.1038/srep25333 (2016).
Robert, J. et al. Comparative pharmacokinetics and metabolism of doxorubicin and epirubicin in patients with metastatic breast cancer. Cancer Treat. Rep. 69, 633–640 (1985).
Hempel, G., Flege, S., Wurthwein, G. & Boos, J. Peak plasma concentrations of doxorubicin in children with acute lymphoblastic leukemia or non-Hodgkin lymphoma. Cancer Chemother. Pharmacol. 49, 133–141. https://doi.org/10.1007/s00280-001-0392-4 (2002).
Bernstein, D. & Burridge, P. Patient-specific pluripotent stem cells in doxorubicin cardiotoxicity: A new window into personalized medicine. Prog. Pediatr. Cardiol. 37, 23–27. https://doi.org/10.1016/j.ppedcard.2014.10.006 (2014).
Konig, J., Seithel, A., Gradhand, U. & Fromm, M. F. Pharmacogenomics of human OATP transporters. Naunyn Schmiedebergs Arch. Pharmacol. 372, 432–443. https://doi.org/10.1007/s00210-006-0040-y (2006).
Okabe, M. et al. Characterization of the organic cation transporter SLC22A16: A doxorubicin importer. Biochem. Biophys. Res. Commun. 333, 754–762. https://doi.org/10.1016/j.bbrc.2005.05.174 (2005).
Bailey, D. G., Dresser, G. K., Leake, B. F. & Kim, R. B. Naringin is a major and selective clinical inhibitor of organic anion-transporting polypeptide 1A2 (OATP1A2) in grapefruit juice. Clin. Pharmacol. Ther. 81, 495–502. https://doi.org/10.1038/sj.clpt.6100104 (2007).
Ahuja, G. et al. Loss of genomic integrity induced by lysosphingolipid imbalance drives ageing in the heart. EMBO Rep. https://doi.org/10.15252/embr.201847407 (2019).
Zhao, X. et al. Comparison of non-human primate versus human induced pluripotent stem cell-derived cardiomyocytes for treatment of myocardial infarction. Stem Cell Rep. 10, 422–435. https://doi.org/10.1016/j.stemcr.2018.01.002 (2018).
Milani-Nejad, N. & Janssen, P. M. Small and large animal models in cardiac contraction research: Advantages and disadvantages. Pharmacol. Ther. 141, 235–249. https://doi.org/10.1016/j.pharmthera.2013.10.007 (2014).
Perlman, R. L. Mouse models of human disease: An evolutionary perspective. Evol. Med. Public Health 2016, 170–176. https://doi.org/10.1093/emph/eow014 (2016).
McGonigle, P. & Ruggeri, B. Animal models of human disease: Challenges in enabling translation. Biochem. Pharmacol. 87, 162–171. https://doi.org/10.1016/j.bcp.2013.08.006 (2014).
Kim, C. iPSC technology—Powerful hand for disease modeling and therapeutic screen. BMB Rep. 48, 256–265. https://doi.org/10.5483/bmbrep.2015.48.5.100 (2015).
Liu, S. J. et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science https://doi.org/10.1126/science.aah7111 (2017).
Xu, J. et al. Genome-wide CRISPR screen identifies ZIC2 as an essential gene that controls the cell fate of early mesodermal precursors to human heart progenitors. Stem Cells https://doi.org/10.1002/stem.3168 (2020).
Li, S. et al. Genetic and chemical screenings identify HDAC3 as a key regulator in hepatic differentiation of human pluripotent stem cells. Stem Cell Rep. 11, 22–31. https://doi.org/10.1016/j.stemcr.2018.05.001 (2018).
Li, Y. et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. U. S. A. 116, 9527–9532. https://doi.org/10.1073/pnas.1900867116 (2019).
Noisa, P., Raivio, T. & Cui, W. Neural progenitor cells derived from human embryonic stem cells as an origin of dopaminergic neurons. Stem Cells Int. 2015, 647437. https://doi.org/10.1155/2015/647437 (2015).
Han, X. et al. Efficient and fast differentiation of human neural stem cells from human embryonic stem cells for cell therapy. Stem Cells Int. 2017, 9405204. https://doi.org/10.1155/2017/9405204 (2017).
Ma, Y., Ramezani, A., Lewis, R., Hawley, R. G. & Thomson, J. A. High-level sustained transgene expression in human embryonic stem cells using lentiviral vectors. Stem Cells 21, 111–117. https://doi.org/10.1634/stemcells.21-1-111 (2003).
Gropp, M. et al. Stable genetic modification of human embryonic stem cells by lentiviral vectors. Mol. Ther. 7, 281–287. https://doi.org/10.1016/s1525-0016(02)00047-3 (2003).
Budde, T. et al. Acute exposure to doxorubicin results in increased cardiac P-glycoprotein expression. J. Pharm. Sci. 100, 3951–3958. https://doi.org/10.1002/jps.22622 (2011).
Durmus, S. et al. In vivo disposition of doxorubicin is affected by mouse Oatp1a/1b and human OATP1A/1B transporters. Int. J. Cancer 135, 1700–1710. https://doi.org/10.1002/ijc.28797 (2014).
Johnson, B. A., Cheang, M. S. & Goldenberg, G. J. Comparison of adriamycin uptake in chick embryo heart and liver cells an murine L5178Y lymphoblasts in vitro: Role of drug uptake in cardiotoxicity. Cancer Res. 46, 218–223 (1986).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. https://doi.org/10.1186/s13059-014-0554-4 (2014).
Luo, B. et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. U. S. A. 105, 20380–20385. https://doi.org/10.1073/pnas.0810485105 (2008).
Acknowledgements
We would like to acknowledge members of the Jain lab for thoughtful discussion of the manuscript and experimental design. This work was supported by Grants from the National Institutes of Health (NIH), including NIH R03HL133720, R01ES027595, and S10OD020025 (to M.J.), R01CA199376 (to S.S.), R01HL128135 and R01HL132511 (to R.L.), K23HL107755 (to E.A.), and K01HL135464 (to A.A.). V.S. was supported by NIH T32GM007752, and G.M. and J.D. were supported by UC San Diego Chancellor’s Research Excellence Scholarships. Imaging experiments performed at UCSD Microscopy Core were supported by P30 NS047101.
Author information
Authors and Affiliations
Contributions
M.J., V.S. and A.A. conceived of and designed the experiments. V.S., G.M. and J.D. performed the experiments. V.S. analyzed the experiments and generated the figures. M.J. and V.S. wrote the paper. E.A., A.A., S.S. and R.L. provided conceptual support and critically revised the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Video 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sapp, V., Aguirre, A., Mainkar, G. et al. Genome-wide CRISPR/Cas9 screening in human iPS derived cardiomyocytes uncovers novel mediators of doxorubicin cardiotoxicity. Sci Rep 11, 13866 (2021). https://doi.org/10.1038/s41598-021-92988-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92988-1
This article is cited by
-
Compact CRISPR genetic screens enabled by improved guide RNA library cloning
Genome Biology (2024)
-
Membrane transporters in drug development and as determinants of precision medicine
Nature Reviews Drug Discovery (2024)
-
A genome-wide CRISPR screen identifies BRD4 as a regulator of cardiomyocyte differentiation
Nature Cardiovascular Research (2024)
-
Recent advances in pluripotent stem cell-derived cardiac organoids and heart-on-chip applications for studying anti-cancer drug-induced cardiotoxicity
Cell Biology and Toxicology (2023)
-
Gene Modulation with CRISPR-based Tools in Human iPSC-Cardiomyocytes
Stem Cell Reviews and Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
