
Abstract
PrPC variation at residue 96 (G/S) plays an important role in the epidemiology of chronic wasting disease (CWD) in exposed white-tailed deer populations. In vivo studies have demonstrated the protective effect of serine at codon 96, which hinders the propagation of common CWD strains when expressed in homozygosis and increases the survival period of S96/wt heterozygous deer after challenge with CWD. Previous in vitro studies of the transmission barrier suggested that following a single amplification step, wt and S96 PrPC were equally susceptible to misfolding when seeded with various CWD prions. When we performed serial prion amplification in vitro using S96-PrPC, we observed a reduction in the efficiency of propagation with the Wisc-1 or CWD2 strains, suggesting these strains cannot stably template their conformations on this PrPC once the primary sequence has changed after the first round of replication. Our data shows the S96-PrPC polymorphism is detrimental to prion conversion of some CWD strains. These data suggests that deer homozygous for S96-PrPC may not sustain prion transmission as compared to a deer expressing G96-PrPC.
Similar content being viewed by others

Introduction
Chronic wasting disease (CWD) is a prion disease affecting multiple species of cervids in North America, South Korea, and Scandinavia. Prion diseases, which also include scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle and Creutzfeldt-Jakob disease (CJD) in humans, are fatal diseases produced by the aberrant misfolding of the cellular prion protein (PrPC) into an infectious isoform (PrPCWD) which accumulates in the brain causing neurodegeneration1. PrPCWD is resistant to protease K (PK) digestion producing a resistant core referred to as PrPRes [reviewed by2]. PrPC is encoded in all mammalian species by the PRNP gene and is expressed in numerous organs with the highest levels found in the central nervous system (CNS)3. Unlike most prion diseases, CWD is highly contagious among farmed and wild animals, spreading through animal-to-animal interactions and exposure to contaminated environments4,5,6,7.
The primary sequence of the prion protein is one of the main determinants of the susceptibility to prion diseases in numerous species, including humans, mice, sheep, goats and cervids. In sheep, the susceptibility to classical scrapie is largely regulated by amino acid variations (polymorphisms) at positions 136, 154 and 171 of the prion protein. The expression of alanine (A), arginine (R), arginine (R) at these positions provides great resistance to the disease8,9,10,11. Accordingly, breeding programs aimed at eliminating animals expressing susceptible alleles and increasing the frequency of animals expressing the A136/R154/R171 haplotype have been implemented in sheep herds from countries where scrapie is endemic. This selective breeding for resistance has resulted in a significant decrease in scrapie prevalence, reducing it to almost zero in some cases12,13,14. A similar approach has been proposed to control and/or eradicate classical scrapie in goat populations. This breeding program, although not yet implemented, is based on the selection of animals expressing the K222 PRNP allele, which provides a level of resistance to scrapie equivalent to ARR in sheep15,16,17,18.
In cervids, there is also a close relationship between polymorphisms of the PRNP gene and CWD infection status. Supplementary Figure 1 shows sequence alignment of PrPC polymorphisms in cervids commonly affected by CWD. The most common PRNP allele found in white-tailed deer populations affected by CWD encodes PrPC molecules with glutamine (Q), glycine (G), alanine (A) and glutamine (Q) at positions 95, 96, 116 and 226, respectively, and is designated as the wild-type (wt)19,20,21,22,23,24,25. Epidemiological studies in CWD endemic areas have shown that white-tailed deer expressing certain amino acid variations such as S96, H95, and G116 are underrepresented among CWD positive animals suggesting a protective effect against the disease26,27,28,29,30. The protective effect of S96 and H95 alleles was further demonstrated by experimental oral infection in white-tailed deer expressing these amino acid substitutions19. Among the alleles of the PRNP gene associated with a lower CWD incidence and extended preclinical phase, S96 has the highest allelic frequency (~ 25%) after the wt allele in several white-tailed deer populations from the United States and Canada26,27,31. Subsequent independent transmission and epidemiological studies have demonstrated that deer homozygous and heterozygous for S96-PrPC are, compared to wt/wt deer, less susceptible to CWD infection, present prolonged survival times, show delayed prion accumulation and are generally at a significantly earlier stage of disease when deer herds are depopulated23,31,32,33.
Intracerebral challenge of mice expressing S96-PrPC with numerous CWD isolates has resulted in no disease21,22 or incomplete attack rates34, corroborating the impact of this polymorphism on CWD infection. The same S96 (tg60) mouse line is, however, susceptible to the challenge with the CWD strain H95+, which originated from the passage of Wisc-1 CWD prions from wt/wt deer into deer expressing H95-PrPC25. Natural infection of white-tailed deer expressing G116-PrPC also resulted in different strains35. These polymorphisms have been reported at relatively lower frequencies in wild and captive deer populations27,28,30,31. By contrast, the S96-PrPC allele could become dominant in white-tailed deer populations exposed to CWD within a short time frame36.
Here, we compared the efficiency with which different CWD strains induce and maintain the in vitro misfolding of white-tailed deer G96- and S96-PrPC molecules. Previously, cell-free conversion assays showed that S96-PrPC can be converted into PrPCWD as efficiently as wt-PrPC when incubated with CWD seeds from different cervid species37. Similarly, we observed that deer wt-PrPC and S96-PrPC are converted by different CWD strains with similar efficiencies after one round of PMCA. However, while all these CWD strains self-propagate efficiently in the wt-PrPC, they could not sustain serial propagation in the S96-PrPC substrate. These results suggest that the most common CWD strains cannot stably template their conformation on S96-PrPC, leading to the extinction of prion conversion after a few rounds of in vitro amplification in this substrate. These results reinforce the importance of the S96 allele as a candidate for selective breeding programs aimed at controlling and eradicating CWD in wild and farmed white-tailed deer populations.
Results
CWD strains replicate with similar efficiencies in a single round of PMCA with wild type and S96-PrPC
Traditionally, the method for quantitatively estimating the infectivity of a prion isolate is end-point dilution titration in animals, generally transgenic mice expressing the prion protein of interest. This method is, however, time-consuming, and expensive. Therefore, numerous studies have relied on protein misfolding cyclic amplification (PMCA) to estimate prion infectivity in vitro. PMCA allows precise titration of prion agents in a few days significantly reducing the number of animals required38,39,40,41.
We used PMCA to estimate the infectivity of different CWD strains by determining their limiting dilution (i.e., the highest dilution capable of producing detectable prion amplification) in brain substrates prepared from tg33 and tg60 mice. Tg33 mice express deer wt-PrPC (i.e., G96-PrPC) at levels ~ onefold compared to deer brain, whereas the PrPC expression level in tg60 mouse brain is about 30% lower than in tg3321,22. Therefore, to obtain more comparable results, we adjusted the PrPC content of the tg33 substrates by diluting the tg33 brain homogenate in brain homogenate from Prnp0/0 mice.
To compare the propagation efficiency of different CWD strains in wt and S96-PrPC, 10% (wt/vol) brain homogenates from cervids infected with Wisc-1, H95+25 or CWD242 strains, and a brain homogenate from a mule deer naturally infected with CWD (MD inoculum) were used to prepare serial tenfold dilutions.
The Wisc-1 and H95+ strains were obtained from two experimentally inoculated white-tailed deer, one wt (Q95G96) homozygous and another expressing different H95/S96 PrPC molecules, respectively19,25. The specific PrPCWD conformations of these two strains have been linked to differences in host range and reproducible neuropathological, biochemical and biophysical signatures upon serial transmission into experimental hosts24,25,43. The CWD2 strain was obtained from orally inoculated elk of the MM132 genotype44 and it was classified as a different strain based on incubation periods and histopathological features in transgenic mice42. The MD inoculum was obtained from a naturally infected free ranging wt/wt mule deer from Alberta, Canada and was included for comparison with the Wisc-1 given the similarity in PrPC primary sequence and PrPRes. Bioassay characterization of this MD isolate is ongoing in tg33 and tg60 mice.
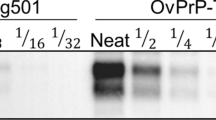
Aliquots from each dilution were used to seed the wt or S96 substrates. After a single round of 24 h of PMCA, reactions were analyzed for PrPCWD amplification. Following one round of PMCA, both Wisc-1 and H95+ prions were detectable up to the 10−5 dilution in the wt substrate and up to the 10−4 dilution in the S96 substrate (Fig. 1). Elk CWD2 prions propagated with the same efficiency (10−4 dilution) in both substrates. MD CWD only propagated in the wt substrate and only up to the 10−3 dilution, suggesting that white-tailed deer prions propagate more efficiently in both substrates. Alternatively, the Wisc-1 prions have higher seeding activity because they were obtained from a terminally ill experimental animal while the MD isolate was from a hunter-harvested deer submitted for CWD surveillance. Interestingly, when compared by western blot, the MD isolate had approximately twice the PrPRes content compared to the Wisc-1 (not shown). Our results suggest that different CWD isolates propagate with similar efficiencies in both wt and S96 substrates after one round of in vitro prion amplification.

PMCA amplification efficiency of different CWD isolates in wild-type (wt) and S96-PrPC substrates. Serial dilutions (10−3 to 10−8) of white-tailed deer CWD strains Wisc1 and H95+, CWD2 strain from elk and a CWD isolate from a naturally infected mule deer (MD) were subjected to PMCA in wt-PrPC or S96-PrPC brain homogenate substrates (tg33 and tg60 mouse brain homogenates, respectively). PMCA products were digested with PK (50 µg/ml) and analyzed by western blot. Wisc-1 and H95+ strains propagated more efficiently in the wt-PrPC substrate. CWD2 strain propagated with the same efficiency in both wt and S96-PrPCsubstrates. A 10-2 and a 10-3 dilution of the CWD2 strain in brain homogenate, not subjected to PMCA, is shown as a control of PMCA amplification. MD CWD only propagated in the substrate expressing wt-PrPC. mAb Sha31 1:10,000.
Wisc-1 and elk CWD2 attenuation through serial PMCA on S96-PrPC
Serial PMCA is a technique that enables replication of the abnormal, pathological prion protein (PrPCWD in CWD) in vitro. After one round of PMCA, an aliquot of the amplified sample is diluted tenfold into fresh brain homogenate substrate and subjected to another round of PMCA. This process can be repeated, obtaining newly converted PrPCWD which is able to induce PrP conversion with similar efficiency as brain-derived PrPCWD45.
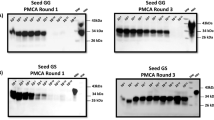
Wisc-1, H95+, CWD2 and MD CWD prions were subjected to 4 rounds of PMCA, in triplicate, in wt or S96-PrPC brain substrates. Analysis of the last PMCA round demonstrated that all CWD isolates propagated efficiently in the wt-PrPC substrate, showing abundant PrPCWD that was biochemically indistinguishable from the original brain-derived seed. Surprisingly, no PrPCWD was detected in the final round of Wisc-1 and CWD2 amplification in the S96-PrPC substrate. These results were unexpected, considering that these isolates propagated with similar efficiencies in wt and S96 substrates in the first round of PMCA. The MD CWD isolate was not able to propagate in the S96 substrate. Seeding of the S96-PrPC with the H95+ strain, however, resulted in successful serial prion propagation (Fig. 2).

Serial PMCA amplification of Wisc-1, H95+, CWD2 and MD CWD prions in wt and S96 PrPC substrates. 10−3 dilutions of the CWD brain homogenates from infected cervids were used to seed wt-PrPC substrate (tg33 mouse brain homogenate) or S96-PrPC brain substrate (tg60 mouse brain homogenate) and subjected to 4 serial rounds of PMCA. Amplification products were digested with PK (50 µg/ml) and analyzed by western blot. Although all CWD strains were serially propagated in the wt-PrPC substrate, only the H95+ strain maintained its amplification efficiency in the S96-PrPC brain substrate through the serial rounds of PMCA. Wisc-1 and CWD2 strains lost their ability to misfold the S96-PrPC after serial rounds of PMCA. MD CWD isolate was not able to propagate in this substrate. mAb Sha31 1:10,000.
Poor prion propagation by PMCA could be due to low amounts of PrPCWD present in the brain used as a seed46. To evaluate the stability of the Wisc-1 and CWD2 strains after passage into transgenic mice, we used brain homogenate from a tg33 mouse challenged with the Wisc-1 strain and brain homogenate from a tgElk mouse47 inoculated with the CWD2 strain as seeds for serial propagation in the S96 substrate through 5 rounds of PMCA. Both animals were euthanized at the terminal stage of the disease and accumulated very similar amounts of PrPCWD in the brain (Supplementary Figure 2). After one round of PMCA, tg33 Wisc-1 material amplified up to the 10−5 dilution when using the S96 substrate, and the tgElk CWD2 propagated up to the 10−4 dilution. When these PMCA products were subjected to a second round of PMCA in the S96 substrate, we observed that PrPCWD amplification was reduced tenfold. After 5 rounds of PMCA, both tg33 Wisc-1 and tgElk CWD2 lost the ability to efficiently propagate in the S96 substrate (Fig. 3).

Serial PMCA amplification of tg33 Wisc-1 and tgElk CWD2 prions in S96-PrPC substrate. Serial dilutions (10−3 to 10−7) of brain homogenate from a tg33 mouse (expressing white-tailed deer wild-type PrP) infected with Wisc-1 CWD strain and a tgElk mouse (expressing elk PrP) infected with the CWD2 strain were serially propagated in the S96-PrPC brain substrate (tg60 mouse brain homogenate) for 5 rounds of PMCA. PMCA products were digested with PK (50 µg/ml) and analyzed by western blotting. Amplification efficiency of both strains was reduced in one log after two rounds of PMCA. After 5 rounds of PMCA no PrPCWD amplification in the S96 substrate is detected with neither of the strains. mAb Sha31 1:10,000.
These results suggest that, although the most common CWD strains found in the wild (Wisc-1 = CWD1; CWD2) can misfold the white-tailed deer S96-PrPC, they apparently lack the ability to maintain prion propagation in this substrate, with PrPCWD amplification disappearing after serial passages in the S96-PrPC background. Only the H95+ strain was able to serially propagate in the S96-PrPC substrate corroborating the results obtained in vivo25.
To evaluate whether the extinction of seeding activity observed with Wisc-1 and CWD2 during serial PMCA in S96-PrPC substrate was related to the loss of affinity for the substrate or due to unstable propagation of alternative conformers competing and blocking each other, we tested for seeding activity in wt-PrPC substrates from tg33 mice. When the products of the 4th PMCA round of Wisc-1 and CWD2 in S96-PrPC substrate were seeded in tg33 wt-PrPC substrate no amplification occurred. Similarly, the products of the 5th PMCA round of tg33-derived Wisc-1 and tgElk-derived CWD2 in the S96 substrate did not amplify or generate protease resistant prion protein in tg33 wt-PrPC.
If these strains persisted during serial propagation in the S96-PrPC or if novel conformers emerged due to heterologous prion conversion, it is likely that these amplification products would seed wt-PrPC substrate in subsequent PMCA rounds since it is a more favorable substrate for these strains24. We did not recover any PrPCWD seeding activity from the 4th round and 5th round products of Wisc-1 and CWD2 in S96 PrPC following two rounds of PMCA in wt-PrPC substrate (tg33). This indicates that no infectivity was present after serial propagation with the S96-PrPC and that no novel conformers were generated by either Wisc-1 or CWD2 during in vitro conversion of S96-PrPC. None of the amplification replicates (n = 3) for the products of the 4th (deer strains) and 5th (mouse strains) rounds of Wisc-1 and CWD2 in S96-PrPC substrate contained seeding activity amplifiable in wt-PrPC, suggesting that Wisc-1 and CWD2 strains gradually lost their ability to misfold the S96-PrPC until replication became extinct and no PrPCWD could be detected. These results contrast with previously reported in vivo data that demonstrated that brains from asymptomatic (PK-res negative) tg60 mice challenged with Wisc-1 strain can still infect tg33 mice albeit with very long incubation periods24.
Discussion
Numerous studies in wild and farmed white-tailed deer populations have demonstrated the importance of the prion protein sequence in the susceptibility to CWD26,27,28,29,30,48. This evidence was complemented with experimental infections in deer or transgenic mice with PrPC polymorphisms that slow disease progression and modulate the degree of neuropathology19,23. One of these polymorphisms, S96-PrPC, is relatively common in white-tailed deer populations and has been suggested to be under selection as a response to CWD epizootics26,27,31,36. We show that the Serine (S) at codon 96 leads to the collapse of prion conversion for the most common CWD strains (Wisc-1 and CWD2). We propose that the interlocking at the N-terminus between the invading PrPCWD strain and the S96-PrPC controls efficient prion conversion, strain adaptation and disease susceptibility, considering that the Serine residue is in the N-terminus of the white-tailed deer PrP. Only the prion strain with the most unfolding-resistant N-terminal structure (i.e., H95+) was serially propagated on S96-PrPC24. Conversely, the Wisc-1 PrPCWD is less structurally stable24. Our experiments suggest that, when encoded in S96-PrPCWD (after round 1), Wisc-1 and CWD2 strains are less functional than H95+ at stably misfolding the S96-PrPC, impairing proper PrPC − PrPCWD domain alignment, misfolding efficiency and leading to extinction of the colonizing prion conformers.
There is no effective vaccine or preventive treatment against CWD prions. Therefore, strategies to mitigate CWD have been aimed at detecting positive cervids and in some regions reducing their numbers to lower disease prevalence and potentially reduce horizontal transmission between cervids and decrease human exposure. Breeding programs directed at increasing the frequency of PRNP alleles associated with resistance to scrapie, implemented by many European countries since 200149, have been very effective for the control of the disease14. These programs are based on the elimination of animals expressing the most susceptible genotypes50 and gradual replacement with more resistant sheep through the introduction of breeding males of the ARR/ARR genotype.
While the ARR haplotype confers almost complete resistance to scrapie51,52, there is no known deer PRNP polymorphism that prevents CWD infection23,31. The S96 allele is, despite this, a good candidate for the genetic selection of deer in herds at risk of CWD exposure. After the depopulation of a CWD positive white-tailed deer farm in Wisconsin, it was found that 88% of the deer were positive for PrPCWD accumulation in at least one tissue, and the age of the positive animals ranged from fawns to adult deer. From this cohort, only two adult deer had the S96/S96 PRNP genotype, with only one of them showing faint PrPCWD accumulation in the retropharyngeal lymph nodes, while the other animal was negative. Further studies have demonstrated that deer expressing the S96 allele in homozygosity not only are less susceptible to CWD infection, but are also found in a significant earlier stage of the disease than deer of the wt genotype31,33. These observations and the older age at which positive S96 deer are detected compared to wt/wt deer, indicate the serine at this codon affects prion conversion resulting in slow disease progression.
These conclusions are consistent with the results of experimental infections under controlled conditions in deer and transgenic mice expressing S96 or wt-PrPC21,22,23,24,25,34. In contrast, previous in vitro replication experiments in which [35S]-labeled recombinant wt or S96 PrPC were exposed to different CWD prions showed that both prion proteins are equally susceptible to misfolding37. While, in vivo, the conversion of S96-PrPC into PK-res PrPCWD during Wisc-1 infection in tg60 mice was inefficient and resulted in low levels of protease-resistant prion conformers24. These novel S96-PrPRes molecules were unable to adapt in vivo and were unstable when subjected to PMCA with S96-PrPC showing altered PrPRes glycotypes24. These results indicated that, although the Wisc-1 strain can persistently replicate, the S96-PrPCWD products are less structurally compatible with the S96-PrPC limiting the adaptation in tg60 mice. Independent transmission experiments support this interpretation for other CWD strains, including S96/S96 deer prions, which transmitted with incomplete attack rates when inoculated into tg33 mice expressing wt-PrPC and failed to adapt in tg60 mice22.
In this study, we further evaluated the capacity of S96-PrPC to support the in vitro replication of different CWD strains (i.e., white-tailed deer Wisc-1 and H95+ strains, elk CWD2 strain and a mule deer (wt/wt) field CWD isolate). We also compared the ability of S96-PrPC and wt-PrPC substrates to support serial prion propagation. After a single round of PMCA, Wisc-1, H95+ and CWD2 strains replicated with similar efficiencies in both substrates, similar results to those obtained in other in vitro replication assays37. CWD prions from a wild mule deer (wt/wt genotype) were unable to misfold the S96-PrPC, which may indicate lower compatibility between this wt/wt mule deer CWD isolate and S96-PrPC compared to Wisc-1, CWD2 and H95+ strains perhaps indicating a different strain in mule deer. Contrastingly, after several rounds of PMCA, there was no detectable amplification in the S96-PrPC reactions seeded with Wisc-1 or CWD2 indicating that these CWD prions lost their replication ability when encoded on S96-PrPCWD. Serial propagation of these two strains in wt-PrPC substrate resulted in efficient amplification. The loss of replicative capacity in the S96-PrPC observed with these two strains (Wisc-1 and CWD2) but not with the control H95+ strain (propagated in parallel and in the same sonicator), is a phenomenon that has not been described during in vitro propagation of prions under standard serial passage conditions.
Previous transmission studies have hypothesized CWD prion strain diversification or differential strain amplification from natural prion strain mixtures following transmission of cervid prions into transgenic mice overexpressing homologous PrPC42. To evaluate these propositions, we repeated the serial replication experiment with S96-PrPC, seeding this time with brain homogenates from transgenic mice that had succumbed to prion disease after inoculation of Wisc-1 and CWD2 from the deer and elk. Each strain was obtained by passage in hosts of homologous PrPC (tg33 and tgElk lines, respectively) that were euthanized with advanced signs of prion disease and contained similar levels of PrPCWD in the brain. The seeding activity in both tg33 and tgElk mice was approximately equivalent as estimated by limiting dilution and a single round of PMCA. Generally, the amount of prions in the input inocula is a major factor influencing the maximum dilution of brain homogenate detectable by PMCA46. Serial PMCA amplification allows the detection of highly diluted prion agents with a sensitivity that could exceed that of the mouse bioassay by 104 to 105-fold40,41,53,54. Serial rounds of PMCA in principle always allow the generation of large amounts of PrPRes which is accompanied by an increase in western blot detection of the limiting dilutions of the original seed54. Here, however, we observed the opposite effect with the S96 substrate. The PMCA efficiency in generating PrPRes decreased with each round for both strains only while serially propagated in S96-PrPC. After 5 rounds of serial PMCA, we did not detect generation of S96-PrPRes with either Wisc-1 or CWD2 confirming the results obtained when the inocula was from deer and elk. No emergence of alternate prion strain conformers (e.g., H95+) was observed suggesting faithful strain replication upon transfer of the prion strains between the brains of two host species (white-tailed deer and elk into transgenic mice) with the same PrPC.
These results confirm that the white-tailed deer Wisc-1 CWD strain, which is favorably propagated in wt-PrP, suffered a gradual loss of replicative capacity when encoded on S96-PrPCWD indicating the residues directly interacting with the S96-PrPC have less affinity than Wisc-1 encoded on wt-PrPCWD. These observations made here reconcile the apparently conflicting results previously obtained by exposing the white-tailed deer S96-PrPC to prions from animals of the wt genotype in vitro and in vivo. We must also consider that in vitro systems cannot reproduce all the pathogenic events occurring in a living organism. Animals can degrade a significant amount of the inoculated prions55,56 and certain neuroinflammatory mechanisms are neuroprotective in the early stage of prion disease57. These phenomena are not present in in vitro assays, making the conversion of PrPC generally easier and/or more efficient.
The Wisc-1 S96-PrPRes and CWD2 S96-PrPRes generated in vitro are unstable and not able to self-propagate when passed serially, leading to the extinction of prion amplification after several passages. This was confirmed by seeding wt-PrPC substrates with material from the last round of Wisc-1 and CWD2 PMCA in S96-PrPC. No amplification was obtained in the wt-PrPC, (not shown) indicating that these strains did not propagate at a low level in the S96-PrPC substrate.
These results suggest that the genetic selection for animals of the S96 genotype could be a simple and efficient strategy for the control of CWD in deer populations. These results, however, should be interpreted with caution. The introduction of breeding males of the ARR/ARR genotype in sheep has been efficient in controlling scrapie because the ARR haplotype provides a high degree of disease resistance even in heterozygous animals58,59. ARR-PrPC, together with other prion proteins, can interfere with prion replication in a dominant negative manner, impairing the misfolding of wt-PrPC when co-expressed in the same organism60. Heterozygous wt/S96 deer are susceptible to CWD and show a similar PrPCWD distribution to that of wt/wt deer at the terminal stage of the disease20, although they present extended incubation periods19. We suggest, therefore, that the beneficial effects of this genetic selection strategy on CWD control would begin to be detected after a certain percentage of S96 homozygotes is reached in the deer population.
Another important factor to consider when proposing strategies for the control of CWD is the prion strain to which the animals would be exposed. The H95+ strain maintains its replicative ability after multiple rounds in S96-PrPC substrate (Fig. 2). In vivo assays have also confirmed that hosts expressing S96-PrPC are susceptible to the challenge with this CWD strain24,25. Genetic selection for the control of a prion disease would not necessarily protect the animal against all the strains present. For example, sheep expressing the ARR allele are strongly resistant to classical scrapie, but susceptible to atypical scrapie61,62,63. Deer selected for the expression of the S96 allele would still be susceptible to the H95+ strain19,24,25,36. It is possible, therefore, that selecting for deer of the S96-PrPC may initially have beneficial effects on CWD control, but this also could lead to the selection of other prion strains, such as H95+, which would be able to propagate between deer of this genotype. The H95+ strain has yet to be described in wild deer populations since this strain emerged following experimental transmission in deer expressing H95 PrPC24,25. In addition, the H95-PRNP allele has been described in low frequency in several CWD enzootic regions of North America26,27,31. An increase in the allele frequencies of protective PRNP alleles in response to chronic CWD exposure can in turn result in better conditions for other strains to emerge25,35,36.
In conclusion, S96 allele plays an important role in CWD resistance, possibly because most common prion strains (Wisc-1 and CWD2) cannot propagate stably misfolded S96-PrPC in vitro. Our data demonstrates that most common CWD prions do not maintain their replicative capacity when serially passaged in S96-PrPC substrates in vitro, which is consistent with in vivo data for the Wisc-1 strain24. This suggests that even if a deer of the S96 genotype could be infected with these strains, transmission to other animals of the same genotype may not occur. Therefore, selective breeding is a potential strategy for controlling the spread of CWD. However, these strategies could favor emergence of less abundant strains with ability to propagate in more resistant genotypes.
Material and methods
CWD isolates
Wisc-1 and H95+ isolates consisted of 10% brain homogenates (wt/vol in water) obtained from the brain of terminally ill, orally infected white-tailed deer (Odocoileus virginianus) expressing the wild-type (Q95/G96) and H95/S96 PrPC, respectively19,25. These isolates have been thoroughly characterized in transgenic mice, wild type mice and hamsters25,43. There is no evidence demonstrating that Wisc-1 and the previously described CWD1 strain are different strains since they show identical glycosylation patterns and similar incubation periods and neuropathological features in transgenic mice25,42.
CWD2 isolate was prepared from a pool of brains from three CWD positive elk (Cervus canadensis) of the MM132 genotype44 and was generously provided by Dr. Catherine Graham. This elk pool was characterized as CWD2 strain in previous studies using transgenic mice expressing deer PrPC42.
Mule deer (Odocoileus hemionus) CWD (MD CWD isolate) was obtained from the obex region of a hunter-harvested deer of the wild type PRNP genotype (animal ID 102565) and provided by Dr. Margo Pybus (Alberta Environment and Parks). This animal was culled in the 2017 Alberta hunting season and tested for CWD by Alberta Environment and Parks. PrPCWD, as detected by western blot, was abundant in the brain material from this deer, showing a glycosylation profile indistinguishable from Wisc-1 strain.
Tg33 Wisc-1 isolate consisted of a 10% brain homogenate (wt/vol in water) from a tg33 transgenic mouse that had developed terminal prion disease after the inoculation of deer Wisc-1 CWD (369 days post-inoculation)25. TgElk CWD2 seed was a 10% (wt/vol) brain homogenate from a transgenic mouse overexpressing elk PrPC47 euthanized at the terminal stage of prion disease (110 days post-inoculation) after the challenge with the CWD2 elk pool.
Experimental procedures in mice were conducted in accordance with the Canadian Council on Animal Care Guidelines, Policies with approval from Animal Care and Use Committee: Health Sciences at the University of Alberta and ARRIVE guidelines.
Protein misfolding cyclic amplification of CWD isolates
PMCA was performed as described previously45. PMCA was performed using brain homogenates from uninfected tg33 and tg60 mice as substrates. Tg33 mice express the wild-type PrPC from white-tailed deer (G96) at levels ~ 1 × than those found in the deer brain. Tg60 mice express the white-tailed deer S96 PrPC, at levels ~ 0.7 × than those detected in the tg33 transgenic line. Both transgenic lines have the same strain background21,22. After euthanasia by isoflurane inhalation, tg33 and tg60 mice were perfused using ice-cold PBS + 5 mM EDTA. Perfused brains were immediately frozen at − 80 ºC. Brain substrate (10% w/v brain homogenate) was prepared using a tissue grinder, and the brains were homogenized in PMCA conversion buffer (PBS + 150 mM NaCl, 1% Triton X-100 + 0.5 M EDTA, 1 × Protease Inhibitor Cocktail). Aliquots of 90 µl were prepared with brain homogenates from tg60 mice and stored at − 80 ºC. Since tg60 mice express 30% less PrPC than tg33 mice, tg33 substrates were diluted with a perfused brain homogenate from Prnp0/0 mice, obtaining substrates with an equivalent PrPC amount.
For the titration of the CWD agents, CWD seeds were diluted from 102 to 107 -fold in conversion buffer. Then, 10 µl of each dilution were used to seed 90 µl of tg33 or tg60 substrate. Three PTFE beads (3/32″; McMaster-Carr) were added to each PMCA reaction to increase the efficiency of prion amplification39. Seeded substrates were placed on the plate holder of a S-4000 Misonix sonicator (QSonica, Newtown, CT, USA) and subjected to one round of PMCA consisting of incubation cycles of 15 min at 37 ºC without shaking, followed by sonication pulses of 30 s at 60% power. After a round of 24 h of PMCA, samples were analyzed for PrPCWD amplification.
Analysis of serial amplification of Wisc-1, CWD2, H95+ and MD deer CWD isolates in wt or S96-PrPC was performed by diluting the CWD seeds 103-fold into 10% normal tg33 or tg60 brain homogenates. Each of these reactions was seeded in triplicate and carried out in the same sonicator, using identical PMCA conditions. Reactions were subjected to PMCA for 24 h. After this first round, 10 µl of the PMCA products were diluted into 90 µl of fresh substrate and sonicated again, repeating this process through 4 rounds of PMCA. Tg33 Wisc-1 and tgElk CWD2 seeds were serially diluted (10−3 to 10−7) in the tg60 substrate and propagated for 5 rounds of PMCA. Both serial PMCA experiments were performed twice obtaining the same results.
Biochemical analysis of PMCA products
PMCA amplified samples were protease digested using 50 µg/ml PK during 1 h at 37 ºC with constant agitation (700 rpm). Digestion was terminated by the addition of 2 × Laemmli sample buffer (150 mM Tris–HCl, pH 6.8, 0.5% bromophenol blue, 25% glycerol, 5% [wt/vol] SDS, 12.5% ß-mercaptoethanol) and boiling at 100 ºC for 10 min. Samples were analyzed by western blot. Immunodetection of PrPCWD was performed with mouse monoclonal antibody Sha31 (1:10,000; Cayman Chemical) and alkaline phosphatase-conjugated goat anti-mouse secondary antibody (1:10,000; Promega). Blots were developed using the AttoPhos AP Fluorescent Substrate System (Promega).
Data availability
Data available within the article and its supplementary materials.
References
Prusiner, S. B. Prions. Proc. Natl. Acad. Sci. USA 95, 13363–13383 (1998).
Caughey, B. & Chesebro, B. Prion protein and the transmissible spongiform encephalopathies. Trends Cell Biol. 7, 56–62 (1997).
Manson, J. et al. The prion protein gene: A role in mouse embryogenesis?. Development 115, 117–122 (1992).
Mathiason, C. K. et al. Infectious prions in pre-clinical deer and transmission of chronic wasting disease solely by environmental exposure. PLoS ONE 4, e5916 (2009).
Miller, M. W. & Wild, M. A. Epidemiology of chronic wasting disease in captive white-tailed and mule deer. J. Wildl. Dis. 40, 320–327 (2004).
Spraker, T. R. et al. Spongiform encephalopathy in free-ranging mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginianus) and Rocky Mountain elk (Cervus elaphus nelsoni) in northcentral Colorado. J. Wildl. Dis. 33, 1–6 (1997).
Williams, E. S. & Young, S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J. Wildl. Dis. 16, 89–98 (1980).
Belt, P. B. et al. Identification of five allelic variants of the sheep PrP gene and their association with natural scrapie. J. Gen. Virol. 76(Pt 3), 509–517 (1995).
Bossers, A., Schreuder, B. E., Muileman, I. H., Belt, P. B. & Smits, M. A. PrP genotype contributes to determining survival times of sheep with natural scrapie. J. Gen. Virol. 77(Pt 10), 2669–2673 (1996).
Goldmann, W. et al. Two alleles of a neural protein gene linked to scrapie in sheep. Proc. Natl. Acad. Sci. USA 87, 2476–2480 (1990).
Westaway, D. et al. Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev. 8, 959–969 (1994).
Arnold, M. & Ortiz-Pelaez, A. The evolution of the prevalence of classical scrapie in sheep in Great Britain using surveillance data between 2005 and 2012. Prev. Vet. Med. 117, 242–250 (2014).
Hagenaars, T. J. et al. Scrapie prevalence in sheep of susceptible genotype is declining in a population subject to breeding for resistance. BMC Vet. Res. 6, 25 (2010).
Nodelijk, G. et al. Breeding with resistant rams leads to rapid control of classical scrapie in affected sheep flocks. Vet. Res. 42, 5 (2011).
Acutis, P. L. et al. Resistance to classical scrapie in experimentally challenged goats carrying mutation K222 of the prion protein gene. Vet. Res. 43, 8 (2012).
Barillet, F. et al. Identification of seven haplotypes of the caprine PrP gene at codons 127, 142, 154, 211, 222 and 240 in French Alpine and Saanen breeds and their association with classical scrapie. J. Gen. Virol. 90, 769–776 (2009).
Hazards EPoB et al. Genetic resistance to transmissible spongiform encephalopathies (TSE) in goats. EFSA J. 15, e04962 (2017).
Sacchi, P. et al. Predicting the impact of selection for scrapie resistance on PRNP genotype frequencies in goats. Vet. Res. 49, 26 (2018).
Johnson, C. J. et al. Prion protein polymorphisms affect chronic wasting disease progression. PLoS ONE 6, e17450 (2011).
Otero, A. et al. Prion protein polymorphisms associated with reduced CWD susceptibility limit peripheral PrP(CWD) deposition in orally infected white-tailed deer. BMC Vet. Res. 15, 50 (2019).
Meade-White, K. et al. Resistance to chronic wasting disease in transgenic mice expressing a naturally occurring allelic variant of deer prion protein. J. Virol. 81, 4533–4539 (2007).
Race, B., Meade-White, K., Miller, M. W., Fox, K. A. & Chesebro, B. In vivo comparison of chronic wasting disease infectivity from deer with variation at prion protein residue 96. J. Virol. 85, 9235–9238 (2011).
Miller, M. W. et al. Survival patterns in white-tailed and mule deer after oral inoculation with a standardized, conspecific prion dose. J. Wildl. Dis. 48, 526–529 (2012).
Duque Velasquez, C. et al. Chronic wasting disease (CWD) prion strains evolve via adaptive diversification of conformers in hosts expressing prion protein polymorphisms. J. Biol. Chem. 295, 4985–5001 (2020).
Duque Velasquez, C. et al. Deer prion proteins modulate the emergence and adaptation of chronic wasting disease strains. J. Virol. 89, 12362–12373 (2015).
Johnson, C., Johnson, J., Clayton, M., McKenzie, D. & Aiken, J. Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J. Wildl. Dis. 39, 576–581 (2003).
Johnson, C. et al. Prion protein polymorphisms in white-tailed deer influence susceptibility to chronic wasting disease. J. Gen. Virol. 87, 2109–2114 (2006).
O’Rourke, K. I. et al. Polymorphisms in the prion precursor functional gene but not the pseudogene are associated with susceptibility to chronic wasting disease in white-tailed deer. J. Gen. Virol. 85, 1339–1346 (2004).
Keane, D. P. et al. Chronic wasting disease in a Wisconsin white-tailed deer farm. J. Vet. Diagn. Invest. 20, 698–703 (2008).
Kelly, A. C. et al. Prion sequence polymorphisms and chronic wasting disease resistance in Illinois white-tailed deer (Odocoileus virginianus). Prion 2, 28–36 (2008).
Haley, N. J. et al. Estimating relative CWD susceptibility and disease progression in farmed white-tailed deer with rare PRNP alleles. PLoS ONE 14, e0224342 (2019).
Wolfe, L. L. et al. PrPCWD in rectal lymphoid tissue of deer (Odocoileus spp.). J. Gen. Virol. 88, 2078–2082 (2007).
Haley, N. J. et al. Antemortem detection of chronic wasting disease prions in nasal brush collections and rectal biopsy specimens from white-tailed deer by real-time quaking-induced conversion. J. Clin. Microbiol. 54, 1108–1116 (2016).
Angers, R. et al. Structural effects of PrP polymorphisms on intra- and interspecies prion transmission. Proc. Natl. Acad. Sci. USA 111, 11169–11174 (2014).
Hannaoui, S. et al. Destabilizing polymorphism in cervid prion protein hydrophobic core determines prion conformation and conversion efficiency. PLoS Pathog. 13, e1006553 (2017).
Robinson, S. J., Samuel, M. D., Johnson, C. J., Adams, M. & McKenzie, D. I. Emerging prion disease drives host selection in a wildlife population. Ecol. Appl. 22, 1050–1059 (2012).
Raymond, G. J. et al. Evidence of a molecular barrier limiting susceptibility of humans, cattle and sheep to chronic wasting disease. EMBO J. 19, 4425–4430 (2000).
Boerner, S., Wagenfuhr, K., Daus, M. L., Thomzig, A. & Beekes, M. Towards further reduction and replacement of animal bioassays in prion research by cell and protein misfolding cyclic amplification assays. Lab. Anim. 47, 106–115 (2013).
Gonzalez-Montalban, N. et al. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 7, e1001277 (2011).
Makarava, N., Savtchenko, R., Alexeeva, I., Rohwer, R. G. & Baskakov, I. V. Fast and ultrasensitive method for quantitating prion infectivity titre. Nat. Commun. 3, 741 (2012).
Moudjou, M. et al. Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. mBio 5, e00829-13 (2013).
Angers, R. C. et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 328, 1154–1158 (2010).
Herbst, A., Velasquez, C. D., Triscott, E., Aiken, J. M. & McKenzie, D. Chronic wasting disease prion strain emergence and host range expansion. Emerg. Infect. Dis. 23, 1598–1600 (2017).
Pushie, M. J., Shaykhutdinov, R., Nazyrova, A., Graham, C. & Vogel, H. J. An NMR metabolomics study of elk inoculated with chronic wasting disease. J. Toxicol. Environ. Health A 74, 1476–1492 (2011).
Castilla, J., Saa, P., Hetz, C. & Soto, C. In vitro generation of infectious scrapie prions. Cell 121, 195–206 (2005).
Lyon, A. et al. Application of PMCA to screen for prion infection in a human cell line used to produce biological therapeutics. Sci. Rep. 9, 4847 (2019).
LaFauci, G. et al. Passage of chronic wasting disease prion into transgenic mice expressing Rocky Mountain elk (Cervus elaphus nelsoni) PrPC. J. Gen. Virol. 87, 3773–3780 (2006).
Abrams, J. et al. Human prion disease mortality rates by occurrence of chronic wasting disease in free-ranging cervids, United States. Prion 14, 182–183 (2018).
Council E. Regulation (EC) No 999/2001 of the European Parliament and of the Council of 22 May 2001 laying down rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies. Off. J. Eur. Union L147 (2001).
Dawson, M., Hoinville, L. J., Hosie, B. D. & Hunter, N. Guidance on the use of PrP genotyping as an aid to the control of clinical scrapie. Scrapie Information Group. Vet. Rec. 142, 623–625 (1998).
Baylis, M. et al. Risk of scrapie in British sheep of different prion protein genotype. J. Gen. Virol. 85, 2735–2740 (2004).
Hunter, N., Goldmann, W., Smith, G. & Hope, J. The association of a codon 136 PrP gene variant with the occurrence of natural scrapie. Arch. Virol. 137, 171–177 (1994).
Saa, P., Castilla, J. & Soto, C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 (2006).
Johnson, C. J., Aiken, J. M., McKenzie, D., Samuel, M. D. & Pedersen, J. A. Highly efficient amplification of chronic wasting disease agent by protein misfolding cyclic amplification with beads (PMCAb). PLoS ONE 7, e35383 (2012).
Safar, J. G. et al. Prion clearance in bigenic mice. J. Gen. Virol. 86, 2913–2923 (2005).
Safar, J. G. et al. Search for a prion-specific nucleic acid. J. Virol. 79, 10796–10806 (2005).
Carroll, J. A., Race, B., Williams, K., Striebel, J. & Chesebro, B. Microglia are critical in host defense against prion disease. J. Virol. 92, e00549–18 (2018).
Caplazi, P. A., O’Rourke, K. I. & Baszler, T. V. Resistance to scrapie in PrP ARR/ARQ heterozygous sheep is not caused by preferential allelic use. J. Clin. Pathol. 57, 647–650 (2004).
Goldmann, W. PrP genetics in ruminant transmissible spongiform encephalopathies. Vet. Res. 39, 30 (2008).
Perrier, V. et al. Dominant-negative inhibition of prion replication in transgenic mice. Proc. Natl. Acad. Sci. USA 99, 13079–13084 (2002).
Arsac, J. N. et al. Similar biochemical signatures and prion protein genotypes in atypical scrapie and Nor98 cases, France and Norway. Emerg. Infect. Dis. 13, 58–65 (2007).
Luhken, G. et al. Epidemiological and genetical differences between classical and atypical scrapie cases. Vet. Res. 38, 65–80 (2007).
Saunders, G. C., Cawthraw, S., Mountjoy, S. J., Hope, J. & Windl, O. PrP genotypes of atypical scrapie cases in Great Britain. J. Gen. Virol. 87, 3141–3149 (2006).
Acknowledgements
We acknowledge funding for this research to Dr. Aiken and Dr. McKenzie from Genome Canada, Alberta Prion Research Institute and Alberta Agriculture and Forestry through Genome Alberta and the University of Alberta in support of the Systems Biology and Molecular Ecology of Chronic Wasting Disease project. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. We would also like to thank all members of the Aiken-McKenzie lab for their constructive comments about our manuscript.
Author information
Authors and Affiliations
Contributions
Conception and design of the work: A.O., C.D.V., J.A. and D.M.; acquisition, analysis and interpretation of the data: A.O. and C.D.V., resources and funding: J.A. and D.M.; writing the original draft: A.O. and C.D.V., revision and edition of the final manuscript: A.O., C.D.V., J.A. and D.M. All authors approved and agreed to the submitted version of the present manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Otero, A., Duque Velásquez, C., Aiken, J. et al. White-tailed deer S96 prion protein does not support stable in vitro propagation of most common CWD strains. Sci Rep 11, 11193 (2021). https://doi.org/10.1038/s41598-021-90606-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-90606-8
This article is cited by
-
Ticks harbor and excrete chronic wasting disease prions
Scientific Reports (2023)
-
Emergence of CWD strains
Cell and Tissue Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
