
Abstract
Although several potent bile acid Farnesoid X receptor (FXR) and Takeda G-protein-coupled receptor 5 (TGR5, GPBAR1) dual agonists such as INT-767 have been reported, no non-bile acid FXR/TGR5 dual agonist has been investigated to date. Therefore, we attempted to discover potent non-bile acid FXR/TGR5 dual agonists and identified some non-bile acid FXR/TGR5 dual agonists, such as isonicotinamide derivatives in vitro assay. Compound 20p was evaluated in C57BL/6J mice, that were administered a choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD) consisting of 60 kcal% fat and 0.1% methionine by weight for one week. Compound 20p dose-dependently induced small heterodimer partner (SHP) mRNA and decreased cytochrome P450 7A1 (CYP7A1) in the liver at 10 and 30 mg/kg, respectively, which were used as FXR agonist markers. Compound 20p significantly increased the plasma levels of GLP-1 as a TGR5 agonist, and a high concentration of GLP-1 lowered blood glucose levels. We confirmed that compound 20p was a non-bile acid FXR/TGR5 dual agonist.
Similar content being viewed by others


Introduction
Type 2 diabetes and nonalcoholic fatty liver disease (NAFLD) are serious hepatic disease events1. Metabolic syndromes, including insulin resistance, hypertension, dyslipidemia, and obesity, are closely associated with the pathogenesis of NAFLD2. Approximately 20–30% NAFLD patients develop nonalcoholic steatohepatitis (NASH)3, which can lead to cirrhosis and hepatocellular carcinoma4,5.
The farnesoid X receptor (FXR, NR1H4), a member of the nuclear receptor (NR) superfamily, is a ligand-induced transcription factor6,7. The expression levels of FXR are high in the liver, intestine, kidney, and adrenal glands8,9,10. The bile acid (BA), chenodeoxycholic acid (CDCA), is an endogenous ligand against FXR. BAs play key roles in regulating cholesterol and bile acid homeostasis11. Activation of FXR in the liver induces a downregulation of CYP7A1 gene transcription. CYP7A1 regulates the conversion of free cholesterol to BAs, and the downregulation of CYP7A1 upregulates a small heterodimer partner (SHP)12.
Obeticholic acid (OCA, INT-747, 2) is the first steroidal FXR agonist. In the combination with ursodeoxycholic acid, it reduces alkaline phosphatase levels in patients with primary biliary cirrhosis13. A proof-of-concept study reported that OCA significantly improves insulin sensitivity and reduces liver inflammation and fibrosis markers, such as alanine aminotransferase (ALT) and enhanced liver fibrosis score, in patients with type 2 diabetes and NAFLD14. OCA can also be used for the treatment of patients with NASH. It has been reported that 25 mg/day of the FXR ligand, OCA, improves NAFLD activity score (NAS) and fibrosis status in the livers of noncirrhotic, nonalcoholic steatohepatitis patients. However, OCA causes pruritus as a clinical adverse event15.
Several potent and selective FXR agonists16,17,18, such as GW4064 (4), have been reported. GW4064 is a selective nonsteroidal FXR agonist19 that significantly reduces free fatty acid and triglyceride levels in db/db mice20.
Takeda G-protein-coupled receptor 5 (TGR5, GPBAR) is a class A G-protein-coupled receptor (GPCR) that is expressed in the liver, skeletal muscle, intestine, and brown adipose tissues21. Activation of TGR5 by BA stimulates glucagon-like peptide-1 (GLP-1) secretion from intestinal enteroendocrine L cells by increasing intracellular cAMP concentration22,23,24. A TGR5 agonist reduces lipopolysaccharide (LPS)-induced release of proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin (IL)-1, IL-6, and IL-8. Thus, TGR5 agonists may have broad therapeutic applications, from the treatment of metabolic disorders to liver inflammatory diseases25.
INT-767 (3), a steroidal FXR/TGR5 dual agonist, decreases serum cholesterol and triglyceride levels in diabetic db/db mice and streptozotocin-induced type 1 diabetes model mouse26. Moreover, INT-767 reduces the production of inflammatory markers, such as IL-1beta, cytokine, and TNF-α, in diabetic db/db mice and apolipoprotein E-deficient mice27,28. Treatment with INT-767 for 16 weeks significantly improves histological parameters as compared to OCA treatment. Furthermore, INT-767 leads to a significant reduction in NAS and fibrosis in ob/ob NASH mice model as compared to OCA29.
Crystallographic studies have enabled a clear understanding of the interaction between FXR agonists and the ligand binding domain (LBD), making FXR an attractive target. A cocrystal structure of GW4064 with the active site of FXR protein has been reported (PDB code 4DCT)30. The isoxazole moiety of GW4064 interacts with Trp454 and His447 in helix 11, forming an edge-to-face stacking interaction with Trp469 located in helix 12 (AF2). The carboxylic acid of GW4064 forms an electrostatic interaction with Arg331 in helix 5. A cocrystal structure of compound 7 with the active site of FXR has been reported (PDB code 1OSH). However, the insertion regions of 15 residues between helices 1 and 3 are completely disordered31. The interactions between FXR LBD and compound 7 can be divided into two sets. The first set of interactions stabilizes the position of hexyl ring, which contacts Ile339 and Leu344. The second set comprises a biaryl ring and dimethylamine group. The dimethylamine group interacts with Trp454. The side chain of Trp454 is shifted by the dimethylamine group as compared to PDB code 3DCT. The biaryl group stabilizes His447 and Trp469.
Several non-bile acid TGR5 agonists have been reported in literature32. An isonicotinamide compound has been identified as a non-bile acid TGR5 agonist (8, Fig. 1)33,34. This compound inhibits TGR5-dependent LPS-induced TNF-α and IL-12 release in mice. These results illustrate the important regulatory roles of TGR5 agonists in controlling inflammation.

FXR and TGR5 ligands.
Several potent bile acid FXR/TGR5 dual agonists have been reported so far, but no non-bile acid FXR/TGR5 dual agonist has been investigated to date16. Therefore, in this study, we attempted to discover potent non-bile acid FXR/TGR5 dual agonists. This study reports our efforts to develop a non-bile acid FXR/TGR5 dual agonists and investigate its in vitro and in vivo activities.
Results
Fexaramine (compound 7) was reported as a specific and potent partial agonist against FXR with no significant agonist activity against TGR531. The indole derivative 8 is a TGR5 agonist (Fig. 1) with no agonist or antagonist activity against FXR33.
Presently, two categories of FXR and TGR5 agonists exist. One is structurally based on steroidal scaffolds, including bile acids, such as cholic acid and lithocholic acid, and semisynthetic steroid derivatives, such as OCA, INT-767, and INT-777, which are FXR and FXR/TGR5 dual or selective TGR5 agonists16. The other category is small molecules. Isoxazole and compound 7 derivatives are small-molecule FXR agonists18, and isoxazole, pyridine, and isonicotinamide derivatives are small molecule TGR5 agonists32,33. We observed that compound 7 showed some structural similarities to isonicotinamide derivative 8. The isonicotinamide group of 8 is linked to the cyclohexyl group of compound 7, and both compounds have an N-benzyl framework. This observation prompted us to synthesize a few chimera compounds between compound 7 and isonicotinamide 8 and to evaluate their FXR/TGR5 dual agonist activities.
To evaluate direct agonist activities against FXR, test compounds were evaluated using a CHO cell-based FXR assay by Indigo Biosciences (State College, PA), in which cells were transfected with an FXR luciferase reporter system. Cells were incubated with the test compounds for 22 h, and potency was assessed using a multi-mode microplate reader. OCA 2 was used as the reference compound for 1 µM, which we set as 100% FXR activation35,36.
TGR5 agonist activity was evaluated by transfecting CHO cells with TGR5 using the Discover X expression kit (cAMP Hunter eXpress GPCR Assay, Discover X Fremont, CA). Cells were incubated with test compounds for 0.5 h, and intracellular cAMP levels were measured using the multi-mode microplate reader. The response to 10 µM INT-767 3 was set as 100% TGR5 activation37.
Compound 7 was divided into four fragments, dimethylaminophenyl group (A), center phenyl linker (B), cyclohexyl group (C), and aniline group (D), to explore the structure activity relationship (SAR). A chimera strategy between compound 7 and isonicotinamide compound 8 enabled the modulation of cyclohexyl (C) and aniline (D) groups to rapidly assess the C/D region of molecule. To investigate the activities of some chimera compounds between compound 7 and isonicotinamide compound 8 C/D phenyl ring, we synthesized compounds 13a-13j, as shown in Fig. 2. Compound 7 was synthesized and optimized to incorporate a 3-methyl acrylate moiety for potent FXR agonist activity38. The 2-methyl isonicotinamide group is important for TGR5 agonist activity33,34, and ortho-substituted anilides, such as 2-methyl-4-fluoroanilide, in the center aniline group exhibit sub-nanomolar TGR5 agonist activities in the cAMP assay33.

(a) FXR agonist acitivity of 4-dimethylamino biphenyl derivatives 13a-13j. Efficacy (%); % vs OCA 1 µM as 100%, OCA; 100% (1 µM), 13.3% (0.1 µM); (b) TGR5 agonist activity of 4-dimethylamino biphenyl derivatives 13a-13j. Efficacy (%); % vs INT-767 10 µM as 100%, INT-767 100% (10 µM), 25.5% (1 µM), 1.7% (0.1 µM). (c) Structures of 4-dimethylamino biphenyl derivatives 13a-13j.
Compound 13a showed a strong FXR agonist activity with full efficacy while compounds 13b and 13c significantly decreased the FXR activity. The methyl group at 2-position and the position of nitrogen in C region are important for FXR agonist activity. Unfortunately, although a methyl acrylate moiety at the meta position in D region showed a potent FXR agonist activity, compounds 13a and 13b were inactive against TGR5. Compound 13e, which is a similar type of compound 8, showed a strong TGR5 agonist activity. Compounds 13f and 13g, however, exhibited decreased TGR5 activities. These results mimic the following SARs of Novartis compounds33,34: (1) the position of methyl group is important and (2) a pyridine isomer leads to a complete activity loss in the C region. To explore the disubstituted analogs in D region, we investigated 2, 3-, 2, 5-, and 3, 5-disubstituted analogs. Compound 13h decreased TGR5 potency; however, 13i showed a weak FXR/TGR5 dual agonist activity. The methyl acrylate group plays a critical role in FXR agonist efficacy, and the 2-methyl group in D region exhibited an agonist activity against TGR5. A significant breakthrough was achieved with the 3, 5-disubstituted aniline analog. Compound 13j exhibited a potent FXR/TGR5 dual agonist activity.
Next, we investigated the effect of substitution pattern on aniline moiety (part D), as shown in Supplementary Fig. S1. A 3, 5-disubstituted pattern was important for FXR/TGR5 dual agonist activity. Compounds 13j and 13k showed dual potencies against both FXR and TGR5. However, 13m, 13n, and 13o exhibited remarkably reduced activities against TGR5. The 2, 5-di- or 2, 3, 5-trisubstituted compounds 13p, 13q, and 13r exhibited less potency in FXR agonist assay whereas they demonstrated good potency against TGR5. The 2-methyl-3-substituted compound 13s showed a moderate FXR/TGR5 agonist activity. The 2-fluoro-3-substituted compounds 13t and 13u only showed strong FXR agonist activities, with a similar potency as OCA. These results suggested that the 3, 5-halogenated substitution pattern was essential for optimal FXR/TGR5 dual agonist potency.
We next sought to improve the dual potency against FXR/TGR5 by altering the substitution patterns on C ring (Supplementary Fig. S2.). Compounds 13v and 13w significantly decreased FXR potencies with an equipotent TGR5 agonist potency. In contrast, the 3-halogenated compounds, 13x and 13y, retained FXR potencies and reduced TGR5 potencies. The trifluoromethylpyridinyl group 13z decreased the potencies against both FXR and TGR5. The 3-methoxy group of 13aa showed a weak dual agonist activity. The pyridazine compound 13ab did not have any activity against either FXR or TGR5. A lack of nitrogen in C ring of compounds 13ac, 13ad, 13ae, 13af, and 13ag showed strong FXR activities. Compound 13ag exhibited an FXR agonist activity of 136.5%, but it demonstrated a significantly low potency against TGR5. The results of Supplementary Figs. S1 and S2 suggest that a combination of substitutions at the C/D ring would create effective selective or dual agonist compounds against FXR and/or TGR5.
Some FXR agonist compounds with cyclohexyl or bicyclo groups in the B ring have been reported39,40,41,42,43,44,45,46,47. We synthesized cyclohexyl and bicyclo groups at the B ring, and we not only evaluated FXR agonist activity but also TGR5 agonist activity (Fig. 3). Although the des-dimethylamino group of 13ah decreased the FXR agonist potency, its TGR5 agonist potency increased. Thus, compound 13ah was a TGR5 full agonist (118.6% vs. INT-767). Compound 20a exhibited a significantly increased FXR agonist activity but decreased TGR5 agonist activity. Similar trends were observed in 20d, 20e, and 20f, which were more potent partial FXR agonists and weak TGR5 agonist. The bicyclo[2.2.2]octyl group fills the hydrophobic pocket in FXR LBD. We assumed that the bicyclic ring system would fill the central portion of binding pocket, and the bulkiness and tight conformation would cause a conformational resistance of Trp454. Merk et al. recently reported that a partial agonist DM175 (9) causes an outward movement of Trp45448. Compound 7 also moved Trp454 outward. LY2562175 (6) is also a partial agonist49.

(a) FXR agonist activity of isonicotinamide derivatives 13j, 13ah, 20a–20g. Efficacy(%); % vs OCA 1 µM as 100%, OCA; 100% (1 µM), 13.3% (0.1 µM); (b) TGR5 agonist activity of isonicotinamide derivatives 13j, 13ah, 20a–20g. Efficacy(%); % vs INT-767 10 µM as 100%, INT-767 100% (10 µM), 25.5% (1 µM), 1.7% (0.1 µM). (c) Structures of isonicotinamide derivatives 13j, 13ah, 20a–20g.
Finally, we targeted the substitution of part A because the substituent would interact with Trp454. Additional SAR investigations extensively explored the substitutions of aryl rings (A, C, and D rings, Fig. 4).

(a) FXR agonist activity of bicyclo[2.2.2]octane-isonicotinamide derivatives 20f–20v. Efficacy(%); % vs OCA 1 µM as 100%, OCA; 100% (1 µM), 13.3% (0.1 µM); (b) TGR agonist activity of bicyclo[2.2.2]octane-isonicotinamide derivatives 20f–20v. Efficacy(%); % vs INT-767 10 µM as 100%, INT-767 100% (10 µM), 25.5% (1 µM), 1.7% (0.1 µM). (c) Structures of bicyclo[2.2.2]octane-isonicotinamide derivatives 20f-20v.
Compound 20f increased the FXR agonist activity to a similar potency as OCA, although it decreased TGR5 agonist potency. To further explore the impact of a bicyclo B ring, we maintained the bicyclo ring in that region and concentrated our efforts on investigating substitutions around the ring. Compound 20h showed a dual agonist activity. The 3, 4-dimethoxyphenyl group of compounds 20k and 20l exhibited decreased activities against FXR. Compounds 20m and 20n demonstrated improved potencies as FXR/TGR5 dual agonists. Compound 20o exhibited a decreased FXR agonist potency, although TGR5 agonist potency was increased, as expected. Substitution of a nitrile moiety at the 3 position of benzene ring (A ring) in 20p and 20q provided a further improvement in FXR/TGR5 dual agonist potency. These compounds exhibited strong dual agonist potencies. Compound 20r showed the most potent TGR5 activity, although its activity against FXR was less potent. Compound 20s exhibited a high potency against FXR. Introduction of a nitrogen atom in A ring (20u) decreased the dual agonist potency. Replacement of the nitrile group with chloro group in compound 20v decreased the agonist activity. Compounds 20p and 20q were promising candidates for FXR/TGR5 dual agonist.
Compound 7 is a potent FXR partial agonist in cell-based assays31. Compound 7 has been reported to possess a crystal structure of human FXR LBD31. The complex has a disordered 15-residue insertion region between helices 1 and 3. Compound 7 and its derivatives are partial agonists of FXR due to their decreased abilities to recruit coactivator SRC-1 as compared to that of the typical full agonist GW4064. The most notable impact of a partial agonist is the outward movement of Trp454 (Trp458 in PDB 1OSH) in compound 7 and the FXR-LBD complex31. The cocrystal structure indicated that this is the mechanistic difference between an agonist and partial agonist. A partial agonist causes an outward movement of Trp454 and destabilizes a loop connecting helix 12. In the agonistic conformation, the ligand interacts with Trp454 and His447, and His447 makes an edge-to-face stacking interaction with Trp469 in helix 12 (AF2), thus, stabilizing it.
Merk et al. recently reported the partial agonist DM175 and its cocrystal structure with FXR LBD48. Binding of DM175 causes an outward movement of Trp454, which is driven by site occupation by the tert-butyl moiety. The binding mode of DM175 is obviously different from the structure of the complex containing full agonists such as OCA, CDCA, and GW4064.
The agonist mechanism of action for FXR LBD is through interaction with Trp469 and His447. Moreover, the interaction with Trp454 plays an important role between FXR LBD and agonist ligands. We examined the computational molecular docking of 20p into the X-ray structure of FXR LBD (PDB 1OSH, Fig. 5a). The cocrystal structure of GW4064 with the active site of FXR protein has been reported (Fig. 5b)30. The isoxazole moiety of GW4064 interacts with Trp454 and His447 in helix 11, and His447 forms an edge-to-face stacking interaction with Trp469 located in helix 12 (AF2), which stabilizes Trp458 and recruits the co-activator. Compound 7 causes an outward movement of Trp458, thus, inducing a partial agonist activity against FXR. Ivermectin, a widely used antiparasitic drug with a distinct chemical structure from GW4064 and compound 7, promoted coactivator recruitment by FXR. Ivermectin enhanced the interaction of FXR with various coactivator LXXLL motifs from the family of steroid receptor coactivators such as SRC1, SRC2, and SRC3. Since its interaction is less than that of GW4064, ivermectin is a partial agonist50. However, ivermectin also induces the recruitment of corepressor NCoR-2 by FXR, which is not exhibited by GW4064. The results indicate a partial agonist activity of ivermectin with unique properties that modulate coregulator recruitment. The cocrystal structure of ivermectin with a corepressor instead of a coactivator showed a disordered helix 12 and loop between helices 11 and 12. Additionally, helix 12 is shortened such that the structure contains no activator but a corepressor. Therefore, the cocrystal structure between ivermectin and FXR-LBD is the antagonist form.

(a) Model showing the docking of compound 20p into the FXR ligand binding domain obtained from the X-ray crystal structure PDB 1OSH. (b) GW4064 (4) is shown for comparison (3DCT).
The compounds 20p and 20q were used to measure the EC50 value. The potencies of 20p (FXR EC50 = 190 nM, Emax = 28%, TGR5 EC50 = 420 nM) and 20q (FXR; EC50 = 79 nM, Emax = 33%, TGR5; EC50 = 790 nM) were stronger than those of OCA and INT-767 (Fig. 6).

Concentration–response curves. (a) OCA FXR EC50 value; (b) INT-767 FXR EC50 value; (c) 20p FXR EC50 value; (d) 20q FXR EC50 value; (e) INT-767 TGR5 EC50 value; (f) 20p TGR5 EC50 value; (g) 20q TGR5 EC value; (h) Structures of 20p and 20q.
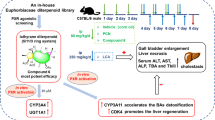
Pharmacokinetic profiles of the bicyclo compounds 20p and 20q were evaluated in rats (Supplementary Table S1). The in vivo pharmacokinetic study revealed that 20p and 20q exhibited a rather low plasma exposure at an oral dose of 3 mg/kg with Cmax values of 50.3 and 3.20 ng/mL and T1/2 values of 1.45 and 4.52 h, respectively. These results suggested that an intestinal exposure to 20p and 20q after oral dosing may be sufficient to activate TGR5 in the intestine, increase GLP-1 secretion, and decrease blood glucose levels. Moreover, Fang et al. recently reported that compound 7 acts against intestinal FXR and activates FXR agonist potency without being absorbed51. Therefore, compounds 20p and 20q were selected for further evaluation in C57BL/6J mice, that were administered a choline-deficient amino acid-defined high-fat diet (CDAHFD) consisting 60 kcal% fat and 0.1% methionine by weight for 1 week52. This model was reported an elevated ALT level, which was a parameter to detect liver injury. Histopathological results indicated hepatocellular steatosis after 1 week. CDAHFD was fed for 6 weeks, and fibrosis developed in C57BL/6J mice.
The FXR target genes, SHP mRNA and CYP7A1, were measured using real-time PCR after a single dose of 20p and 20q (100, 30, and 10 mg/kg). Compound 7 is an FXR agonist, and it increases SHP mRNA in the liver of mice53. Both compounds, 20p and 20q, dose-dependently induced SHP mRNA in the liver at 100, 30, and 10 mg/kg (Fig. 7a,b) and decreased CYP7A1 in the liver at 30 and 10 mg/kg (Fig. 7c). These results indicated that 20p and 20q are in vivo FXR agonists.

(a) Induction of SHP mRNA in mice liver by 20p and 20q following a single oral dose (100 mg/kg), compared with fexaramine 7 (100 mg/kg). (b) Induction of SHP mRNA in mice liver by 20p and 20q (10, 30 mg/kg). #p < 0.05, ##p < 0.01, significant difference from a control group, as analyzed by Student’s t-test., **p < 0.01 significant difference from a control group, as analyzed by Dunnett’s multiple comparison test. n = 5, control n = 10. (c) Repression of CYP7A1 in mice liver by 20p and 20q following a single oral dose (10, 30 mg/kg). **p < 0.01 significant difference from the control group, as analyzed by Dunnett’s multiple comparison test. n = 5, control n = 10.
After identifying the compounds with a high potency against TGR5, 20p and 20q were evaluated for in vivo GLP-1 secretion in C57BL/6J mice. A single oral administration of 20p and 20q at a dose of 100 mg/kg increased plasma GLP-1 levels, as assessed by an oral glucose tolerance test (OGTT). 20p and 20q significantly increased the plasma levels of GLP-1 (Fig. 8a).

(a) GLP-1 secretion study of 20p and 20q in C57 mice. (b) Blood glucose levels of 20p and 20q on OGTT in C57 mice. #p < 0.05, ##p < 0.01, significant difference from the control group, as analyzed by Student’s t-test.
Compounds 20p and 20q elevated the plasma levels of GLP-1, and a high concentration of GLP-1 could lower blood glucose levels. The favorable GLP-1 secretion results encouraged us to further evaluate the in vivo efficacy in C57 mice. A single oral dose of 20p robustly lowered the blood glucose level, although a single oral dose of 20q did not reduce the blood glucose level (Fig. 8b).
Compound 20p was screened against a broad panel of NRs and GPCRs to determine its agonist activity in vitro. Compound 20p showed no significant off-target activity against GPCRs and NRs. Binding assays for RXR alpha, ER alpha, and ER beta (EC50 > 30 µM) and cellular and NR functional assays for GPR40, GPR120, GPR119, LXR alpha, LXR beta, CAR, PPAR alpha, PPAR delta, PPAR gamma, and PXR (EC50 > 30 µM) are included in additional data of S4 Supplementary information.
Discussion
We identified 20p (FXR EC50 = 190 nM, Emax = 28%, TGR5 EC50 = 420 nM) as a non-bile acid FXR/TGR5 dual agonist. The potency of 20p was stronger than those of OCA and INT-767 in vitro assay. Compound 20p was evaluated in C57BL/6 J mice, that were administered CDAHFD consisting of 60 kcal% fat and 0.1% methionine by weight for one week. Elevated ALT levels were observed in the model after one week of initiation of feeding, and histopathological results indicated hepatocellular steatosis after 1 week52. Moreover, after feeding CDAHFD for 6 weeks, fibrosis was developed in C57BL/6J mice52. Compound 20p dose-dependently induced SHP mRNA and decreased CYP7A1 expression in the liver. The compound 20p significantly increased the GLP-1 level in C57BL/6J mice. The in vivo glucose-lowing study indicated that a single oral dose of 20p significantly reduced blood glucose levels. These results indicate that 20p is an FXR/TGR5 dual agonist.
Although several potent steroidal FXR/TGR5 dual agonists have been reported, no non-steroidal FXR/TGR5 dual agonist has been reported to date. A variety of isonicotinamide derivatives were designed, synthesized, and evaluated as a series of highly potent non-bile acid FXR/TGR5 dual agonists. The EC50 values of agonist activities against FXR and TGR5 for some isonicotinamide derivatives were below 1 µM. An in vivo glucose-lowering study reported that a single oral dose of 20p significantly reduces blood glucose levels. Compound 20p significantly increased the GLP-1 level. Intestine-restricted FXR agonist compound 7 is known to stimulate serum GLP-1 secretion in wild-type C57BL/6J mice for 7 days but not in TGR5-/- or FXR-/- mice54. In our model, a single dose of compound 7 did not increase serum GLP-1 in the wild-type mice. It has been reported that compound 7 modulates gut microbiomes and turns taurochenodeoxycholic acid into lithocholic acid (LCA) by the gut microbiomes, and the LCA stimulates TGR5 expression and TGR5-mediated cAMP signaling and increases GLP-1 secretion54. The GLP-1 secretion of compounds 20p and 20q would be a direct action of TGR5 agonist activities via TGR5.
Methods
In vitro FXR agonist assay protocol
To directly evaluate the in vitro agonist activity towards FXR, test compounds were evaluated in CHO cells transfected with FXR responsive luciferase reporter (Nuclear Receptor & In Vitro Toxicology Solutions, Indigo Biosciences, State College, PA, USA). Cells were incubated with the test compounds for 22 h, and potency was assessed using Multi-Mode Microplate Reader (FlexStation 3, Molecular Devices Inc., San Jose, CA, USA). Efficacies are reported relative to OCA, which was set as 100% FXR activation at a concentration of 1 µM. Each compound was tested in duplicate, and the average value was reported35,36.
The cell recovery medium (CRM, Human Farnesoid X Receptor (NR1H4, FXR reporter assay system, Indigo Biosciences, State College, PA, USA) and compound screening medium (CSM, Human Farnesoid X Receptor (NR1H4, FXR reporter assay system, Indigo Biosciences, State College, PA, USA) were removed from the freezer and thawed in a water bath at 37 °C. Test compounds were dissolved in dimethyl sulfoxide (DMSO, FUJIFILM Wako Pure Chemical Corporation, Osaka city, Osaka, Japan), and the treatment medium was prepared and diluted with CSM to achieve a final concentration of 0.2% total DMSO. The reporter cells were thawed by transferring 3.3 mL CRM at 37 °C into tubes of frozen cells. The tube containing reporter cells was recapped, and it was immediately place in a 37 °C water bath for 5 min. The reporter cells were gently inverted several times, and 100 µL cell suspension was dispensed into each well. One hundred microliters of treatment media was added into wells. Subsequently, the assay plate was kept at 37 °C, and it was incubated with 5% CO2 for 22 h. To prepare luciferase detection reagent (LDR), the detection substrate was gently mixed with detection buffer, media contents were removed from each well, and 100 µL LDR was added to each well of the assay plate. The assay plate was allowed to rest at room temperature for 5 min, and luminescence was quantified using the Multi-Mode Microplate Reader (FlexStation 3, Molecular Devices Inc., San Jose, CA, USA)36.
In vitro TGR5 agonist assay protocol
TGR5 agonist activity was evaluated by transfecting CHO cells with TGR5 using the Discover X expression kit (cAMP Hunter eXpress Assay Kit, Discover X Fremont, CA, USA). Cells were incubated with the test compounds for 0.5 h, and intracellular cAMP levels were measured using Multi-Mode Microplate Reader (FlexStation 3, Molecular Devices Inc., San Jose, CA, USA). The response to 10 µM INT-767 3 was set as 100% TGR5 activation37.
Each compound was tested at concentrations of 10, 1, or 0.1 µM in duplicate (n = 2), with the average value shown. EC50 value of compounds 20p and 20q were determined by using a 4 parameter logistic equation, Y = min + (max − min)/(1 + 10^(n * (log10(EC50) − log10(conc)))), in nonlinear least squares fitting using the Microsoft Excel Solver55.
In vivo assay protocol (FXR)
Test compounds were orally administered to 7-week old male C57BL/6J mice (Nippon Charles River Co., Ltd., Yokohama City, Kanagawa, Japan), that were administered CDAHFD (#A06071302, Research Diets, 20 Jules Lane, New Brunswick, NJ, USA) consisting 60 kcal% fat and 0.1% methionine by weight for 7 days. After four hours of administration, the liver was removed, and total RNA was extracted using a Pure Link RNA Mini Kit (Invitrogen, Thermo Fisher Scientific K. K., Minato-ku, Tokyo, Japan). The concentration of total RNA was measured using Micro Sample Spectrophotometer (SimpliNano, GE Healthcare UK Ltd, Amersham Place Little Chalfont Buckinghamshire, HP7 9NA UK), and cDNA was synthesized using ReverTra Ace qPCR RT Master Mix (Toyobo Co., Ltd., Osaka City, Osaka, Japan). Real-time PCR was performed using a CFX96 Touch Real-Time PCR Analysis System (CFX Manager, Bio-Rad, 1000 Alfred Nobel Drive, Hercules, CA, USA), and the mRNA expression levels of SHP and CYP7A1, the target genes of FXR, were measured using the intercalator method52.
In vivo assay protocol (GLP-1)
Measurement of GLP-1 secretion: To measure plasma GLP-1 levels, 100 mg/kg of compounds 20p and 20q were administered orally to overnight-fasted C57 mice (n = 5 animals/group). One hour later, all mice were challenged with 2 g/kg glucose, and blood samples were collected after 15 min of glucose challenge test. Plasma GLP-1 levels were measured using the GLP-1 ELISA Kit Wako, High Sensitive (FUJIFILM Wako Pure Chemical Corporation, Osaka city, Osaka, Japan)56.
In vivo assay protocol (OGTT test)
To measure blood glucose levels, 100 mg/kg of test compounds 20p and 20q or vehicle were administered orally to overnight-fasted C57 mice (n = 5 mice/group, control; n = 10). One hour later, all mice were challenged with 2 g/kg glucose, and blood samples were collected after 15 min of glucose loading. The blood glucose levels were measured using LabAssay Glucose (FUJIFILM Wako Pure Chemical Corporation, Osaka City, Osaka, Japan)56.
In vivo in-house animal guidelines
Study protocols were designed and refined by taking reduction of animal use into consideration. The study protocol was approved by the Animal Care and Utilization Committee of Fuji Yakuhin Research Laboratories, and all methods complied. We carried out in the compliance with ARRIVE guidelines. A statement to confirm that all methods were carried out in accordance with relevant guidelines and regulations.
General synthesis
General synthesis procedures were conducted as described in Supplementary S5.
The general synthesis of isonicotinamide derivative 13 was conducted as described in Supplementary Fig. S3.
Substituted-phenyl-cycloalkyl compounds were synthesized as described in Supplementary Fig. S440.
Substituted-phenyl-bicyclo[2,2,2]octane analogs were synthesized as described in Supplementary Fig. S5 (route 1)43. Some substituted-phenyl-bicyclo[2,2,2]octane compounds were synthesized43,44,45,46,47.
The direct cross-coupling of substituted aromatic ring (A region) and bicyclo[2,2,2]octane (B region) is effective for the synthesis and evaluation of bicyclo[2,2,2]octane compounds. Baran et al. recently reported a cross-coupling method of Redox-Active Ester (RAE) with organometallic species such as a borane, nickel, and iron-catalyst57,58,59,60,61. Using an iron-based organometallic catalyst, such as tris(2,4-pentanedionato)iron (III) (Fe(acac)3), enables the formation of the C–C bond between tertiary alkyl carboxylic acids and aryl magnesium coupling59,61. The iron-catalyzed cross-coupling of RAE was a good opportunity for us to optimize the A ring region with bicyclo[2,2,2]octane derivatives. We synthesized C–C coupling of the N-hydroxyphthalimide-based ester compound 35 derived from 4-(methoxycarbonyl) bicyclo[2,2,2]octane-1-carboxylic acid 31 with Fe(acac)3 and aryl magnesium bromide 36 in the presence of 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, as shown in Supplementary Fig. S6.
References
Marchesini, G. et al. Nonalcoholic fatty liver disease A feature of the metabolic syndrome. Diabetes 50, 1844–1850 (2001).
Farrell, G. C. The liver and the waistline: Fifty years of growth. J. Gastroenterol. Hepatol. 24, S105–S118 (2009).
Sanyal, A. J. NASH: A global health problem. Hepatol. Res. 41, 670–674 (2011).
Singh, S. et al. Fibrosis progression in nonalcoholic fatty liver versus nonalcoholic steatohepatitis: A systematic review and metaanalysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 13, 643–654 (2015).
Vernon, G., Baranova, A. & Younossi, Z. M. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharamacol. Ther. 34, 274–285 (2011).
Forman, B. M. et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell 81, 687–693 (1995).
Mangelsdorf, D. J. & Evans, R. M. The RXR heterodimers and orphan receptors. Cell 83, 841–850 (1995).
Makishima, M. et al. Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 (1999).
Parks, D. J. et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 284, 1365–1368 (1999).
Wang, H., Chen, J., Hollister, K., Sowers, L. C. & Forman, B. M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553 (1999).
Sinal, C. J. et al. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 102, 731–744 (2000).
Lu, T. T. et al. Molecular Basis for Feedback Regulation of Bile Acid Synthesis by Nuclear Receptors. Mol. Cell 6, 507–515 (2000).
Hirschfield, G. M. et al. Efficacy of Obeticholic Acid in Patients With Primary Biliary Cirrhosis and Inadequate Response to Ursodeoxycholic Acid. Gastroenterology 148, 751–761 (2015).
Mudaliar, S. et al. Efficacy and safety of the farnesoid x receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 (2013).
Neuschwander-Tetri, B. A. et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicenter, randomized, placebo-controlled trial. Lancet 385, 956–965 (2015).
Xu, P. Recent progress on bile acid receptor modulators for treatment of metabolic diseases. J. Med. Chem. 59, 6553–6579 (2016).
Gege, C., Kinzel, O., Steeneck, C., Schulz, A. & Kremoser, C. Knocking on FXR’s door: The “Hammerhead”-structure series of FXR agonists—amphiphilic isoxazoles with potent in vitro and in vivo activities. Curr. Top. Med. Chem. 14, 2143–2158 (2014).
Carotti, A. et al. Beyond bile acids: Targeting farnesoid X receptor (FXR) with natural and synthetic ligands. Curr. Top. Med. Chem. 14, 2129–2142 (2014).
Maloney, P. R. et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J. Med. Chem. 43, 2971–2974 (2000).
Jiang, T. et al. Farnesoid X receptor modulates renal lipid metabolism fibrosis, and diabetic nephropathy. Diabetes 56, 2485–2493 (2007).
Vassileva, G. et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem. J. 398, 423–430 (2006).
Watanabe, M. et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormoneactivation. Nature 439, 484–489 (2006).
Maruyama, T. et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J. Endocrinol. 191, 197–205 (2006).
Katsuma, S., Hirasawa, A. & Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 329, 386–390 (2005).
Keitel, V., Donner, M., Winandy, S., Kubitz, R. & Haussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 372, 78–84 (2008).
Rizzo, G. et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol. Pharamacol. 78, 617–620 (2010).
McMahan, R. H. et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J. Biol. Chem. 288, 11761–21170 (2013).
Miyazaki-Anzai, S., Masuda, M., Levi, M., Keenan, A. L. & Miyazaki, M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS ONE 9, e108270 (2014).
Roth, J. D. et al. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J. Gastroenterol 24, 195–210 (2018).
Akwabi-Ameyaw, A. et al. Conformationally constrained farnesoid X receptor (FXR) agonists: Naphthoic acid-based analogs of GW 4064. Bioorg. Med. Chem. Lett. 18, 4339–4343 (2008).
Downes, M. et al. A chemical, genetic, and structural analysis of the nuclear bile acid receptor FXR. Mol. Cell 11, 1079–1092 (2003).
Gioiello, A., Rosatelli, E., Nuti, R., Macchiarulo, A. & Pellicciari, R. Patented TGR5 modulators: A review (2006−present). Expert Opin. Ther. Pat. 22, 1399–1414 (2012).
Högenauer, K. et al. G-protein-coupled bile acid receptor 1 (GPBAR1, TGR5) agonists reduce the production of proinflammatory cytokines and stabilize the alternative macrophage phenotype. J. Med. Chem. 57, 10343–10354 (2014).
Arista, L., Högenauer, K., Scmiedeberg, N., Werner, G. & Jaksche H. Heterocyclic Amides for Use as Pharmaceuticals. WO2007/110237 (2007).
Mary, C. et al. Characterization of EDP−305, a highly potent and selective Farnesoid X Receptor agonist, for the treatment of non-alcoholic steatohepatitis. Int. J. Gastroenterol. 3, 4–16 (2019).
https://indigobiosciences.com/indigo-kits-services/farnesoid-x-receptor-fxr-nr1h4/, Indigo biosciences,Human FXR Reporter Assay System, 1 x 96-well format assays kit.
Nicolaou, K. C. et al. Discovery and optimization of non-steroidal FXR agonists from natural product-like libraries. Org. Biomol. Chem. 21, 908–920 (2003).
Smith. N. D., Govek, S. P. & Nagasawa. J. Y. Farnesoid X Receptor Agonist and Uses Thereof. WO2017/049172 (2017).
Smith. N. D., Govek, S. P. & Nagasawa. J. Y. Farnesoid X Receptor Agonist and Uses Thereof. WO2017/049173 (2017).
Smith, N. D., Govek, S. P. & Douglas, K. L. Farnesoid X Receptor Agonist and Uses Thereof. WO2017/049176 (2017).
Smith, N. D. & Govek S. P. Farnesoid X Receptor Agonist and Uses Thereof. WO2017/049177 (2017).
Smith, N. D., Govek, S. P., Nagasawa, J. Y. & Douglas, K. L. Farnesoid X Receptor Agonist and Uses Thereof. WO2018/170165 (2018).
Smith, N. D., Govek, S. P. & Nagasawa, J. Y. Farnesoid X Receptor Agonist and Uses Thereof. WO2018/170166. (2018).
Smith, N. D., Govek, S. P. & Nagasawa J. Y., Farnesoid X Receptor Agonist and Uses Thereof. WO2018/170167. (2018).
Smith, N. D., Govek, S. P., Nagasawa, J. Y. & Lai. A.; Farnesoid X Receptor Agonist and Uses Thereof. WO2018/170173 (2018).
Smith, N. D., Govek, S. P., Nagasawa, J. Y, Douglas, K. L, Lai, A. G. Farnesoid X Receptor Agonist and Uses Thereof. WO2018/170182, 20, September (2018).
Merk, D. et al. Molecular tuning of farnesoid X receptor partial agonism. Nat. Commun. 10, 2915 (2019).
Genin, M. J. et al. Discovery of 6-(4-{[5-Cyclopropyl-3-(2,6-dichlorophenyl)isoxazol-4-yl]methoxy}piperidin-1-yl)-1-methyl-1H-indole-3-carboxylic Acid: A novel FXR agonist for the treatment of dyslipidemia. J. Med. Chem. 58, 9768–9772 (2015).
Jin, L. et al. The antiparasitic drug ivermectin is a novel FXR ligand that regulates metabolism. Nat. Commun. 4, 1937 (2013).
Fang, S. et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 21, 159–165 (2015).
Matsumoto, M. et al. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 94, 93–103 (2013).
Liu, Y. et al. Farnesoid X receptor agonist decreases lipid accumulation by promoting hepatic fatty acid oxidation in db/db mice. Int. J. Mol. Med. 42, 1723–1731 (2018).
Pathak, P. et al. Instetine farnesoid X receptor agonist and gut microbiota active G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 68, 1574–1588 (2018).
Kemmer, G. & Keller, S. Nonlinear least-squares data fitting in Excel spreadsheets. Nat. Protoc. 5, 267–281 (2010).
Duan, H. et al. Design, synthesis, and antidiabetic activity of 4-phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists. J. Med. Chem. 55, 10475–10489 (2012).
Ni, S. et al. A radical approach to anionic chemistry: synthesis of ketones, alcohols, and amines. J. Am. Chem. Soc. 141, 6726–6739 (2019).
Chen, T. G. et al. Quaternary centers by nickel-catalyzed cross-coupling of tertiary carboxylic acids and (hetero)aryl zinc reagents. Angew. Chem. Int. Ed. Engl. 58, 2454–2458 (2019).
Sandfort, F., O’Neill, M. J., Cornella, J., Wimmer, L. & Baran, P. S. Alkyl-(hetero)aryl bond formation via decarboxylative cross-coupling: A systematic analysis. Angew. Chem. Int. Ed. Engl. 56, 3319–3323 (2017).
Smith, J. M. et al. Decarboxylative alkynylation. Angew. Chem. Int. Ed. Engl. 56, 11906–11910 (2017).
Toriyama, F. et al. Redox-active esters in Fe-catalyzed C-C coupling. J. Am. Chem. Soc. 138, 11132–11135 (2016).
Acknowledgements
Dr. N. Ashizawa, Mr. N. Kurita and Mr. J. Uda for their generous support.
Author information
Authors and Affiliations
Contributions
S. M. and Y. K. contributed equally to this study as first authors. S. M., Y. K., Y. M. and R. T. designed and performed synthesis; M. S., M. M., N. C., Y. W. and T. T. designed and performed pharmacological experiments. D. M. carried out the pharmacokinetic studies. All authors contributed to results analysis and manuscript writing. S.M. and R.T. wrote the main manuscript text. S.M. and R.T. prepared Fig. 1, S.M., M.M. and R.T. prepared Figs. 2, 3 and 4, R.T. prepared Fig. 5, S.M. and M.M. prepared Fig. 6, S.M., N.C. and R.T prepared Figs. 7 and 8. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miyata, S., Kawashima, Y., Sakai, M. et al. Discovery, optimization, and evaluation of non-bile acid FXR/TGR5 dual agonists. Sci Rep 11, 9196 (2021). https://doi.org/10.1038/s41598-021-88493-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88493-0
This article is cited by
-
Effect of FXR agonist GW4064 in the treatment of hilar cholangiocarcinoma in rats
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
