
Abstract
Soft tissue sarcoma represents about 1% of all adult cancers. Occurrence of multiple sarcomas in a same individual cannot be fortuitous. A 72-year-old patient had between 2007 and 2016 a glomangiopericytal tumor of the right forearm and a succession of sarcomas of the extremities: a leiomyosarcoma of the left buttock, a myxofibrosarcoma (MFS) of the right forearm, a MFS of the left scapula, a left latero-thoracic MFS and two undifferentiated sarcomas on the left forearm. Pathological examination of the six locations was not in favor of disease with local/distant recurrences but could not confirm different diseases. An extensive molecular analysis including DNA-array, RNA-sequencing and DNA-Sanger-sequencing, was thus performed to determine the link between them. The genomic profile of the glomangiopericytal tumor and the six sarcomas revealed that five sarcomas were different diseases and one was the local recurrence of the glomangiopericytal tumor. While the chromosomal alterations in the six tumors were different, a common somatic CDKN2A/CDKN2B deletion was identified. RNA-sequencing of five tumors identified mutations in GLT8D1, GATAD2A and SLC25A39 in all samples. The germline origin of these mutations was confirmed by Sanger-sequencing. Innovative molecular analysis methods have made possible a better understanding of the complex tumorigenesis of multiple sarcomas.
Similar content being viewed by others

Introduction
Soft tissue sarcomas (STS) are rare (1% of all cancers, 3.6 per 100,000)1. The STS classification is historically based on histological subtypes and grading2. However, there is a wide variety of different histological subtypes of sarcomas and they may be difficult to differentiate3. In recent decades, advanced molecular techniques and genetic profiling have revolutionized the approach to sarcoma classification4. One third of all STS are characterized by recurrent specific chromosomal translocations, resulting in fusions of specific genes, usually encoding aberrant chimeric transcription factors. The other two-thirds of STS have no genetic signature and are characterized by numerous aberrations, including chromosomal losses and gains5. These sarcomas are more frequent and are generally high-grade, including undifferentiated pleomorphic sarcomas (UPS) and leiomyosarcomas (LMS). This group of tumors has a high prevalence of p53 control point alterations, including TP53 inactivating mutations and homozygous deletion of CDKN2A6. Several genetic syndromes are associated with an increased risk of STS, particularly Li-Fraumeni syndrome7. In the present study, we report the unique case of a 72-year-old patient who presented with glomangiopericytal tumor followed by six sarcomas of the extremities between 2007 and 2016. Pathological examination of the tumors could not separate a unique disease with local/distant recurrences or a succession of different diseases. An extensive molecular and genetic analysis was then performed to determine the tumorigenesis in this patient.
Material and methods
Patient and samples
Molecular analysis was based on the case of a 72-year-old patient with a history of ear melanoma and pulmonary sarcoidosis with mediastinal lymph nodes, who presented a glomangiopericytal tumor and a succession of six sarcomas of the extremities (Figs. 1, 2 and Table S1). In July 2007, he synchronously developed a tumor of the right forearm and a tumor of the left buttock. The former was surgically removed and proved to be a glomangiopericytal tumor of uncertain potential (named in our study S2007A). The tumor of the left buttock was widely excised and the diagnosis was a grade 3 (FNCLCC Grading System) LMS (S2007B). In October 2008, he presented with a slowly enlarging mass on the right forearm. Histological analysis revealed features of fibroblastic differentiation with spindle-shaped cells containing elongated nuclei within a myxoid matrix. The diagnosis was a grade 1 fibrosarcoma or myxofibrosarcoma (MFS, S2008). In February 2012, he presented with a subcutaneous tumor on the left scapula. Histological analysis revealed spindle-shaped cells with high mitotic activity, darkly staining nuclei with variably prominent nucleoli and eosinophilic cytoplasm. The stroma contained variable collagen with myxoid and fibrotic areas. The diagnosis of grade 3 MFS was made (S2012A). In October 2012, he presented with an enlarging left latero-thoracic sub-cutaneous mass that was considered a superficial grade 2 MFS (S2012B). In April 2016, he presented a mass on the left forearm. Histological analysis revealed pleomorphic spindle-shaped cells with high mitotic activity. The diagnosis of grade 2 UPS was made (S2016A). In June 2016, he presented a grade 2 UPS in the left forearm (S2016B). The patient finally died in June 2018 from diffuse metastatic evolution (including bone and lung metastases) that did not respond to chemotherapy. No material was available for the melanoma and for the bone and lung metastases. Before performing the analyses, the patient provided written informed consent for use of all specimens for the purpose of the study. All cases have been reviewed by an expert pathologist (SLG) of the French Sarcoma Group according to the World Health Organization3. All experimental protocols were approved by the ethical committee of the University Hospital Center of Amiens. All methods were carried out in accordance with relevant guidelines and regulations.

Location and histological subtypes of the seven soft tissue tumors. LMS leiomyosarcoma, MFS myxofibrosarcoma, UPS undifferentiated pleomorphic sarcoma.

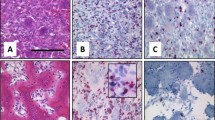
Histological characteristics of the soft tissue tumors. (A) S2007A: glomangiopericytal tumor with uncertain potential: ovoid cells embedded in a myxoid background and concentrically arranged around rounded vascular structures. The tumor is classified according to the WHO classification as a tumor with uncertain potential because of its size. (B) S2007B: grade 3 LMS: proliferation consisting of long bundles composed of spindle cells with abundant eosinophilic cytoplasm. Black arrow: atypical mitosis. (C) S2008: Low grade MFS: slightly atypical fusiform cells embedded in myxoid background. Numerous thin-walled vessels. (D) S2012A: MFS: High Power magnification of atypical spindle cells. Black arrow: atypical mitosis. (E) S2012B: MFS: At low magnification power, juxtaposition of dense spindle cell area and paucicellular myxoid proliferation. (F) S2016A: grade 2 UPS: proliferation of spindle cells dissecting adipose tissue (at the bottom). (G) S2016B: grade 2 UPS: atypical spindle and pleomorphic cells on a myxoid background. Black arrow: atypical mitosis. LMS leiomyosarcoma, HE hematoxylin and eosin staining, MFS myxofibrosarcoma, UPS undifferentiated pleomorphic sarcoma. Scale bar: 100 µm.
DNA extraction
Areas of high tumor cellularity (> 80%) or healthy tissue were defined by a pathologist on a hematoxylin and eosin-stained histologic section and were transferred to a formalin-fixed paraffin-embedded (FFPE) tumor block. DNA was extracted using the QIAamp DSP DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s recommendations. Genomic DNA was then quantified using a Nanodrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and a Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) using the Qubit dsDNA BR Assay Kit according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA).
DNA array
Genomic profiling was performed using the Affymetrix OncoScan CNV Arrays (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Profiles were visualized and analyzed with the Chromosome Analysis Suite Software (Thermo Fisher Scientific, Waltham, MA, USA) and the annotations of the genome version GRCH37 (hg19). Ploidy was evaluated by analyzing allele difference, according to the manufacturer’s instructions (P/N CL00731).
RNA extraction
RNA extraction and quality assessment of FFPE tissue were performed as described in Lesluyes et al.8.
RNA sequencing
The RNA sequencing libraries from FFPE tissue sample total RNA were prepared at the Centre Nacional d'Anàlisi Genòmica (CNAG, Barcelona, Spain) using a modified TruSeq RNA Sample Prep Kit v2 protocol (Illumina, Inc., San Diego, CA, USA) as previously described in Lesluyes et al.8. Each library was sequenced using TruSeq SBS Kit v3-HS in paired end mode with the read length 2 × 76 bp on HiSeq2000 (Illumina, Inc., San Diego, CA, USA) following the manufacturer’s protocol, as described in Lesluyes et al.8.
Bioinformatics analysis pipeline for RNA sequencing
RNA sequences were aligned using STAR v2.6.0c9 with default parameters on the Human Genome version hg38. Thus, duplicated PCR reads were removed with PicardTools v2.18.2118 (http://broadinstitute.github.io/picard/index.html). SNV were detected by BCFtools mpileup v1.6 with a minimum 20 of phred quality (-Q 20) and by BCFtools call-Ac10. Thus, variants with fewer than 5 total reads and 2 alternative reads were filtered out. Finally, they were annotated with Annovar v20160201 tool11.
Filtering of RNA-seq data
After the RNA-seq pipeline treatment, 650,497 variants were detected. First, we kept only the exonic and splicing mutations that were not synonymous or not identified in dbSNP or in the 1000 Genomes databases, i.e. 4507 mutations. Second, to remove mutations potentially due to fixation artefacts, mutations also found in at least 8/17 of the 17 FFPE samples from a previously published cohort8 were removed after conversion of coordinates with LiftOver (hg19 to hg38; https://genome.ucsc.edu/cgi-bin/hgLiftOver). Thus, only 6 mutations were removed. Finally, only exonic mutations detected in all the five studied tumors were kept for further analysis, i.e. 77 exonic mutations (Tables S2 and S3). All mutations were then visualized in the five samples (S2007A, S2007B, S2008, S2012A and S2016A) using the Integrative Genomics Viewer (IGV_2.6.3)12. The Ensemble Genome Browser was consulted to ensure that the mutations were not referenced in the dbSNP database and alternative reads were aligned on the genome in some cases using the blat function on the UCSC website. Thus 53/77 (68.8%) variations were removed since they were found in dbSNP, 7/77 (9.1%) mutations were excluded because of a wrong alignment or multiple alignments in other genes, 2/77 (2.6%) mutations were not considered due to a sequencing error and 9/77 (11.7%) mutations were excluded because of a low percentage of alternative reads in most cases. Therefore, only 6 mutations were kept for further validation (7.8%) (Tables S2 and S3).
Fusion transcript analysis
DeFuse (v0.6.2) was used with ENSEMBL GRCh37.74 annotations and candidate fusions were filtered as previously described13.
PCR on genomic DNA and Sanger sequencing
To analyze the sequences of p14ARF and p16INK4A, we used the primers described by Iwato et al. for exons 1 and the common exon 214. For common exon 3 and CDKN2A IVS2-105A/G, we used the primers described by Laud et al.15. PCR primers were designed using the Primer 3 program (http://frodo.wi.mit.edu/primer3/) for CDKN2B mutation screening and for validation of GLT8D1, GATAD2A, SLC25A39, AZIN1, COG3 and COPA mutations on genomic DNA (see Table S4). Regarding CDKN2A and CDKN2B screening, all exons were sequenced in HT2013 and Sarc2011 and only those of CDKN2A in HT2012 (insufficient material to sequence all exons of both genes). All PCR were performed on 50 ng of gDNA using AmpliTaqGold DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions and using the following Touch-down PCR program: 2 cycles at 60 °C, followed by 2 cycles at 59 °C, 2 cycles at 58 °C, 3 cycles at 57 °C, 3 cycles at 56 °C, 4 cycles at 55 °C, 4 cycles at 54 °C, 5 cycles at 53 °C and finally 10 cycles at 52 °C. Sanger sequencing was performed by Genoscreen (Lille, France). Sequence electrophoregrams were obtained using FinchTV software (version 1.4.0, Geospiza Inc., Seattle, WA, USA).
Results
Tumor genomic profiles
To assess whether the tumors were genetically related to each other, we first characterized the glomangiopericytal tumor and the six sarcomas by DNA array. The seven tumors presented losses and more rarely gains, mainly involving whole chromosomes or chromosome arms (Fig. 3). According to their genomic profiles and allelic status, the tumors developed between 2007 and 2012 had diploid profiles whereas those developed in 2016 were mainly tetraploid. The losses of part of chromosomes 1p, 2p, 8p, 9p, 10p, 17p and 20p were detected in two or more tumors. However, close inspection of the boundaries showed that all alteration breakpoints were different between the tumors, except between S2007A and S2008 (Figure S1). The latter shared several breakpoints (on chromosomes 1p, 2p, 3p 4q, 6q, 7p, 9p, 9q, 13 and 18p), suggesting that S2008 was a local recurrence of S2007A. Genomic profiles of six out of seven tumors were thus very different overall, so they were likely independent.

Tumor genomic profiles. Copy number variations (CNVs) and allele frequency differences, plotted on the upper and lower lane of each panel respectively, demonstrate that all tumors present different genomic profiles except for S2007A and S2008 which have alterations in common. Deletion of CDKN2A and CDKN2B genes on the chromosome 9 is highlited. Ploidie of each tumor is indicated near each profile and the status of the CDKN2A/CDKN2B deletion in each tumor is defined. x axis: chromosome 1 to chromosome Y; y axis: weighted log2(ratio) (upper lane) and allele difference (lower lane).
Following our hypothesis of a constitutional alteration leading to the development of these tumors, we first studied the most frequently altered gene in sarcomas with complex genetics: TP5316. It showed a heterozygous loss in all tumors except in S2012A and S2016B, again with different breakpoints between the tumors. Regarding other frequently altered genes in sarcomas, one copy of chromosome 13 carrying RB1 was lost only in S2012B and S2016A and one copy of PTEN was lost only in S2008 and S2016A, indicating that these alterations were probably not the primary genetic alterations (Fig. 3).
However, even though the alterations were different between all primary tumors, those affecting the short arm of chromosome 9 have the same consequence in all tumors: the loss of CDKN2A and CDKN2B genes (Fig. 3). Breakpoints for these losses were different between the tumors, except for S2007A and S2008 (Figure S2A). The upstream breakpoints in S2007A/S2008 and S2012A were located at different positions in the MLLT3 gene (Figure S2A). According to the weighted Log2ratio and allele difference, the loss of CDKN2A and CDKN2B was homozygous in the tumors developed between 2007 and 2012 (Fig. 3 and Figures S2A and B). Regarding the tetraploid tumors developed in 2016, the loss was heterozygous with two copies left.
Constitutional analysis of CDKN2A/CDKN2B
Constitutional CDKN2A/B deletions have already been observed in patients with Melanoma-Astrocytoma Syndrome17 and in one patient with Li-Fraumeni syndrome18. Thus, DNA array was performed in five non-tumor tissues (Table S1). All five genomic profiles showed no such deletion on chromosome 9 nor any copy number variations (Figure S3). Furthermore, both genes were sequenced at the DNA level in non-tumor tissues. No mutation was found in the two genes (data not shown), definitively ruling out any constitutional origin of the CDKN2A/2B alteration.
Fusion transcript and mutation analyses by RNA sequencing
RNA-seq was performed to explore the presence of fusion transcripts as well as the expressed mutational profile in five tumors (S2007A, S2007B, S2008, S2012A, and S2016A).
No fusion transcript shared by all or several tumors was detected, except EEF1DP3-FRY which is actually a read-through (Table S5)19.
Regarding mutations, bioinformatic analysis detected 650,497 variations present in at least one tumor. After applying several filters to reduce the number of mutations, only six variations present in all samples were kept as potential real mutations and not polymorphisms or artefacts (Tables S2 and S3). These variations affected the following genes: AZIN1 (c.A1099G), COG3 (c.A1903G), COPA (c.A490G), GATAD2A (c.A65G), GLT8D1 (c.C955G) and SLC25A39 (c.C809T), and were all verified by Sanger sequencing on tumors and non-tumor DNA. The variations predicted in AZIN1, COG3 and COPA were not found by Sanger sequencing (Table S2), suggesting that they were artefacts. The variations in GATAD2A (c.A65G), GLT8D1 (c.C955G) and SLC25A39 (c.C809T) were validated in all sequenced cases both in non-tumor and tumor samples (Fig. 4 and Figure S4), showing that these variations were constitutive mutations.

Validated genomic mutations. Sequence chromatograms showing GLT8D1, GATAD2A and SLC25A39 mutations observed on genomic DNA in two healthy tissues (HT2012, HT2013) and two tumors (S2008, S2012A) (Sequence viewer: FinchTV, Geospiza). Frames indicate mutation sites (hg38, GLT8D1: chr3:g.52695006G > C, GATAD2A: chr19:g.19465410A > G, SLC25A39: chr17:g.44320429C > T). Allelic status is indicated for each case and number of copies of each gene, determined according to DNA-array data, in each tumor is also presented. MUT mutated allele, WT wild-type allele.
GATAD2A and SLC25A39 mutations were heterozygous in all cases (Fig. 4 and Figure S4) and both alleles were expressed according to RNA-seq data (Table S3). Interestingly, the mutation in GLT8D1 was associated with a deletion (distinct from one sample to another) of one copy in five of the seven tumors. It was homozygous in four diploid cases (S2007A, S2007B, S2008 and S2012A) and was identified in two copies in the tetraploid S2016A (with one copy of the WT allele) (Fig. 4, Figure S4 and Figure S5). In addition, only the mutated allele was expressed in the five RNA-sequenced cases, and even in S2016A which still retained WT alleles (alternative allele quantification ranges from 82.6% to100% with very low remaining expression of the WT allele likely due to contaminating normal cells) (Table S3).
The three mutations were missense. Substitution in GATAD2A (D22G) changed an uncharged polar amino acid (asparagine) to a non-polar amino acid (glycine), while the one in GLT8D1 (Q319E) led to the replacement of an uncharged polar amino acid (glutamine) by a negatively charged polar amino acid (glutamic acid). Finally, the mutation in SLC25A39 (A270V) gave rise to the change of an aliphatic hydrophobic non-polar amino acid (alanine) into an amino acid sharing the same features (valine) (Table S3). According to PhosphoSitePlus (v6.5.9.1v6.5.9.1) all three changed amino acids are maintained between human and mouse and are not related to post-translational modifications.
The effect of the missense mutations on the function of the altered gene was therefore unclear. Functional prediction algorithms almost all agreed that the mutation in GATAD2A (D22G) was neutral. Predictions about the mutation in GLT8D1 (Q319E) were more ambiguous with four algorithms in favor of a deleterious effect and five others suggesting that the effect is minor. Finally, nine out of the 10 algorithms used presented the mutation in SL25A39 (A270V) as deleterious (Table S3). However, regarding the GLT8D1 mutation, its association with a deletion in five cases and a loss of WT allele expression in all studied tumors suggested a loss of function of the GLT8D1 gene.
Discussion
For this unique case of a 72-year-old patient who first presented with glomangiopericytal tumor and then six sarcomas of the extremities between 2007 and 2016, histological examination of the tumors could not discriminate between too clinically distinct situations, i.e. a unique disease with local/distant recurrences or a succession of different diseases.
The first issue was thus to establish whether all the tumors were clonally related or independent from each other. Genomic profile analysis showed that the S2008 tumor was the local recurrence of the S2007A tumor and that the other tumors were not clonally related.
Second, the presence of six different sarcomas in the same patient prompted us to look for a common tumorigenic alteration that might be constitutional. The only shared alteration between all tumors was the loss of the CDKN2A/2B genes that resulted from chromosome 9 rearrangements in all non-related tumors, but which is not constitutional because it is not observed in the non-tumor tissues. This alteration has already been described in sarcomas with complex genetics6,20. Interestingly, although the breakpoints for these losses are different in each tumor, they all occur in a narrowed region around CDKN2A/2B genes, and for three tumors the breaks occurred in the MLLT3 gene. MLLT3 is one of the most common fusion partner genes of the MLL gene resulting in the t(9;11)(p22;q23) detected in acute myelogenous leukemia (AML) and in acute lymphocytic leukemia (ALL)21. Strissel et al. identified several common structural DNA elements between MLLT3 and MLL genes and proposed a DNA breakage and repair model in which a non-homologous chromosomal recombination with subsequent DNA repair, could result in translocations between the two genes21. Thus, a particular chromatin state of this region may have promoted the rearrangement in MLLT3 or its surrounding region, leading to the loss of CDKN2A/2B loci.
Third, no fusion transcript shared by all tumors was found and only variations in three genes were detected both in tumor and non-tumor tissues: SLC25A39 (c.C809T), GLT8D1 (c.C955G) and GATAD2A (c.A65G). Analysis in cBioPortal revealed that mutations in these genes have been very rarely reported in sarcomas22,23. In the 255 sarcomas of the TCGA PanCancer Atlas Studies, alterations were found for SLC25A39, GLT8D1 and GATAD2A in 0.78%, 1.57%, and 2.75% of cases, respectively24,25.
SLC25A39 is located on chromosome 17q21.31 and encodes a protein required for normal heme biosynthesis26. Little is known about the functions of SLC25A39 and only its role in erythropoiesis and neural functions have been described26,27. The SLC25A39 missense mutation observed in the patient (A270V) was heterozygous with both alleles expressed and occurred in one of the mitochondrial carrier domains of the protein28, potentially modifying the function of the protein.
GATAD2A (GATA Zinc Finger Domain Containing 2A), located on chromosome 19p13.11, codes for the p66α protein, which is a subunit of the nucleosome remodeling and histone deacetylation (NuRD) complex, itself implicated in transcription regulation through chromatin compaction and decompaction29. At the transcriptional level, the NuRD complex is recruited by tissue-specific oncogenic transcription factors to repress the expression of tumor suppressor genes, while at the post-translational level, it has been shown to deacetylate p53 to inactivate p53-induced apoptosis30. It has also been detected at replication forks and ensures proper DNA replication, cellular proliferation and protection of genome integrity31. Moreover, it has been shown that GATAD2A/NuRD can be recruited to sites of DNA damage to promote repair by homologous recombination32. The mutation found here (D22G) occurred at an amino acid not located in an identified functional domain33. Functional prediction algorithms almost all agree that the mutation in GATAD2A is neutral. While the NuRD complex is known to play several important emerging roles in cancer biology31, the involvement of GATAD2A in cancer is still poorly understood. Nevertheless, a genome-wide meta-analysis of breast, ovarian and prostate cancers identified three cancer susceptibility loci associated with intronic variants of GATAD2A30.
GLT8D1 (glycosyltransferase 8 domain containing 1), located on chromosome 3p21.1, encodes a glycosyltransferase enzyme of unknown function, ubiquitously expressed and localized in the Golgi apparatus34. Amyotrophic Lateral Sclerosis (ALS), a severe neurodegenerative disorder, has been ascribed to missense mutations in GLT8D135. All mutations found in familial ALS cases and in early-onset sporadic ALS arise in GLT8D1 exon 4, which encodes the substrate-binding domain of GLT8D1 and is associated with reduced enzymatic activity. Thus, pathogenic GLT8D1 mutations are thought to be autosomal dominant mutations that are associated with haploinsufficiency and/or a dominant-negative effect34. The missense GLT8D1 mutation found in the patient (Q319E) is located in exon 10 and the modified amino acid is located in the glycosyltransferase domain of the protein, like ALS mutations, but in a more C-terminal part35. GLT8D1 overexpression was also recently reported in melanoma to be associated with worse overall survival and progression-free survival36. Unfortunately, no material was available from the melanoma in our patient. Consensus is lacking about the deleterious effect of this mutation. Deletion of GLT8D1 in five tumors and homozygous expression of the mutated allele in all of them, is consistent with a loss of function of the GLT8D1 gene. The deletion could thus be an early event acquired during the first steps of the oncogenic process, suggesting that its functional loss participates in the tumor inception.
Finally, a potential role for SLC25A39 and GATAD2A in tumor development in this patient is weakly supported by literature and the most likely candidate gene appears to be GLT8D1.
The predisposing impact of this mutation must be moderate because, even if the patient developed many tumors, they all occurred late in life. It’s more likely that it could deregulate aging cells. In addition, the cell type in which this deregulation occurred might be at the origin of the different types of cancer developed by the patient. For example, if melanocytes were to be affected, this would result in a melanoma37. Likewise, if different mesenchymal cell types are impacted, it could lead to the development of LMS, UPS or MFS38,39.
Obviously, a thorough experimental evaluation is mandatory to conclude on the definitive involvement of one of these candidates in sarcoma’s, predisposition/inception.
References
Ducimetière, F. et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS ONE 6, e20294 (2011).
Coindre, J.-M. Grading of soft tissue sarcomas. Arch. Pathol. Lab. Med. 130, 6 (2006).
Jo, V. Y. & Fletcher, C. D. M. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 46, 95–104 (2014).
Norberg, S. M. & Movva, S. Role of genetic and molecular profiling in sarcomas. Curr. Treat. Options Oncol. 16, 24 (2015).
Guillou, L. & Aurias, A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 456, 201–217 (2010).
Pérot, G. et al. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am. J. Pathol. 177, 2080–2090 (2010).
Ognjanovic, S. et al. Sarcomas in TP53 germline mutation carriers. Cancer 118, 1387–1396 (2012).
Lesluyes, T. et al. RNA sequencing validation of the complexity INdex in SARComas prognostic signature. Eur. J. Cancer 57, 104–111 (2016).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Delespaul, L. et al. Recurrent TRIO fusion in nontranslocation-related sarcomas. Clin. Cancer Res. 23, 857–867 (2017).
Iwato, M., Tachibana, O., Tohma, Y., et al. Alterations of the INK4a/ARF Locus in Human Intracranial Germ Cell Tumors, Vol. 4.
Laud, K. et al. Comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J. Med. Genet. 43, 39–47 (2006).
Hui, J. Y. C. Epidemiology and etiology of sarcomas. Surg. Clin. N. Am. 96, 901–914 (2016).
Soura, E. et al. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Dermatol. 74, 395–407 (2016) ((quiz 408–410)).
Chan, S. H. et al. Germline hemizygous deletion of CDKN2A–CDKN2B locus in a patient presenting with Li–Fraumeni syndrome. NPJ Genom. Med. 1, 16015 (2016).
Kim, J. et al. Recurrent fusion transcripts detected by whole-transcriptome sequencing of 120 primary breast cancer samples. Genes Chromosomes Cancer 54, 681–691 (2015).
Ogura, K. et al. Integrated genetic and epigenetic analysis of myxofibrosarcoma. Nat. Commun. https://doi.org/10.1038/s41467-018-03891-9 (2018).
Strissel, P. L. et al. DNA structural properties of AF9 are similar to MLL and could act as recombination hot spots resulting in MLL/AF9 translocations and leukemogenesis. Hum. Mol. Genet. 9, 1671–1679 (2000).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Cerami, E. et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Sanchez-Vega, F. et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 173(321–337), e10 (2018).
Hoadley, K. A. et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173(291–304), e6 (2018).
Nilsson, R. et al. Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab. 10, 119–130 (2009).
Haitina, T. et al. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 88, 779–790 (2006).
Yu, X. X. et al. Overexpression of the human 2-oxoglutarate carrier lowers mitochondrial membrane potential in HEK-293 cells: Contrast with the unique cold-induced mitochondrial carrier CGI-69. Biochem. J. 353, 369–375 (2001).
Torchy, M. P., Hamiche, A. & Klaholz, B. P. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol. Life Sci. 72, 2491–2507 (2015).
Kar, S. P. et al. Genome-wide meta-analyses of breast, ovarian and prostate cancer association studies identify multiple new susceptibility loci shared by at least two cancer types. Cancer Discov. 6, 1052–1067 (2016).
Lai, A. Y. & Wade, P. A. NuRD: A multi-faceted chromatin remodeling complex in regulating cancer biology. Nat. Rev. Cancer 11, 588–596 (2011).
Gong, F. et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 29, 197–211 (2015).
Brackertz, M. et al. p66alpha and p66beta of the Mi-2/NuRD complex mediate MBD2 and histone interaction. Nucleic Acids Res. 34, 397–406 (2006).
Moll, T., Shaw, P. J. & Cooper-Knock, J. Disrupted glycosylation of lipids and proteins is a cause of neurodegeneration. Brain 143, 1332–1340 (2020).
Cooper-Knock, J. et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 26(2298–2306), e5 (2019).
Hu, H. et al. GLT8D1 overexpression as a novel prognostic biomarker in human cutaneous melanoma. Melanoma Res. 29, 612–620 (2019).
van der Kooij, M. K. et al. Uveal versus cutaneous melanoma; same origin, very distinct tumor types. Cancers (Basel). https://doi.org/10.3390/cancers11060845 (2019).
Chibon, F., Darbo, E. & Pérot, G. Leiomyosarcomas: Whole genome sequencing for a whole biology characterization. Curr. Opin. Oncol. 31, 317–321 (2019).
Soini, Y. Epigenetic and genetic changes in soft tissue sarcomas: A review. APMIS 124, 925–934 (2016).
Acknowledgements
The authors would like to thank the Centre Nacional d'Anàlisi Genòmica (CNAG, Barcelona, Spain) for RNA sequencing. They also thank Dr. Ray Cooke for copyediting the manuscript and the department of Clinical Research of Amiens University Hospital for funding.
Funding
Department of Clinical Research, Amiens University Hospital, France.
Author information
Authors and Affiliations
Contributions
The authors contributed to this research study as follows: Conception and design of the study: A.B., G.P., B.C. and F.C. Histological analysis of tumors: H.S. and S.L.G. Experiments realization: G.P. Bioinformatic analysis: N.T. Analysis and interpretation of the data: A.B., G.P., B.C., and F.C. Funding acquisition: A.C. and B.C. Manuscript writing: A.B. and G.P. Manuscript review and editing: A.B., G.P., B.C., F.C., N.T., S.L.G., H.S. and A.C. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Beddok, A., Pérot, G., Le Guellec, S. et al. Germinal GLT8D1, GATAD2A and SLC25A39 mutations in a patient with a glomangiopericytal tumor and five different sarcomas over a 10-year period. Sci Rep 11, 9765 (2021). https://doi.org/10.1038/s41598-021-88671-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88671-0
This article is cited by
-
Glycosyltransferase 8 domain-containing protein 1 (GLT8D1) is a UDP-dependent galactosyltransferase
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
