
Abstract
Effective agents to treat coronavirus infection are urgently required, not only to treat COVID-19, but to prepare for future outbreaks. Repurposed anti-virals such as remdesivir and human anti-inflammatories such as barcitinib have received emergency approval but their overall benefits remain unclear. Vaccines are the most promising prospect for COVID-19, but will need to be redeveloped for any future coronavirus outbreak. Protecting against future outbreaks requires the identification of targets that are conserved between coronavirus strains and amenable to drug discovery. Two such targets are the main protease (Mpro) and the papain-like protease (PLpro) which are essential for the coronavirus replication cycle. We describe the discovery of two non-antiviral therapeutic agents, the caspase-1 inhibitor SDZ 224015 and Tarloxotinib that target Mpro and PLpro, respectively. These were identified through extensive experimental screens of the drug repurposing ReFRAME library of 12,000 therapeutic agents. The caspase-1 inhibitor SDZ 224015, was found to be a potent irreversible inhibitor of Mpro (IC50 30 nM) while Tarloxotinib, a clinical stage epidermal growth factor receptor inhibitor, is a sub micromolar inhibitor of PLpro (IC50 300 nM, Ki 200 nM) and is the first reported PLpro inhibitor with drug-like properties. SDZ 224015 and Tarloxotinib have both undergone safety evaluation in humans and hence are candidates for COVID-19 clinical evaluation.
Similar content being viewed by others

Introduction
The Coronavirus disease 2019 (COVID-19) pandemic caused by Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) is the largest global health emergency to emerge this century1. Severely affected patients can display sepsis, through inappropriate recruitment and expansion of the innate immune response and even patients who have cleared the virus may continue to suffer, in part due to fibrotic lesions2. To date there are limited direct antiviral medicines approved for treatment of COVID19, thus we aimed to determine whether any molecules previously approved for clinical study could be repurposed for the treatment of COVID19.
Vaccines against COVID-19 are reducing COVID19 outbreaks and mortality3, although viral mutations may compromise the longevity of current vaccines4 and may not protect against future coronaviral disease. In response to this there are calls to develop a ‘universal vaccine’5. Outside of vaccination small molecule inhibitors may play a role suppressing viral proliferation in patients already infected at an early stage in disease and thus reduce overall disease burden6 and have so far shown broad spectrum activity against several coronavirus variants7,8.
Existing small molecules for treating COVID19 include the anti-viral RNA polymerase inhibitor remdesivir9 (originally developed for Hepatitis C Virus) as well as compounds with anti-inflammatory and immunosuppressant effects such as dexamethasone10 and baricitinib11. Unfortunately, none of these medicines have delivered meaningful benefits to patient populations, execpt dexamethasone which provides benefits in only the most advanced stages of the disease10, although combinations of remdesivir and baricitinib show promising results11. Thus there is strong motivation for the discovery of new therapeutics not only for the treatment of acute disease. Antivirals offer the potential of prophylaxis, reduction of transmissibility, treatment of unvaccinated patients and suppression of emergent coronaviruses. Considering the lengthy timescales required to develop and approve new therapeutic agents, repurposing of known drugs can potentially reduce the time to develop new treatments.
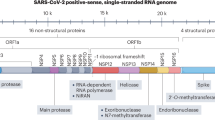
The SARS-CoV-2 genome encodes small molecule druggable targets including the main protease12 (Mpro) encoded by non-structural protein 5 (nsp5) and the papain-like protease8 (PLpro) which is part of non-structural protein 3 (nsp3)13. As both proteins are essential for viral replication they present attractive targets for drug repurposing efforts. These cysteine proteases are encoded along with the other 14 nsps by the 5′-terminal open reading frame 1a/b (ORF1a/b), which takes up approximately two-thirds of the viral genome. This leads to the expression of the two large replicase polyproteins pp1a and pp1ab. Mpro and PLpro are responsible for the proteolytic cleavage of pp1a and pp1ab which consist of nsps 1–11 and 1–16, respectively14. Mpro cleaves at 11 sites releasing the functional nsps 4–16, while PLpro cleaves at 3 sites releasing nsps 1–315. Additionally, PLpro acts as a deubiquitinase and deISGylase which may modulate the host anti-viral response via suppression of type-I interferon production16. Recent developments have seen a rationally designed Mpro inhibitor enter clinical testing17.
To rapidly identify potential anti–coronavirus therapeutics, we undertook extensive experimental screens of the drug repurposing ReFRAME library, that consists of 12,000 therapeutic agents against Mpro and PLpro to identify clinically-viable agents that may be repositioned to treat COVID-19 and future outbreaks. One compound, SDZ 224015, is a potent irreversible inhibitor of Mpro, whilst a second, Tarloxotinib, is a sub micromolar inhibitor of PLpro.
Results
ReFRAME library screening and hit triage
The ReFRAME library comprises 12,000 molecules that have been previously approved for clinical investigation in humans, including all currently approved medicines18. Despite the overall quality and relevance of this library, it contains some older compounds with properties less attractive for modern drug discovery, such as polyphenol groups, flavonoids and catechols (sennosides)19, reactive Michael acceptors (oxantel)20, unattractive molecular weights and poor solubility, many of which can also cause assay interference21.
Mpro and PLpro have the potential to exist in several conformationally distinct states, each of which may favour the binding of different inhibitors22. Mpro substrate velocity titrations revealed evidence for catalytically distinct monomeric and dimeric forms23. Dimer dependent catalysis manifests as the observation of enzyme concentration dependent sigmoidal substrate velocity plots (Fig. 1a) as observed for Mpro from SARS-CoV-124. The parameters describing the midpoint or slope of the sigmodal substrate velocity plots displayed a bell-shaped relationship with enzyme concertation (Fig. 1b,c), whereas the maximum velocity displayed a sigmoidal relationship with enzyme concentration (Fig. 1d). These results strongly indicate dimerization may be induced either by increasing enzyme or substrate concentration, at high concentrations (> 300 nM) Mpro spontaneously dimerizes and low concentration (< 3 nM) Mpro behaves as a monomer. These data indicate the potential for inhibitors to bind to these catalytically distinct forms25 as well as the potential for binding at the dimer interface24. Consequently, the Mpro HTS assay was designed to balance these forms.

Mpro enzymology. Panel (a) shows the initial rate of substrate cleavage by Mpro at different enzyme concentrations. Data are plotted as an average of four replicates, shown as black circles with error bars representing the standard deviation; a fit of an allosteric sigmoidal model is shown as a black line. Panels (b) and (c) show a bell-shaped relationship for both the Hill-factor and Khalf obtained for Mpro-substrate kinetics at differing concentrations of Mpro, fitted results are shown as black circles, with lines between the points. Panel (d) shows a log–log plot of [Mpro] vs Vmax; circles show the fitted results.
PLpro was found to require the presence of high concentrations of anionic Hoffmeister salts to catalyse hydrolysis of small peptide substrates, but not large ubiquitin mimetics (Fig. 2). This indicated that the PLpro active site is not accessible in isotonic buffer, as previously observed for SARS-CoV-1 PLpro26. This requirement may relate to the subcellular location of nsp3 expressed during viral infection. Nsp3 is expressed on the surface of the endoplasmic reticulum and in combination with nsp4 creates double membraned vesicles27. For the papain-like protease of the betacoronavirus murine hepatitis virus, PLpro did not process the viral polyprotein unless expressed on the ER membrane28. Thus, to maximise the discovery of inhibitors, the HTS was run in the presence of 0.8 M citrate.

PLpro enzymology. The upper two panels show the initial rate of reaction for ubiquitin-rhodamine cleavage by PLpro in either tris-saline buffer or 0.8 M citrate. The lower two panels show the initial rate of substrate cleavage of a 7-mer peptide corresponding to the C-terminal of ubiquitin in either tris-saline buffer or 0.8 M citrate. The data points show the average of 4-replicates with error bars showing the standard deviation, with either a straight line or Michaelis–Menten fit shown as a solid black line.
In order to minimise the number of HTS false positives, confirmatory screens were run, followed by a counter screen of Mpro and PLpro hits against one another, taking advantage that despite minimal homology both have nucleophilic cysteine containing active sites but recognise distinct peptide substrate sequences.
An initial single point screen of the ReFRAME library performed well for Mpro and adequately for PLpro (Supplementary Fig. 1). Following single point screening, a hit triage consisting of repeat confirmation and selectivity counter screening (Supplementary Fig. 2), twenty-one Mpro and thirty-five PLpro compounds remained (Supplementary Table 1). After filtering to remove undesirable chemical structures, such as pan assay interference compounds etc., two novel Mpro and a single PLpro hit were selected for further analysis.
SARS-CoV-2 Mpro inhibitors
The two selected Mpro hits from the ReFRAME screen are compounds 1 and 4 (Fig. 3). 4 has an IC50 of 30 nM (the biochemical limit of the assay), however contains a less attractive peptide backbone scaffold, whilst 1 exhibits a weaker IC50 of 3 µM and structure consistent with drug-like absorption, distribution, metabolism, and excretion (ADME) properties29. 1 is a derivative of the investigational compound ABT-95730 (2, Fig. 3, Supplementary Table 1) a calpain 1 & 2 inhibitor, differing only by the presence of a pyridinyl group instead of the ABT-957 cyclopropyl-group. This substitution is responsible for the Mpro inhibition potency of 1 compared to 2 (IC50 > 100 µM, Fig. 3). Inhibition of Mpro does not seem to be a general property of calpain inhibitors, as the tool calpain inhibitor Z-L-Abu-CONH-ethyl (3, Fig. 3) does not inhibit. 1 was confirmed to bind to Mpro in a SPR assay, giving a KD of 1 µM (Supplementary Fig. 3). Furthermore, in HUH7 cells 1 was non cytotoxic at concentrations up to 100 µM and showed an antiviral effect of 75% at 100 µM (Supplementary Fig. 5).

Potent inhibitors of SARS-CoV-2 Mpro. Structures of the compounds tested against Mpro are shown to the left of the graphs. Compounds 1–3 are calpain inhibitors and 4–7 are caspase-1 inhibitors. 1 is a pyridine analogue of ABT-957 (2), 4 is the ester prodrug SDZ 224015 with 5 Caspase-1 active acid version of 4. The upper graph shows titrations of the compounds plotted against inhibition of Mpro, and the lower graph shows the same compounds against PLpro. Data are singlicate and representative of at least four repeats on separate occasions.
Compound 4 is the investigational caspase 1 inhibitor prodrug SDZ-22401531, which is cleaved by esterases in vivo to yield 5, where the free aspartic acid is revealed (Fig. 3). In contrast to the exquisite potency of 4, 5 has limited potency for Mpro, yielding only 50% inhibition at 100 µM. Inhibition of Mpro did not seem to be a general property of caspase 1 inhibitors, as the tool tetrapeptide Ac-YVAD-AOM (6, Fig. 3) and the investigational caspase 1 drug belnecasan (7, Fig. 3) did not substantially inhibit Mpro. 4 was confirmed to bind to Mpro in an SPR assay, although due to the mechanism of action a KD cannot be reported (Supplementary Fig. 3). Compound 4 is a suicide inhibitor which is cleaved by Mpro, releasing a dichlorobenozic acid leaving group and forming an irreversible covalent adduct by reaction with the nucleophilic cysteine (Supplementary Fig. 4).
Due to the presence of three esters, 4 is unstable in aqueous media so is unsuitable for the long incubations used in antiviral assays. However, when the dose was refreshed daily, 4 was found to be non-cytotoxic in HUH7 cells at concentrations at or below 10 µM and showed an antiviral effect of 50% at 10 µM (Fig. 4). Refreshing the dose more frequently is likely to further increase the apparent potency.

Anti-viral effect of compounds 4 and 8. The left hand chart shows anti-viral effect of compounds 4 and 8 in HUH7_mCherry cells. An anti-viral effect was established after a 4-day incubation with SARS CoV-2 virus at concentrations which did not cause significant cytotoxic effects. The anti-viral effect was established by an increase in fluorescent cells counted compared to an untreated control. The right-hand chart shows cell viability after 4-days treatment with compounds 4 and 8 in HUH7_mCherry cells. Viability was established by counting the number of fluorescent cells. Data are shown as the average of two technical repeats with error bars representing the range. Data are representative of two technical repeats.
SARS CoV-2 PLpro inhibitors
The ReFRAME screen revealed a single inhibitor, tarloxotinib as a potent PLpro inhibitor (Table 1, 8) which has an IC50 of 300 nM. 8 is also a prodrug, activated in hypoxic conditions in vivo by STEAP432 to yield the equipotent compound 9 (Table 1) which was designed to target the kinase domain of EGFR33. Whilst both 8 and 9 contain a 4-anilinoquinazoline core that is present in several approved drugs34, the tested related molecules proved to be less potent than tarloxotinib (Table 1, 10–13).
Analysis of the other 4-anilinoquinazoline approved medicines revealed the presence of the α,β-unsaturated amide warhead on 8 and 9 was not necessary or sufficient for activity, as 13 achieved weak activity without the warhead whilst the presence of a nitrile on the 3 position of 10 completely removes potency despite the presence of the warhead. These observations show that inhibition is not solely due to intrinsic reactivity of the molecules but requires specific molecular recognition.
8 was found to be competitive with the peptide substrate (Supplementary Fig. 6) and despite the conditions required for its discovery, the potency of 8 was not dependent on the use of high concentrations of anionic Hoffmeister salts (Supplementary Fig. 7). In HUH7 cells 8 was non-cytotoxic at concentrations up to 10 µM, and showed an antiviral effect of 25% at 10 µM (Fig. 4).
Crystallography of the ReFRAME hits
X-ray crystal structures were attempted for compounds 1,4,5 and 8 and obtained for 1 and 5 with Mpro (Fig. 5, PDB codes: 7AEH & 7AEG), at 1.3 Å and 1.8 Å resolution, respectively (Supplementary Table 2). The Mpro dimer is shown in ribbon representation with 1 and 5 bound at the active site (Fig. 5a,b). Both 1 and 5 bind covalently to the catalytic cysteine (Cys145) with well-defined electron density and form hydrogen bonding networks with the Mpro active site (Fig. 5c,d, Supplementary Fig. 8). Despite both 1 and 5 interacting with Cys145, they have substantially different binding modes. 5 extends from P1 to P5 of the Mpro active site (Fig. 5b). By contrast 1 binds from P1 to P1′ across the region occupied by the catalytic cysteine, with the two inhibitor benzyl groups π-stacking together to fill the space of the P1′ pocket (Fig. 5b). Electron density for the 2,6-dichlorobenzoate leaving group of 5 was not observed, providing evidence of the proposed mechanism of inhibition (Supplementary Fig. 4 and 8). 1 forms electrostatic interactions with Gly143, Ser144, Cys145, and His41 as well as a water-mediated interaction with His164 whereas 5 makes electrostatic interactions with Gly143, Cys145, His163, His164, and Glu166 together with a water mediated interaction with Gln189 (Fig. 5).

Crystal structures of SARS-CoV-2 Mpro in complex with ReFRAME inhibitors. (a) Ribbon representation with transparent surface of the Mpro dimer coloured in red and blue to delineate each protomer. The structures of Mpro in complex with 1 and 5 (sticks with yellow and green transparent surface, respectively) reveal that both bind in the Mpro substrate binding pocket. (b) Surface representation showing the overall binding modes of compound 1 and 5 (green and yellow transparent surfaces, respectively). (c) and (d) Stick representations of compounds 1 and 5 showing interactions (hydrogen bonds as dashed lines) within the Mpro binding pocket. Structures are deposited in the pdb as 7AEH for 1 and 7AEG for 5. Figure generated with PyMOL, The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC (https://pymol.org/2/).
Active site plasticity is important in accommodation of the inhibitors. The P1′ pocket expands on binding of 1, with the alpha carbons of Thr25 and Thr26 shifting by 0.9 Å and 0.7 Å respectively. Similarly, the P5 pocket expands upon binding to 5, with Pro168 and Thr190 moving by 1.3 Å and 0.8 Å respectively. Additionally, there is a 1.9 Å shift between Ala191 in the 1 and 5 complexes. In both cases plasticity in response to ligand binding is also observed for the P2 pocket35 (Fig. 6).

Flexibility induced in the active site of SARS-CoV-2 Mpro by compounds 1 and 5 from the ReFRAME library. Grey–Ligand-free Mpro (pdb 5r8t), teal–Mpro in complex with 1, orange–Mpro in complex with 5. Figure generated with PyMOL, The PyMOL Molecular Graphics System, Version 1.8.0.5 Schrödinger, LLC (https://pymol.org/2/).
The pyridine of 1 and the aspartate of 5 extend into the P1 pocket and interact with His163 (Fig. 5c,d). Modelling suggests that the cyclopropyl ring sidechain of 2 is unable to make this interaction and as a consequence does not bind to Mpro. Similarly, the acid of the aspartate in 5 is in close proximity to Glu166 residue which may cause a charge clash explaining the loss in potency of 5 compared to 4.
Prospects for molecular design
Combining the information from the Mpro structures of 1 and 5 could be the starting point to design more potent, drug-like inhibitors. As inhibition of Caspase 1 would inhibit inflammation via suppression of the IL-1β, this could provide additional clinical benefit in the treatment of COVID-1936,37. Thus inhibitors which possess dual anti-inflammatory and antiviral properties may be desirable. A docking analysis38,39of the binding pose of 5 in Mpro and the caspase-1 active site reveals multiple shared interactions, indicating that further, more drug-like molecules could be developed which share the potential dual anti-viral/anti-inflammatory polypharmacology of SDZ 224015 (Supplementary Fig. 9).
For PLpro, the 4-anilinoquinazoline core is one of the most common scaffolds for generation of tyrosine kinase inhibitors. Thus, it should be possible to rapidly expand from 8 and 9, to discover new, potentially more potent PLpro inhibitors with the potential to remove kinase activity all together, whilst retaining drug-like properties.
Discussion
The emergence of COVID-19 has emphasised the need for multiple approaches to tackle viral infections. To bridge the gap between the need to rapidly address a new disease and the time required to safely develop an entirely new medicine, repurposing existing drugs is an attractive alternative40.
There have been several clinical efforts to assess the benefits of existing drugs for COVID-19 treatment. Trials have broadly focused on repurposed anti-viral drugs to reduce infection such as remdesivir, as well as the use of existing anti-inflammatory and immunosuppressant compounds to help the body better manage its subsequent response to the infection. Virtual screening has also been used23,35, but has only identified boceprevir, an HCV protease inhibitor, as an Mpro inhibitor35. This molecule was identified in our screen but has disappointing activity (IC50 of 3 µM) (Supplementary Table 1).
By contrast, the results described here identify highly potent inhibitors of Mpro and for the first time a potent inhibitor of PLpro with drug-like properties. This is also the first description of a non-antiviral molecule to show repurposed PLpro activity. Neither of the two Mpro inhibitors discovered in this study were proposed by docking efforts. 4 is a suicide inhibitor which uses a complex mechanism that is difficult to predict41. Further, the conformation of 1 within the P1’ pocket of Mpro which is driven by intramolecular pi-pi stacking (Fig. 3c) is unusual and not readily predicted by in silico approaches42.
A recent crystallographic screen of 5000 compounds discovered several compounds which crystallised with Mpro43. One such hit was the EGFR inhibitor pelitinib but disappointingly it only subsequently shows micromolar activity in a cellular screen and was not determined to display significant biochemical inhibition of Mpro in this study. In contrast to the Mpro crystallographic screen which was also restricted to a single structural form, we were able to study both Mpro and PLpro in solution where multiple conformations and oligiomeric forms are present. Pelitinib is compound 10 in our study, but our results show that the 4-aminoquinazoline class of EGFR inhibitors 8 and 9 are more promising; and importantly operate as potent PLpro, rather than Mpro, inhibitors.
By employing optimised screens, we specifically interrogated the two essential SARS-CoV-2 viral proteases, discovering compounds not identified in previous phenotypic screens despite possessing anti-viral activity. The ReFRAME collection has been screened in phenotypic viral-replication44 assays. In spite of counter screens, without deconvolution, the results from phenotypic screens can artificially prioritise highly potent compounds such as transcription inhibitors and cytotoxic compounds that have undesirable mechanisms of action precluding therapeutic development45 along with undervaluing the potency of viable compounds. Compounds that require revised assay protocols to observe activity, such as 4, were therefore not previously identified.
In vitro viral replication assays are of limited value for predicting the in vivo pharmacodynamics of candidate molecules46 where multi-day assays often underestimate the true in vivo potency. For compounds such as 4, where a confounding factor is the aqueous stability of the molecule, in vitro data serve to support the mechanism rather than predict in vivo efficacy, where administration frequency, route and immune clearance would positively influence potency47,48.
Similarly, potencies of anti-viral activity can vary drastically depending on the methods used. A recent study of the PLpro tool inhibitor GRL-06178 saw potency vary by two orders of magnitude between biochemical (IC50 of 2 µM), cytopathic-effect (30 µM), viral RNA detection (> 50 µM) and FFU (IC50 > 100 µM) assays. Consequently, there is a prospect that the potencies of both 4 and 8 in vivo may be better than implied by the cytopathic-effect antiviral measure used in this work.
There have been three widespread outbreaks of fatal novel respiratory coronavirus mediated disease in the last two decades49. Retrospectively, the dangers of further outbreaks were evident following the first50. To avoid the debilitating effects of future coronavirus pandemics or even escape from immune protection, a range of treatments are necessary in which effective antiviral drugs will be a critical component. Protease inhibitors have been highly successful in combating other viral infections51. The high conservation of Mpro and PLpro between the three strains of coronavirus which cause greatest impact on human health suggest that these are excellent target opportunities for developing small-molecule anti-viral therapeutics.
Anti-viral efforts aim to treat patients who are already infected and halt progression to severe disease6. This serves to reduce the burden of disease on fragile health care systems, but must also be employed alongside vaccination3 and containment efforts52. Vaccination and containment serve to prevent the potential for infection, whereas anti-viral aim to treat those already infected. To be truly useful anti-viral medicines must be broad spectrum and stockpiled prior to an outbreak as suggested for influenza53.
Our studies describe the discovery of potent, drug-like inhibitors for both Mpro and PLpro. These inhibitors display in vitro antiviral activity and have already been shown to be safe for clinical investigation for other therapeutic areas. Given their existing preclinical safety profiles these compounds have the potential for rapid progression towards a clinical setting.
Methods
Materials
The ReFRAME library was received from Calibr, Scripps Research, as compounds dissolved to 10 mM in DMSO, spotted in 30 nL volumes in black 384 well plates. All peptides used were prepared with C-terminal amides from Cambridge Research Biochemicals (Billingham, UK) and provided at > 95% purity. Cambridge Research Biochemicals (Billingham, UK) synthesized the ester and acid forms of SDZ-224015 (compounds 4 & 5) used in follow-up studies, provided at > 95% purity. Additional compound 4 was synthesised as described below. pelitinib (10), afatinib (11), dacomitinib (12) and gefitinib (13) were obtained from Tocris (Bristol, UK). The active form of Tarloxotinib (9) was from Molport (Riga, Latvia). Compound 3 was from Santa Cruz Biotechnology (Dallas, Texas, USA), Compounds 2, 7 and 8 were from MedChem Express (Sweden); Compound 6 was from Bachem (Bubendorf, Switzerland).
The African monkey kidney cell line Vero E6-GFP was a gift kindly provided by M. van Loock, Janssen Pharmaceutica, Beerse, Belgium.
The hepatocellular carcinoma cell line Huh7 was a gift kindly provided by Ralf Bartenschlager, University of Heidelberg, Germany.
All compounds were obtained at a manufacturer specification of > 98% purity.
Unless otherwise stated all other reagents were from Sigma Aldrich (Poole, UK).
Construct design and construction
The Mpro coding sequence was codon optimised for expression in E. coli and synthesised by Integrated DNA technologies (IDT). The Mpro expression construct used for crystallization comprises an N-terminal GST region, an Mpro autocleavage site, the Mpro coding sequence, a hybrid cleavage site recognizable by 3C HRV protease and a C-terminal 6-Histidine tag54. The overall construct was flanked by In-Fusion compatible ends for insertion into BamHI-XhoI cleaved pGEX-6P-1 (Sigma). An additional Mpro construct was generated with an extended 10-Histidine tag, for enhanced binding to the sensor surface in SPR assays. This construct was amplified by PCR from the above version, with the C-terminal primer incorporating a further 4-Histidines. The resulting amplicon was then inserted into BamHI-XhoI cleaved pGEX-6P-1 by In-Fusion cloning.
The PLpro expression construct was similarly optimised and synthesised and comprised an N-terminal 10 Histidine tag followed by the PLpro sequence (Nsp3 region E746-K1060). This was then directly inserted into NcoI-HindIII digested pOPINF via In-Fusion compatible ends. pOPINF was a gift from Ray Owens (University of Oxford)55 (Addgene plasmid # 26042 ; http://n2t.net/addgene:26042 ; RRID:Addgene_26042).
Protein expression of Mpro with authentic termini
The plasmids were used to transform a competent E. coli expression cell line based on BL21(DE3)-R3-pRARE. The cells were plated on LB-agar plates containing 50 µg/ml carbenicillin and incubated overnight at 37 °C. The next day multiple colonies were picked and use to inoculate a series of consecutive starter cultures (LB, 50 µg/ml Carbenicillin) of 1 ml, 10 ml and 100 ml. At each stage the culture was grown to the exponential phase (OD600 0.6–2, 200 rpm, 37 °C) before using the total volume of culture to inoculate the next, where the inoculate comprised 10% of the volume of the next culture in the series. Once 100 ml of exponential culture was achieved, 10 ml of this was used to inoculate 1 L of Auto Induction medium (Formedium, Terrific broth base including trace elements, prepared to manufacturer’s instructions with addition of 10 ml glycerol and 50 µg/ml carbenicillin). Cultures were grown for 5 h at 37 °C, 200 rpm, followed by 15–20 h at 18 °C, 200 rpm. Cells were harvested by centrifugation and stored at -80 °C.
Protein purification of Mpro with authentic termini for crystallographic analysis
Cells were resuspended in lysis buffer, 50 mM Tris pH 8, 300 mM NaCl, 10 mM imidazole, 0.03 μg/ml Benzonase, and lysed using an Emulsiflex homogeniser (3 passes, 30 kpsi, 4 °C). Insoluble material was removed by centrifugation (50,000 g, 4 °C). Tagged Mpro protein was captured using Nickel-NTA (Takara His60 Superflow Resin) washed with 50 mM Tris pH 8, 300 mM NaCl, 25 mM imidazole, and eluted with 50 mM Tris pH 8, 300 mM NaCl, 500 mM imidazole. To remove the Mpro poly-histidine tag, N-terminal His tagged HRV 3C protease was added to the eluted Mpro fractions at a ratio of 1 mg 3C protease: 10 mg Mpro. The mixture was dialysed overnight into 50 mM Tris pH 8, 300 mM NaCl, 0.5 mM TCEP at 4 °C and purified by reverse Nickel-NTA. Gel filtration was performed using a 16/600 Superdex S200 pg column (GE Healthcare) equilibrated in 50 mM Tris pH 8, 300 mM NaCl buffer. Mpro was concentrated to 36 mg/ml using a centrifugal filter device with a 10 kDa molecular weight cut off prior to flash freezing using liquid nitrogen.
Expression and purification of Mpro-His10
Mpro-His10 was prepared as for Mpro with authentic termini with the following modifications. The HRV 3C protease cleavage and reverse Ni–NTA steps were omitted. Instead, the Ni–NTA purified tagged Mpro was dialysed into 50 mM Tris pH 8.5, 100 mM NaCl, 0.5 mM TCEP at 4 °C overnight. The dialysed sample was rapidly diluted with 50 mM Tris pH 8.5 buffer to achieve a final NaCl concentration of 25 mM. The protein was purified by anion exchange chromatography using a 5 ml HiTrap Q HP column (GE Healthcare) on a NaCl concentration gradient between 25 mM to 0.5 M NaCl. Ion exchange chromatography was followed by a gel filtration purification step as described above.
Expression and purification of cleaved PLpro
Cleaved PLpro was prepared as for Mpro with authentic termini with the following modifications. A tunable T7 expression strain based on Lemo21 (DE3) was utilised in the expression of PLpro PLpro and 34 μg/ml chloramphenicol was added to all solid and liquid media to maintain its pLemo plasmid. 2 mM Rhamnose was included in the medium in the overnight and sub-culture stages. 0.5 mM Rhamnose was included in the auto induction medium. Instead of the HRV 3C protease, TEV protease was used to cleave the 10 Histidine tag of PLpro at the same mass ratio as described above. A 16/600 Superdex S75 pg column (GE Healthcare) was used for gel filtration chromatography.
Expression and purification of His10-PLpro
His10-PLpro was prepared as for cleaved PLpro with the following modifications. The TEV protease cleavage and reverse Ni–NTA steps were omitted. Instead, the Ni–NTA purified tagged PLpro was dialysed into 50 mM Tris pH 8.8, 100 mM NaCl, 0.5 mM TCEP at 4 °C overnight. The dialysed sample was rapidly diluted with 50 mM Tris pH 8.8 buffer to achieve a final NaCl concentration of 25 mM. The protein was purified by anion exchange using a 5 ml HiTrap Q HP column (GE Healthcare) with a NaCl concentration gradient between 25 mM to 0.5 M NaCl. The ion exchange chromatography was followed by a gel filtration purification step as described above.
Mpro characterisation
The Mpro substrate, [5-TAMRA]-AVLQSGFR-[Lys(BHQ-2)]-K-amide was disolved in DMSO (10 mM). This solution was diluted in buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA, 0.5 mM TCEP, 0.05% (v/v) Triton X-100, pH 7.6), then mixed with Mpro solution, to give a final substrate concentration range of 0.8–100 µM and Mpro concentration range of 0.3–300 nM. Initial rates were measured using a Pherastar FSX plate reader equipped with a TAMRA filter set.
Mpro inhibition assays
Compounds were received for ReFRAME screening at 10 mM in DMSO at 30 nL volumes in black 384 wells plates; otherwise, dilutions were performed in DMSO from 10 mM stock solutions; 300 nL was transferred to a black 384 well plate using a Mosquito liquid handler (SPT Labtech, Melbourne, UK). 10 µL of the Assay Buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA, 0.5 mM TCEP, 0.05% (v/v) Triton X-100, pH 7.6) was added to the assay plates to solubilise compounds. 10 µL Mpro, 90 nM in Assay Buffer, was added to the appropriate wells and incubated with the compounds for 60 min at room temperature. The reaction was started via the addition of 10 µL of the substrate [5-TAMRA]-AVLQSGFR-[Lys(BHQ-2)]-K-amide (where BHQ is Black hole quencher 2), 18 µM in assay buffer. This resulted in final assay conditions of 30 nM Mpro and 6 µM substrate, with either 0.1% or 1% DMSO (v/v). The plates were incubated for a further 60 min at room temperature; assays employed a Pherastar FSX plate reader (BMG Labtech, Aylesbury, UK) using a TAMRA filter set. Assay quality was established using Z’, where a low control consisted for substrate alone with balanced DMSO, and the high control was enzyme and substrate without inhibitors but balanced DMSO. The raw fluorescence data was converted to percent inhibition using the same controls as for Z’.
Mpro SPR assay
Cytiva Biacore S200 and T200 machines were used for all SPR experiments. Data were collected at a constant temperature of 20 °C. Mpro-10His was captured on an NTA chip using standard protocols in running buffer: 20 mM Hepes (pH 7.5), 150 mM NaCl, 50 µM EDTA, 0.05% (v/v) Tween 20 and either 1 or 3% (v/v) DMSO at ~ 8500 RU. The compounds were screened at concentrations ranging from 23 nM to 50 µM adjusted appropriately for each compound, injecting from the lowest to highest concentrations. Scrubber 2 (Biologic software) was used to process and analyse SPR data. Kinetics were fitted using a 1:1 binding model with local Rmax for each concentration where required. Data for the inhibitors were referenced to those for a blank surface and blank injections to normalize for non-specific binding and drift. A DMSO calibration was run to remove excluded volume effect of binding responses between reference and target surface.
Mpro protein observed mass spectroscopy
Protein MS-analyses were performed as described56 using a RapidFire RF 365 high-throughput sampling robot (Agilent) attached to an iFunnel Agilent 6550 accurate mass quadrupole time-of-flight (Q-TOF) mass spectrometer operating in the positive ionization mode with the parameters: capillary voltage (4000 V), nozzle voltage (1000 V), fragmentor voltage (365 V), gas temperature (225 °C), gas flow (13 L/min), sheath gas temperature (350 °C), sheath gas flow (12 L/min). The reaction was initiated either by adding SDZ-224015 (compound 4; 10 mM in DMSO) to a final concentration of 2.5 µM into reaction buffer (20 mM HEPES, pH 7.5, 50 mM NaCl) containing 2.5 µM Mpro or by adding Mpro (15 µM in 20 mM HEPES, pH 7.5, 300 mM NaCl) to a final concentration of 1 µM into reaction buffer (20 mM HEPES, pH 7.5, 50 mM NaCl) containing SDZ-224015 (5 µM). A sample from the reaction mixture was directly aspirated under vacuum (0.6 s) and loaded onto a C4 solid phase extraction (SPE) cartridge. After loading, the C4 SPE cartridge was washed with 0.1% (v/v) aqueous formic acid to remove non-volatile buffer salts (5.5 s, 1.5 mL/min) and the protein was then eluted from the SPE cartridge with 0.1% (v/v) aqueous formic acid in 85/15 (v/v) acetonitrile/water into the mass spectrometer (5.5 s, 1.25 mL/min). The SPE was cartridge re-equilibrated with 0.1% (v/v) aqueous formic acid (0.5 s, 1.25 mL/min) and a blank water sample was injected before the next reaction sample was aspirated from the assay mixture. Protein spectra were deconvoluted (mass range: 10–60 kDa, m/z range: 950–1300 Da, mass step: 1 Da) using the MaxEnt1 function in Agilent MassHunter Version 7 (Agilent), normalised, and plotted using Graphpad Prism 5.
Crystallisation and structure determination of Mpro in complex with compounds 1 and 5
Mpro was thawed and diluted to 6 mg/ml using 20 mM Hepes pH 7.5, 50 mM NaCl. The ligand of interest was dissolved in DMSO to 10 mM and then diluted into the protein solution to a final concentration of 1 mM. The ligand was then allowed to incubate with the protein for two hours at room temperature prior to dispensing plates. The drop composition was 0.15 µL protein ligand solution, 0.3 µL 11% (v/v) PEG 4 K, 0.1 M MES pH 6.5, and 0.05 µL Mpro crystal seed stock. The Mpro crystal seed stock was prepared by crushing Mpro crystals with a pipette tip, suspending them in 30% PEG 4 K, 5% (v/v) DMSO, 0.1 M MES pH 6.5, and vortexing for 60 s with approximately 10 glass beads (1.0 mm diameter, BioSpec products). Reservoir solution was 11% (v/v) PEG 4 K, 5% (v/v) DMSO, 0.1 M MES pH 6.5. Crystals were grown using the sitting drop vapor diffusion method at 20 °C and appeared within 24 h, reaching full size within 36 h.
Data collection and structure determination
All diffraction data were collected from crystals cryo-cooled to 100 K at Diamond Light Source. X-ray diffraction data for the Mpro compound 1 complex were collected at beamline I04-1 at a wavelength of 0.9126 Å and data for the Mpro compound 5 complex were collected at I24 at 0.9999 Å. Data were processed using Dials57 via Xia258. The datasets were phased using Molrep59 and the Mpro apo structure60. Ligand restraints were generated using GRADE (Global Phasing Ltd) and AceDRG61. Crystal structures were manually rebuilt in Coot62 and refined using Refmac63 and Buster64.
PLpro enzyme characterisation
50 nM PLpro was incubated with titrations of either Ubiquitin-Rhodamine (RnD Systems, Abingdon, UK) (0.1–10 µM) or [5-TAMRA]- VLRLRGG-[Lys(BHQ-2)]-amide (1–100 µM) in either 150 mM NaCl, 50 mM Tris, 1 mM EDTA, 0.5 mM TCEP, 0.05% (v/v) Triton X-100, pH 7.6 or 800 mM sodium citrate, 200 mM Tris, 0.5 mM TCEP, 0.05% (v/v) Triton X-100, pH 7.6. Initial rate was established using a Pherastar FSX plate reader using either a FITC (Ubiquti-Rhodamine) or a TARMA ([5-TAMRA]- VLRLRGG-[Lys(BHQ-2)]-amide) filter set.
PLpro biochemical assay
Compound plates were prepared as for the Mpro biochemical assay. 10 µL assay buffer (800 mM sodium citrate, 200 mM Tris, 0.5 mM TCEP, 0.05% (v/v) Triton X-100, pH 7.6) was added to the assay plates to solubilise the compound. 10 µL of PLpro at 75 nM in assay buffer, was added to the appropriate wells and incubated with the compounds for 60 min at room temperature. The reaction was started via the addition of 10 µL of the substrate [5-TAMRA]- VLRLRGG-[Lys(BHQ-2)]-amide at 6 µM in assay buffer. This resulted in final assay conditions of 25 nM PLpro and 2 µM substrate, with either 0.1% or 1% DMSO (v/v). The plates were incubated for a further 60 min at room temperature and the assay was read on a Pherastar FSX plate reader (BMG Labtech, Aylesbury, UK) using a TAMRA filter set. Assay quality was established as for the Mpro biochemical assay. Data were converted to percent inhibition in the same manner as the Mpro assay.
PLpro kinetic assays
Assays employed the same conditions as the PLpro inhibition assays, except the compound and substrate were prepared in the assay plate and the assay was started with an injection of 10 µL of enzyme using a Pherastar FSX plate reader. Fluorescence was measured every 15 s post injection using a TAMRA filter set for 60 min. Data were zeroed to the date measured at time of injection and fit to a two-state inhibition model.
Effect of hoffmeister salt concentration on PLpro activity
[5-TAMRA]- VLRLRGG-[Lys(BHQ-2)]-amide was dissolved in buffers with increasing concentration of sodium phosphate (0.15–1.5 M sodium phosphate, 1 mM EDTA, 0.5 mM TCEP, 0.05% Triton-X100, pH 7.4). This was then mixed with PLpro in a matched buffer to give final conditions of: 2 µM substrate and 30 nM enzyme. The initial rate was established using a Pherastar FSX plate reader using either a FITC (Ubiqutin-Rhodamine) or a TARMA ([5-TAMRA]-VLRLRGG-[Lys(BHQ-2)]-amide) filter set.
Effect of Hoffmeister salt concentration on tarloxotinib bromide inhibition
This assay was performed in the same manner as the PLpro biochemical assay, except the buffer was either 0.15, 0.75, 1 or 1.5 M sodium phosphate, 1 mM EDTA, 0.5 mM TCEP, 0.05% Triton-X100 pH7.4.
Cell culture
Huh-7 cells stably expressing H2B-mCherry were generated using lentiviral vectors containing a CMV-H2B-mCherry-P2A-BlastR cassette. The Huh-7 mCherry cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% v/v fetal calf serum (FCS; Biowest), 10 ml HEPES, 5 ml NEAA, and 1 × Pen-strep (Gibco) and kept under 5% CO2 on 37 °C. Assay medium contained only 4% FCS.
Virus culture
SARS-CoV-2 strain BetaCov/Belgium/GHB-03021/2020 recovered from a nasopharyngeal swab taken from an asymptomatic patient returning from Wuhan, China at the beginning of February 2020 was sequenced on a MinION platform (Oxford Nanopore). After serial passaging on Huh7 and Vero E6 cells, infectious content of the virus stock was determined by titration on HUH7 cells using the Spearman-Kärber method. All virus-related work was carried out in certified, high-containment biosafety level-3 facilities of KU Leuven Rega institute.
Antiviral assay
To measure inhibition of the SARS-CoV-2 cytopathic effect, 96-well plates (Corning 3300) were plated with HUH7_mCherry cells at 6000 cells/well in 100 µl. The day after (Day 0), compound was added in a dilution series for concentration response studies. After two hours, addition of virus dilution (final MOI 0.004) was performed and plates were left for incubation at 37 °C, 5% CO2 for four days.
Cytotoxicity was assessed in parallel using the same protocol, albeit without the addition of virus dilution. Plates were imaged on an Arrayscan XTI, Thermofisher.
Image acquisition and analysis
At day four post-infection, mCherry signal was captured using wide field fluorescence imaging by exciting at 560_25 nm and emitting with the BGRFRN filter set. A 5 X objective sufficed to capture 65–70% of an entire well on a 96well plate (4 pictures in total). The optimal exposure time was determined based on fluorescence intensity and was set on 0.09 s. A 2 × 2 binning was used and autofocus plane count was reduced to increase image acquisition speed. An image analysis protocol was developed in-house by using the SpotDetector bioapplication (Cellomics, Thermofisher). After background reduction on the raw image files, a fixed fluorescent intensity threshold was determined for the identification of mCherry cells. Afterwards, the number of fluorescent cells (‘object count’) was calculated per well and compared to the positive (cell control) and negative (virus) control.
Synthesis of SDZ-224015 (compound 4)
Commercially-sourced reagents (Sigma-Aldrich, Inc.; Fluorochem Ltd; Bachem AG) were used as received. Reactions were performed in anhydrous solvents (Sigma-Aldrich Inc.). Purifications, reaction work-ups, and extractions were performed using HPLC grade solvents (Sigma-Aldrich Inc.). A Stuart SMP-40 automated melting point apparatus was used to determine melting points (MP). A Bruker Tensor-27 Fourier transform infrared spectrometer was used for infrared (IR) spectroscopy. A Unipol (Schmidt Haensch) polarimeter was used for optical rotation (α) measurements. A Thermo Scientific Exactive mass spectrometer (ThermoFisher Scientific) operated in the positive ionization mode was employed for high-resolution mass spectrometry (HRMS) using electrospray ionization (ESI) mass spectrometry (MS); data are presented as a mass-to-charge ratio (m/z). A Bruker AVANCE AVIIIHD 600 spectrometer equipped with a 5 mm BB-F/1H Prodigy N2 cryoprobe was used for nuclear magnetic resonance (NMR) spectroscopy. Proton chemical shifts are reported in parts per million (ppm) downfield from tetramethylsilane, the residual protium in the NMR solvent is used as a reference (DMSO-d6: δ = 2.49 ppm). Carbon chemical shifts are reported in parts per million (ppm) in the scale relative to the NMR solvent (DMSO-d6: δ = 39.52 ppm). NMR data are reported as: chemical shift, multiplicity (m: multiplet, s: singlet, d: doublet, dd: doublet of doublets, t: triplet, q: quartet), coupling constant (J, Hz), and integration.
Ethyl (5S,8S,11S)-11-(2-((2,6-dichlorobenzoyl)oxy)acetyl)-5-isopropyl-8-methyl-3,6,9-trioxo-1-phenyl-2-oxa-4,7,10-triazatridecan-13-oate (SDZ-224015, Z-VAD-DCB, compound 4) was synthesized from ethyl (S)-3-((R)-2,2-dimethyl-1,3-dioxolan-4-yl)-3-(phenylamino)propanoate65 and Z-Val-Ala-OH in five steps as reported66. However, the final oxidation reaction of the reported synthesis of 466 was modified, due to the insolubility of the starting material in pure dichloromethane, the optimised protocol is given below:
A solution of ethyl (5S,8S,11S)-11-((R)-2-((2,6-dichlorobenzoyl)oxy)-1-hydroxyethyl)-5-isopropyl-8-methyl-3,6,9-trioxo-1-phenyl-2-oxa-4,7,10-triazatridecan-13-oate65 (1.4 g, 2.0 mmol, 1.0 equiv.) in DMSO (3.0 mL) was diluted with dichloromethane (40 mL). To the resulting clear solution, Dess-Martin periodinane67 (3.39 g, 8.0 mmol, 4.0 equiv.) was added at 0° C under ambient atmosphere; the reaction mixture was stirred at 0 °C for 2 h, then for 2 h at ambient temperature, before aqueous phosphate buffer (100 mL, 0.1 M, pH 7) containing sodium metabisulfite (6 g) was added at 0 °C. The resulting mixture was vigorously stirred at ambient temperature for 30 min, then five times extracted with dichloromethane. The combined organic extracts were washed with saturated aqueous NaHCO3 solution, dried over anhydrous Na2SO4, filtered, evaporated; the residue was then purified by reverse phase HPLC (20 mL/min; linear gradient over 39 min: 2% → 98% acetonitrile in water, each containing 0.1% (v/v) formic acid; tR = 27.0 min) using a Shimadzu HPLC purification system (composed of DGU-20A, 2 LC-20AR, CBM-20A, SPD-20A, and FRC-10A units) equipped with a C18 Grace VYDAC 218TP101522 column (Grace Davison Discovery Sciences) to afford 199 mg (15%) of purified SDZ-224015 (compound 4). The analytical data are in agreement with those reported66. White solid, m.p.: 178–180 °C; 1H NMR (600 MHz, 300 K, DMSO-d6): δ = 8.62 (d, J = 7.5 Hz, 1H), 8.12 (d, J = 6.6 Hz, 1H), 7.60 − 7.59 (m, 2H), 7.55 (dd, J = 9.2, 6.9 Hz, 1H), 7.36 − 7.34 (m, 4H), 7.32 − 7.29 (m, 1H), 7.26 (d, J = 8.7 Hz, 1H), 5.18 (d, J = 17.2 Hz, 1H), 5.14 (d, J = 16.4 Hz, 1H), 5.03 (d, J = 12.7 Hz, 1H), 5.00 (d, J = 12.7 Hz, 1H), 4.64 (q, J = 6.8 Hz, 1H), 4.27 (app. pent., J = 7.2 Hz, 1H), 4.05 (q, J = 7.1 Hz, 2H), 3.87 (dd, J = 7.9, 7.4 Hz, 1H), 2.85 (dd, J = 16.5, 5.8 Hz, 1H), 2.65 (dd, J = 16.5, 7.2 Hz, 1H), 1.98 − 1.92 (m, 1H), 1.22 (d, J = 7.0 Hz, 3H), 1.16 (t, J = 7.1 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H), 0.81 ppm (d, J = 6.7 Hz, 3H); 13C NMR (150 MHz, 300 K, DMSO-d6): δ = 199.5, 172.9, 171.0, 170.1, 163.2, 156.1, 137.0, 132.6, 132.0, 130.8, 128.5, 128.3, 127.7, 127.6, 67.7, 65.4, 60.3, 59.8, 52.8, 48.1, 34.2, 30.3, 19.2, 18.0, 17.7, 13.9 ppm; IR (film): ṽ = 3293, 3067, 2964, 2936, 1733, 1689, 1639, 1538, 1434, 1374, 1287, 1247, 1195, 1148, 1040 cm–1; HRMS (ESI): m/z calculated for C30H36O9N3Cl2 [M + H]+: 652.1823, found: 652.1823; \({[\alpha ]}_{D}^{25}\)= − 47.0 (c = 1.0, acetone).
Hot spot comparison between Mpro and caspase 1
Fragment Hotspot Maps were calculated for structures 6YB7 and 1SC4 using the Hotspots API38,39. The method uses molecular probes, atomic interaction propensity and a local buriedness measure to highlight key hotspots within the binding site.
Research ethics statement
All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by either, an internal research committee at Exscientia. Ltd, the ReFRAME committee or the CARE consortium. Viral swabs were obtained with prior patient's written informed consent for use in research.
Code availably statement
References pointing the reader towards code used for hotspot mapping are provided in the methods section.
Change history
28 September 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-99041-1
References
Coronavirus Disease (COVID-19) Pandemic UNFPA Global Response Plan | UNFPA - United Nations Population Fund. https://www.unfpa.org/resources/coronavirus-disease-covid-19-pandemic-unfpa-global-response-plan (n.d.).
Matsuyama, T., Kubli, S. P., Yoshinaga, S. K., Pfeffer, K. & Mak, T. W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 27, 3209–3225 (2020).
Moghadas, S. M. et al. The impact of vaccination on COVID-19 outbreaks in the United States. Medrxiv 2020 https://doi.org/10.1101/2020.11.27.20240051 (2021).
Consortium, C.-19 G. U. (COG-U. et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 1–16 (2021) https://doi.org/10.1038/s41579-021-00573-0.
Burton, D. R. & Topol, E. J. Variant-proof vaccines—invest now for the next pandemic. Nature 590, 386–388 (2021).
Villamagna, A. H., Gore, S. J., Lewis, J. S. & Doggett, J. S. The need for antiviral drugs for pandemic coronaviruses from a global health perspective. Front. Med. 7, 596587 (2020).
Frecer, V. & Miertus, S. Antiviral agents against COVID-19: structure-based design of specific peptidomimetic inhibitors of SARS-CoV-2 main protease. Rsc Adv. 10, 40244–40263 (2020).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Beigel, J. H. et al. Remdesivir for the treatment of Covid-19 - full report. New Engl. J. Med. 383, 1813–1826 (2021).
Group, R. C. et al. Dexamethasone in hospitalized patients with Covid-19. New Engl. J. Med. 384, 693–704 (2021).
Kalil, A. C. et al. Baricitinib plus Remdesivir for hospitalized adults with Covid-19. New Engl. J. Med. 384, 795–807 (2021).
Ullrich, S. & Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 30, 127377 (2020).
Cárdenas-Conejo, Y., Liñan-Rico, A., García-Rodríguez, D. A., Centeno-Leija, S. & Serrano-Posada, H. An exclusive 42 amino acid signature in pp1ab protein provides insights into the evolutive history of the 2019 novel human-pathogenic coronavirus (SARS-CoV-2). J. Med. Virol. 92, 688–692 (2020).
Kim, D. et al. The architecture of SARS-CoV-2 transcriptome. Cell 181, 914-921.e10 (2020).
Fehr, A. R. & Perlman, S. Coronaviruses, methods and protocols. Methods Mol. Biol. 1282, 1–23 (2015).
Lei, J., Kusov, Y. & Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir Res. 149, 58–74 (2018).
Dolgin, E. The race for antiviral drugs to beat COVID—and the next pandemic. Nature 592, 340–343 (2021).
Janes, J. et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. 115, 201810137 (2018).
Jasial, S., Hu, Y. & Bajorath, J. How frequently are pan-assay interference compounds active? Large-scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J. Med. Chem. 60, 3879–3886 (2017).
Matlock, M. K., Hughes, T. B., Dahlin, J. L. & Swamidass, S. J. Modeling small-molecule reactivity identifies promiscuous bioactive compounds. J. Chem. Inf. Model. 58, 1483–1500 (2018).
Baell, J. & Walters, M. A. Chemistry: chemical con artists foil drug discovery. Nat. News 513, 481 (2014).
Yang, J., Copeland, R. A. & Lai, Z. Defining balanced conditions for inhibitor screening assays that target bisubstrate enzymes. J. Biomol. Screen 14, 111–120 (2009).
Jin, Z. et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature https://doi.org/10.1038/s41586-020-2223-y (2020).
Cheng, S.-C., Chang, G.-G. & Chou, C.-Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS Coronavirus main protease. Biophys. J. 98, 1327–1336 (2010).
Datta, D., McClendon, C. L., Jacobson, M. P. & Wells, J. A. Substrate and inhibitor-induced dimerization and cooperativity in caspase-1 but not caspase-3. J. Biol. Chem. 288, 9971–9981 (2013).
Drag, M. et al. Positional-scanning fluorigenic substrate libraries reveal unexpected specificity determinants of DUBs (deubiquitinating enzymes). Biochem. J. 415, 367–375 (2008).
Snijder, E. J. et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80, 5927–5940 (2006).
Kanjanahaluethai, A., Chen, Z., Jukneliene, D. & Baker, S. C. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 361, 391–401 (2007).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90 (2012).
Jantos, K. et al. Discovery of ABT-957: 1-Benzyl-5-oxopyrrolidine-2-carboxamides as selective calpain inhibitors with enhanced metabolic stability. Bioorg. Med. Chem. Lett. 29, 1968–1973 (2019).
Elford, P. R., Heng, R., Révész, L. & MacKenzie, A. R. Reduction of inflammation and pyrexia in the rat by oral administration of SDZ 224-015, an inhibitor of the interleukin-1β converting enzyme. Brit. J. Pharmacol. 115, 601–606 (1995).
Yu, H. et al. Abstract 4025: STEAP4 ISH and IHC diagnostics for Tarloxotinib activation in EGFR/HER2 mutant cancers. 4025–4025 (2019) https://doi.org/10.1158/1538-7445.am2019-4025.
Liu, S. V. et al. Phase 2 study of tarloxotinib bromide (TRLX) in patients (pts) with EGFR-Mutant, T790M-Negative NSCLC progressing on an EGFR TKI. J. Clin. Oncol. 34, TPS9100–TPS9100 (2016).
Ismail, R. S. M., Ismail, N. S. M., Abuserii, S. & Ella, D. A. A. E. Recent advances in 4-aminoquinazoline based scaffold derivatives targeting EGFR kinases as anticancer agents. Futur. J. Pharm. Sci. 2, 9–19 (2016).
Ma, C. et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 30, 678–692 (2020).
Dinarello, C. A., Simon, A. & van der Meer, J. W. M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 11, 633–652 (2012).
King, A. et al. Anakinra in COVID-19: important considerations for clinical trials. Lancet Rheumatol. 2, e379–e381 (2020).
Curran, P. R. et al. Hotspots API: a python package for the detection of small molecule binding hotspots and application to structure-based drug design. J. Chem. Inf. Model. https://doi.org/10.1021/acs.jcim.9b00996 (2020).
Radoux, C. J., Olsson, T. S. G., Pitt, W. R., Groom, C. R. & Blundell, T. L. Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325 (2016).
Tarighi, P. et al. A review of potential suggested drugs for coronavirus disease (COVID-19) treatment. Eur. J. Pharmacol. 895, 173890 (2021).
Kumalo, H. M., Bhakat, S. & Soliman, M. E. S. Theory and applications of covalent docking in drug discovery: merits and pitfalls. Molecules 20, 1984–2000 (2015).
Ramírez, D. & Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data?. Molecules 23, 1038 (2018).
Günther, S. et al. Inhibition of SARS-CoV-2 main protease by allosteric drug-binding. Biorxiv 2020 https://doi.org/10.1101/2020.11.12.378422 (2020).
Riva, L. et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature https://doi.org/10.1038/s41586-020-2577-1 (2020).
Moffat, J. G. Turning the light on in the phenotypic drug discovery black box. Cell Chem. Biol. 24, 545–547 (2017).
Ferguson, N. M., Fraser, C. & Anderson, R. M. Viral dynamics and anti-viral pharmacodynamics: rethinking in vitro measures of drug potency. Trends Pharmacol. Sci. 22, 97–100 (2001).
Benet, L. Z. Effect of route of administration and distribution on drug action. J. Pharmacokinet. Biop. 6, 559–585 (1978).
Berti, A., Rose, W., Nizet, V. & Sakoulas, G. Antibiotics and innate immunity: a cooperative effort towards the successful treatment of infections. Open Forum Infect. Dis. 7, 302 (2020).
Zhu, Z. et al. From SARS and MERS to COVID-19: a brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 21, 224 (2020).
Cheng, V. C. C., Lau, S. K. P., Woo, P. C. Y. & Yuen, K. Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 20, 660–694 (2007).
Patick, A. K. & Potts, K. E. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 11, 614–627 (1998).
Murano, Y. et al. Impact of domestic travel restrictions on transmission of COVID-19 infection using public transportation network approach. Sci. Rep. UK 11, 3109 (2021).
Monto, A. S. The role of antivirals in the control of influenza. Vaccine 21, 1796–1800 (2003).
Xue, X. et al. Production of authentic SARS-CoV Mpro with enhanced activity: application as a novel tag-cleavage endopeptidase for protein overproduction. J. Mol. Biol. 366, 965–975 (2007).
Berrow, N. S. et al. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 35, e45–e45 (2007).
Malla, T. R. et al. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 Mpro inhibitors. Chem. Comms. 57, 1430–1433 (2021).
Winter, G. et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. Sect. D Struct. Biol. 74, 85–97 (2018).
Winter, G., Lobley, C. M. C. & Prince, S. M. Decision making in xia2. Acta Crystallogr. Sect. D Biol. Crystallogr. 69, 1260–1273 (2013).
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 22–25 (2010).
Alice, et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Commun. 11(1), https://doi.org/10.1038/s41467-020-18709-w (2020)
Long, F. et al. AceDRG: a stereochemical description generator for ligands. Acta Crystallogr. Sect. D Struct. Biol. 73, 112–122 (2017).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 355–367 (2011).
G. B. et al. Buster v. 2.10.13. (2017).
Matsunaga, H., Sakamaki, T., Nagaoka, H. & Yamada, Y. Enantioselective synthesis of (R)- and (S)-4-[(methoxycarbonyl)-methyl]-2-azetidinones from D-glyceraldehyde acetonide. Tetrahedron Lett. 24, 3009–3012 (1983).
Revesz, L. et al. Synthesis of P1 aspartate-based peptide acyloxymethyl and fluoromethyl ketones as inhibitors of interleukin-1β-converting enzyme. Tetrahedron Lett. 35, 9693–9696 (1994).
Dess, D. B. & Martin, J. C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 48, 4155–4156 (1983).
Acknowledgements
We would like to thank Diamond MX group in general and in particular here I24 and I04-1 beamline staff for support and access to the MX beamlines at Diamond Light Source. Part of this research work was performed using the 'Caps-It' research infrastructure (project ZW13-02) that was financially supported by the Hercules Foundation and Rega Foundation, KU Leuven.
Author information
Authors and Affiliations
Contributions
M.A.R Supervised and coordinated research, designed HTS screens and analysed biochemical data, enzyme kinetics, writing original draft and review, editing of manuscript. M.A.W. Supervised and coordinated research, analysed crystallographic data, writing original draft and review, editing of manuscript. L.B. Synthesis of Compound 4, coordinated protein observed MS, writing original draft and review, editing of manuscript. A.H.C. Carried out HTS and biochemical follow-up experiments. C.D.O. Crystallographic data collection, structure solution and data analysis, writing original draft and review, editing of manuscript. P.L. & C.S-D. Cloning, protein production, data analysis, writing original draft and review, editing of manuscript. I.N. S.R. & P.S. Performed & Analysed SPR experiments. P.C. & C.R. Analysed crystallographic data, hot spot analysis, writing original draft and review, editing of manuscript. T.R.M. & A.T. Performed protein observed MS experiments. L.V. Performed the anti-viral assays. T.V. & H.J. generated the cell line. P.L. Supervised the anti-viral assays. T.N & M.H. Supplied the compounds. D.S & C.J.S. Supervised and coordinated research, writing original draft and review, editing of manuscript. A.H., M.S., & D.H. Conceived the work, Hit Selection, writing original draft and review, editing of manuscript. D.F. crystallographic data collection, data analysis. A.D., D.F & F.v.D. Resources.
Corresponding authors
Ethics declarations
Competing interests
Exscientia Ltd. holds patents corresponding to the use of 1, 4, 5 and 8–13 for the treatment of coronavirus infection. The other authours have no competing interests to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained errors in Table 1, where the molecules were incorrectly displayed.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Redhead, M.A., Owen, C.D., Brewitz, L. et al. Bispecific repurposed medicines targeting the viral and immunological arms of COVID-19. Sci Rep 11, 13208 (2021). https://doi.org/10.1038/s41598-021-92416-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-92416-4
This article is cited by
-
A machine learning method for the identification and characterization of novel COVID-19 drug targets
Scientific Reports (2023)
-
Ligand-based and structure-based studies to develop predictive models for SARS-CoV-2 main protease inhibitors through the 3d-qsar.com portal
Journal of Computer-Aided Molecular Design (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
