
Abstract
Antibiotic resistance is a major problem of tuberculosis treatment. This provides the stimulus for the search of novel molecular targets and approaches to reduce or forestall resistance emergence in Mycobacterium tuberculosis. Earlier, we discovered a novel small-molecular inhibitor among 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazoles targeting simultaneously two enzymes—mycobacterial leucyl-tRNA synthetase (LeuRS) and methionyl-tRNA synthetase (MetRS), which are promising molecular targets for antibiotic development. Unfortunately, the identified inhibitor does not reveal antibacterial activity toward M. tuberculosis. This study aims to develop novel aminoacyl-tRNA synthetase inhibitors among this chemical class with antibacterial activity toward resistant strains of M. tuberculosis. We performed molecular docking of the library of 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives and selected 41 compounds for investigation of their inhibitory activity toward MetRS and LeuRS in aminoacylation assay and antibacterial activity toward M. tuberculosis strains using microdilution assay. In vitro screening resulted in 10 compounds active against MetRS and 3 compounds active against LeuRS. Structure-related relationships (SAR) were established. The antibacterial screening revealed 4 compounds active toward M. tuberculosis mono-resistant strains in the range of concentrations 2–20 mg/L. Among these compounds, only one compound 27 has significant enzyme inhibitory activity toward mycobacterial MetRS (IC50 = 148.5 µM). The MIC for this compound toward M. tuberculosis H37Rv strain is 12.5 µM. This compound is not cytotoxic to human HEK293 and HepG2 cell lines. Therefore, 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives can be used for further chemical optimization and biological research to find non-toxic antituberculosis agents with a novel mechanism of action.
Similar content being viewed by others

Introduction
Tuberculosis is one of the most dangerous infectious diseases and a serious social problem. The main problem of tuberculosis treatment is multidrug resistance to existing antibiotics. Therefore, the search for novel molecular targets and the development of novel strategies to overcome M. tuberculosis resistance became a challenging task for modern science.
Nowadays, aminoacyl-tRNA synthetases (ARSases) represent promising molecular targets for antibiotic development1,2,3,4. These enzymes catalyze the covalent attachment of amino acid residues to cognate tRNAs, playing a key role in the first stage of protein biosynthesis. Aminoacyl-tRNA synthetases have a highly conservative structure among different pathogens, which increases the chances of antibiotic development with a broad spectrum of action. At the same time, aminoacyl-tRNA synthetases possess some structural divergence between prokaryotes and eukaryotes which can be sufficient for the development of inhibitors with higher selectivity toward pathogen enzymes. Besides, several pro- and eukaryotic aminoacyl-tRNA synthetases also have differences in aminoacyl-adenylate binding sites in comparison with human mitochondrial ARSases, this may significantly reduce the toxicity of targeting aminoacyl-adenylate binding site inhibitors during antibiotic therapy.
In particular, M. tuberculosis leucyl-tRNA synthetase (LeuRS) has significant differences in the amino acid sequences of the active sites compared to the human enzyme5. Recently, an inhibitor of mycobacterial LeuRS was found among benzoxaborole derivatives – GSK656, which is highly selective toward mycobacterial LeuRS (IC50 = 0.2 µM) compared to human cytoplasmic (IC50 > 300 µM) and mitochondrial LeuRS (IC50 > 300 µM). Besides, GSK656 is effective in an animal model of tuberculosis and is recommended for clinical trials, indicating that M. tuberculosis LeuRS is a validated molecular target for the development of anti-tuberculosis drugs6.
M. tuberculosis methionyl-tRNA synthetase (MetRS) differs in amino acid residues of aminoacyl-adenylate binding sites by 44% and 22% from human cytosolic and mitochondrial MetRS, correspondingly. It should be noted that inhibitor of Staphylococcus aureus MetRS – REP8839, has a 1000-fold higher affinity for the bacterial enzyme than for human mitochondrial MetRS7, while the amino acid difference in the active site between S. aureus and human mitochondrial MetRS is the same as in the case of mycobacterial and human mitochondrial MetRS. With the fact that considerable selectivity between S. aureus and human mitochondrial enzymes was achieved, there are opportunities to develop inhibitors with higher selectivity toward M. tuberculosis MetRS than for both human homologs. Besides, recently several crystal structures of M. tuberculosis MetRS were reported8,9, which makes this enzyme promising for the receptor-oriented rational design of inhibitors with antituberculosis activity. Unfortunately, the frequency of resistance occurrence to aaRSs inhibitors is very high (10–7–10–8) due to point mutations in the gene encoding enzyme which consequently affects ligand-binding interactions.
Several experimental studies suggest that MetRS is a validated molecular target for antibiotic design10,11,12,13,14,15,16,17,18,19,20. However, to date, there are only several published reports of small molecular inhibitors for mycobacterial MetRS among some adenylate analogs16 and previously published by us N-benzylidene-N′-thiazol-2-yl-hydrazine21 and 3-Phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole22 derivatives. Moreover, these derivatives represent first-published dual-target inhibitors of mycobacterial ARSes, inhibiting also LeuRS. However, ARSases inhibitors among N-benzylidene-N′-thiazol-2-yl-hydrazines have poor solubility and reported inhibitor 3-(3-chloro-4-methoxy-phenyl)-5-[3-(4-fuoro-phenyl)-[1,2,4]oxadiazol5-yl]-3H-[1,2,3]triazol-4-ylamine does not reveal antibacterial activity. Therefore, the search for novel M. tuberculosis MetRS inhibitors with antibacterial activity is very important.
Recently, as an approach to overcome resistance, we have proposed an idea for the development of dual-targeted inhibitors toward M. tuberculosis LeuRS and MetRS since mutations are required in all targets to confer resistance to the drug21. We identified the first dual-targeted inhibitors toward mycobacterial LeuRS and MetRS among N-benzylidene-N’-thiazol-2-yl-hydrazine21, which previously were reported as inhibitors of LeuRS5. Moreover, using the method of ligand-based pharmacophore modeling we have found 5 novel hit compounds possessing inhibitory activity simultaneously toward M. tuberculosis LeuRS and MetRS22. The most active compound—3-(3-chloro-4-methoxy-phenyl)-5-[3-(4-fluoro-phenyl)-[1,2,4]oxadiazol-5-yl]-3H-[1,2,3]triazol-4-ylamine inhibits activity of mycobacterial LeuRS and MetRS with IC50 values of 13 µM and 13.8 µM, respectively. Unfortunately, this compound does not have antibacterial activity toward M. tuberculosis. Therefore, the search for novel LeuRS and MetRS inhibitors among this chemical class with antituberculosis activity is of great interest.
Results
To find novel MetRS and LeuRS inhibitors with the antituberculosis activity we performed molecular docking of the pre-selected compound library of 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives into active sites of investigated enzymes. According to molecular docking results and visual analysis of the best-scored complexes, we selected 41 compounds for investigation of their inhibitory activity toward recombinant mycobacterial LeuRS and MetRS. The results of testing are presented in Table 1.
Recombinant M. tuberculosis LeuRS and MetRS were expressed in Escherichia coli and purified according to previously described methods: for LeuRS and MetRS22, respectively. 3-Phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives were provided by OTAVA Ltd (Kyiv, Ukraine). The activity of compounds toward mycobacterial LeuRS and MetRS was determined in aminoacylation assay using BIOMOL GREEN reagent (Enzo Life Sciences)21 . According to screening data, 10 compounds were decreasing the activity of mycobacterial MetRS and 3 compounds targeting mycobacterial LeuRS with an inhibition cut-off taken as 50%. For compounds that inhibited both synthetases IC50 values were determined (Table 2). Noticeably, compounds 2, 13, 27 had higher values than previously reported for compound 17.
All these compounds were tested for antibacterial activity toward M. tuberculosis H37Rv, a rifampin-resistant strain H37Rv (rpoBS450L) (ATCC #35,838), an isoniazid-resistant H37Rv (katGdel) (MS015), a moxifloxacin-resistant strain H37Rv (gyrAD94K) (MOX3), Mycobacterium avium (ATCC 700,891 MAC 101) and Mycobacterium abscessus (ATCC 19,977) in microdilution assays. Compounds were screened in high-throughput way at 20 mg/L, 2 mg/L, and 0.2 mg/L concentrations and activity determined as > 80% reduction in growth. Growth inhibition values for all tested compounds are listed in Table 3.
According to the data from Tables 3, 4 compounds have activity between 2 and 20 mg/L toward several mono-resistant strains. Among these compounds, only one compound 27 (with growth inhibition cutoff 5.17–51.7 µM) has significant enzyme inhibitory activity toward mycobacterial MetRS. Compounds 40 (5.22–52.2 µM cutoff) and 20 (5.7–57 µM) have slight dual-target inhibitory activity toward both M. tuberculosis MetRS and LeuRS.
The compound 27 was taken for MIC determination toward H37Rv strain in Middlebrook 7H9-based media containing either glucose or cholesterol/dipalmitoylphosphatidylcholine (DPPC) as a carbon source in the presence of bovine serum albumin (BSA) or casitone and supplemented with Tyloxapol. MIC1 and MIC2 values were determined after incubation for 7 and 14 days, correspondingly. As it can be seen from the Table 4, compound 27 inhibits the growth of M. tuberculosis H37Rv in the medium containing glucose as a carbon source with 1-week MIC value of 12.5 µM and 2-week MIC value of 25 µM and in the medium containing DPPC as a carbon source with 1-week and 2-week MIC values of 25 µM. In the media containing BSA, the compounds did not inhibit the growth of M. tuberculosis H37Rv. It may be explained by the potency them to bind with hydrophobic pockets of free BSA which leads to the decrease of the efficient concentrations of compounds capable to bind with aminoacyl-tRNA synthetases. Similar insights were shown with molecular simulations of human serum albumin (HSA) and three clinically promising squalenoylated drugs (gemcitabine-squalene, adenine-squalene, and doxorubicin-squalene). Data suggest that these drugs may accumulate by HSA and inside low-density lipoproteins23. DPPC forms spontaneously vesicle-like structures24, thus it may act as the scavenger for different hydrophobic and amphiphilic molecules, similar to lipoproteins23.
We have tested compound 27 for cytotoxicity toward human cell lines HEK293 (human embryonic kidney 293) and HepG2 (human hepatocellular carcinoma cell line) using standard MTT assay. According to the results of in cellulo testing, this compound did not affect cell viability in the range of concentrations from 3.125 µM to 50 µM, suggesting that CC50, concentration to cause 50% cytotoxicity, for HEK293 and HepG2 > 50 µM.
Discussion
In this study, we have investigated 41 derivatives of 3-Phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole for inhibitory activity toward M. tuberculosis MetRS and LeuRS and established several structure–activity relationships (SAR). It has been observed that the nature of R1 substituent significantly influences the compound’s inhibitory activity toward M. tuberculosis LeuRS and MetRS. We found that compounds with R1 = bromine atom are more active than those with an ethoxy group, fluorine, or hydrogen in this position. It can be seen from the comparison of activity for compounds 34, 39, 23, and 1 and compound pairs such as 36, 30, and 35, 26. From the obtained results, the order of potency for the substituent R1 could be proposed as following: Br ≥ CH3CH2COOH > F > H. Fluorine and bromine atoms have almost equal activity toward mycobacterial LeuRS and MetRS for compound pair 12 and 32. In the case of compound pair 15 and 33, the bromine atom is more favored than the fluorine atom for inhibitory activity toward M. tuberculosis MetRS but in a case of mycobacterial LeuRS, a fluorine atom is more profitable.
It was revealed that the R3 significantly affects the compound’s inhibitory activity toward mycobacterial MetRS and LeuRS. The introduction of fluorine atom instead of Hydrogen in this position leads to a significant decrease of inhibitory activity toward LeuRS and MetRS. To see this effect one can refer to compound pair 2 and 3.
We have investigated that the substituent R4 has also influenced the inhibitory activity toward mycobacterial LeuRS and MetRS. For a pair of compounds with R1 = F and R7 = Cl, R4 = methoxy group (27) is more favored than the methyl group (25). For a pair of compounds with R1 = F and R7 = H, R4 = ethoxy group (30) is more profitable than the methoxy group (26) for both enzymes. In a series of compounds with R1 = Br (34–36), the order of inhibitory activity for both enzymes is the following: F < OCH3 < OCH2CH3. For a pair of compounds with R1 = OCH2CH3 (39–40), the Cl atom at the position R4 plays an important role for dual-targeted inhibition activity toward LeuRS and MetRS and consequently for antibacterial activity toward M. tuberculosis resistant strains.
It was found that R5 also impacts the compound’s inhibitory activity toward mycobacterial MetRS and LeuRS. The presence of a methoxy group (compound 20) is more favored than the methyl group (compound 18) in this position for inhibitory activity toward both enzymes. The introduction of substituents CF3 (19) or Cl (16) in this position leads to an increase of inhibitory activity toward MetRS and almost complete loss of activity or even activation of LeuRS and vice versa, the fluorine atom (15) in this position causes increase of inhibitory activity toward LeuRS and activation of MetRS. Therefore, the order of potency of substituent R5 for MetRS could be proposed as following: CF3 < Cl < OCH3 < CH3 < F and for LeuRS as following: F < OCH3 < CH3 < CF3 < Cl. It should be noted, that the R5 = methoxy group (compound 20) is important for membrane permeability since the substitution of OCH3 with any other group such as CH3, CF3, F or Cl leads to complete loss of antibacterial activity toward M. tuberculosis resistant strains.
According to structure–activity relationship (SAR) studies of compounds 9–14, the order of R6 substituent inhibitory efficiency for MetRS is following: F < CH3CH2 < OCH3 < H < Cl < Br, and for LeuRS: OCH3 < CH3CH2 < F < H < Cl < Br. For a pair of compounds with R5 = OCH3 (20, 21), the introduction of R6 = methoxy group instead of Hydrogen leads to complete loss, even activation of both LeuRS and MetRS. Moreover, this substitution causes a complete loss of antibacterial activity toward M. tuberculosis resistant strain. The nature of the R7 substituent for compounds 26–29 has a similar effect for both enzymes: Cl < CH3 < OCH3 < H. The presence of a Cl atom at this position is important for antimycobacterial activity.
Therefore, the chemical structure of all investigated substituents R1-R7 of 3-Phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives influence inhibitory activity toward M. tuberculosis LeuRS and MetRS. Summarizing SAR data of the most active 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives, we can make some conclusions. So, for dual-targeted inhibitory activity toward M. tuberculosis LeuRS and MetRS, the substituents R4 = H (compound 2) and R6 = OCH3 (compounds 13 and 17) are the most profitable. For MetRS inhibitory activity the substituents R4 = F (compounds 31 and 34), R5 = CF3 (compound 19), R6 = F (compounds 12, 22, 41), R7 = Cl (compound 27) possess the highest efficiency. For LeuRS inhibitory activity very important role plays fluorine atom as R5 substituent (compounds 7 and 15). The established SAR can be useful for further chemical optimization to improve inhibitory activity toward both investigated ARSases as well as antibacterial activity toward M. tuberculosis pathogenic strains.
The established SAR of 3-Phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives can be useful for further chemical optimization to improve inhibitory activity toward M. tuberculosis LeuRS and MetRS.
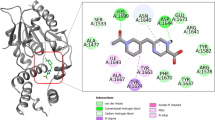
Molecular docking results revealed that compounds in this class have similar binding modes with both active sites of M. tuberculosis LeuRS and MetRS. The complexes of compound 27 with amino acid residues of MetRS and LeuRS active sites are presented in Fig. 1a and Fig. 1b respectively. Despite compound 27 has better inhibitory activity toward MetRS, than toward LeuRS, binding modes for both enzymes are similar: 2-methoxy-5-chloro-phenyl interacts with the adenine-binding region and 4-fluoro phenyl ring binds to the amino acid binding pocket (Fig. 1).

The complexes of compound 27 in the active site of M. tuberculosis MetRS (a) and LeuRS (b), obtained with molecular docking. Hydrogen bonds are shown by the green dotted lines, sulfur–π interaction is presented by the yellow dotted line and hydrophobic interactions are indicated by magenta colors.
We have calculated free energy binding (ΔG) for this compound with aminoacyl adenylate binding pocket of mycobacterial MetRS and LeuRS using umbrella sampling algorithm and Weighted Histogram Analysis Method (WHAM). ΔGb has been determined from the PMF curve as the difference between the PMF with the ligand-bound minus the ligand when it is unbound (Fig. 2).

Potential of Mean Force (PMF) for unbinding of the complex of M. tuberculosis MetRS with compound 27.
According to umbrella sampling calculations, the free energy binding of compound 27 with MetRS is—2 kcal/mol, while for LeuRS it was impossible to build the PMF curve. The profiles of PMF curves for MetRS and LeuRS are available as Supplementary Notes. It seems that the umbrella sampling algorithm is more sensitive in silico method to predict the binding affinity of ligands with receptors than docking and can be a useful approach for structure-based optimization of compounds within one chemical class.
Materials and methods
Molecular docking
Molecular docking of the ligands into the aminoacyl-adenylate binding sites of MetRS crystal structure (PDB ID: 6AX8) and LeuRS homology model, which was obtained by us earlier5, was performed with DOCK 4.0 program25,26,27,28. The water and ligand molecules were deleted from MetRS PDB-file. Ligand geometry was evaluated in the YFF force field29. Partial atomic charges for compounds were set using Kirchhoff method30. Docking of the ligands into MetRS and LeuRS active sites was carried out using previously described parameters21. The complexes of ligands with MetRS and LeuRS active sites were visually analyzed by Discovery Studio Visualizer31.
Free energy calculation by umbrella sampling
Molecular dynamics simulations were carried out with GROMACS v.4.532,33,34. The umbrella sampling algorithm and Weighted Histogram Analysis Method (WHAM)35 were used to calculate the free energy profile for the separation of MetRS-inhibitor and LeuRS–inhibitor complexes. The starting coordinates used for simulations were from docking complexes.
The topology file for ligand was generated using the web-site Automated Topology Builder (ATB)36. Topology files for aminoacyl-tRNA synthetases have been generated from PDB-files using pdb2gmx command. The system was set up using the Gromos96 53a6 force field and solvated with the SPC water model.
The center of mass of the receptor-ligand complex has been placed at (4.0, 4.0, 4.0) in a box of dimensions 12 × 12 × 12 using editconf command. Then, the system was solvated with genbox command and neutralized using Na+ or Cl- ions according to the charge of the system using genion command. The periodic boundary conditions and the particle mesh Ewald method were used with a nonbonded cutoff of 9 Å. Each system was first energy minimized using 5000 steps of steepest descent method followed by NPT equilibration for 100 ps. Using the make_ndx command we have defined a custom index group for pulling simulation. The pulling of ligands for each system has been performed in Y-dimension using a force constant of 1000 kJ/(mol nm2). A series of configurations along the Y-axis has been generated corresponding to each of the frames saved in the continuous pulling simulation. To measure the distance between protein and ligand on all of these frames, we have used Perl script to iteratively call the g_dist command. The total path with a length of 4.5 nm was divided into 0.1 nm wide equidistant windows. Each coordinate file that is required to obtain 0.1-nm spacing has been prepared for umbrella sampling simulations. At first, NPT equilibration in each window was performed. Then, each input file was passed to the umbrella sampling simulation. Then, using WHAM we have extracted the potential of mean force (PMF), which yields the ΔG for the binding/unbinding process.
The file with a list of commands and MDP files used during umbrella sampling simulation for MetRS-inhibitor and LeuRS–inhibitor complexes are available as Supplementary Methods.
Purification of recombinant M. tuberculosis aminoacyl-tRNA synthetases and in vitro aminoacylation reaction with compounds
The pET28a bacterial expression vector encoding the gene for M. tuberculosis LeuRS was a kind gift from Stephen Cusack and Andres Palencia (EMBL, Grenoble). Purification of M. tuberculosis LeuRS was performed according to the protocol described previously. The plasmid construct pET28b-MetRS M. tuberculosis obtained by our group earlier was used for the expression and purification of the target enzyme. The primary activity of both enzymes and their residual activity after incubation with potential inhibitors were checked in the aminoacylation reaction with Met/Leu and total E. coli MRE 600 tRNA (Roche) using the BIOMOL GREEN reagent21.
Growth inhibition determination against M. tuberculosis H37Rv and clinically relevant resistant strains
Percent growth inhibition of compounds was determined against M. tuberculosis strains using detection of reduction in resazurin (calorimetric growth indicator) compared to untreated growth controls. MIC was determined using optical density (OD) and a calorimetric growth indicator. Compound concentration cutoff values for all test compounds that showed > 80% growth reduction when compared to untreated growth control.
Antibacterial activity of compounds was determined in Middlebrook 7H9 complete medium which was prepared by the following procedure: 4.7 g of 7H9 powder (Difco; Becton Dickinson) was supplemented with 2 mL glycerol (Fisher) and with Milli-Q-water to the final volume 900 mL and mixed until dissolved. To this volume 100 ml of ADC (5 g bovine serum albumin (BSA) (Sigma), 2 g dextrose (Fisher), and 3 mg catalase (Sigma) dissolved in Milli-Q-water to final volume 100 mL) was added. Then, 10 mL of sterile 5% [v:v] polysorbate 80 (tween 80) (Sigma) was added to yield 0.05% [v:v] tween 80 final. Inoculums were prepared with M. tuberculosis strains H37Rv, a rifampin-resistant (RIF: rpoBS450L) strain (ATCC #35,838), an isoniazid-resistant (INH: katGdel) strain (MS015), a moxifloxacin resistant (MOX: gyrD94K) strain (MOX3), M. avium (ATCC 700,891 MAC 101) and M. abscessus (ATCC 19,977) strains. The target adjusted final concentration of bacteria was 5 × 105 CFU/mL.
Compounds were dissolved in 100% DMSO. Three drug concentrations were prepared – 20 mg/L, 2 mg/L, and 0.2 mg/L. Assay plates (96-well clear round-bottom plates) were prepared by adding 2 µL of the compound solution to each well at a specific concentration leaving two empty wells untreated growth controls. 100 µL of inoculum was added to all wells. Sealed assay plates were incubated at 37 ºC. On day 7, 10 µL Alamar Blue dye was added to each analytical well. Plates were incubated for 3 days and OD readings were taken at 570 nm and 600 nm. The following formula was used to determine % growth inhibition:
where λ1 = 570, λ2 = 600, (ɛox)λ2 = 117,216, (ɛox)λ1 = 80,586.
Minimum inhibitor concentrations (MICs) were established in four Middlebrook 7H9-based media according to the procedure described earlier by us21.
Cytotoxicity assay
Compound cytotoxicity was determined by standard MTT assay37 using human embryonic kidney 293 (HEK293) and human hepatocellular carcinoma (HepG2) cell lines according to the method described previously38. HEK 293 cell line was obtained from the Russian Cell Culture Collection (Institute of Cytology of the Russian Academy of Science, St.Petersburg, Russia) and HepG2 – from the Bank of Cell Lines from human and animal tissue (R. E. Kavetsky Institute of Experimental Pathology, Oncology and Radiobiology, NAS of Ukraine, Kyiv, Ukraine).
Chemical synthesis
3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives with R8 = NH2 (compounds 1–3, 9–37, 39–41) were synthesized as described earlier39,40 according to scheme presented in Fig. 3, a. 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives with R8 = CH3 (compounds 4–8, 38) were synthesized using previously reported method41 according to scheme presented in Fig. 3, b.

The synthesis pathway for 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives with R8 = NH2 (compounds 1–3, 9–37, 39–41) (a) and general synthetic procedure for 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives with R8 = CH3 (compounds 4–8, 38) (b).
The structures of the compounds were confirmed using NMR spectroscopy and LC–MS analysis. Nuclear magnetic resonance spectra were recorded on a Varian Mercury VRX-400 spectrometer using DMSO-d6 as solvent and tetramethylsilane as internal standard. Chemical shift values (δ) are quoted in ppm and coupling constants (J) in Hz. Liquid chromatography-mass spectra (LC–MS) analyzes were performed using the Agilent 1100 LC/MSD SL (Agilent Technologies) separations module and Mass Quad G1956B mass detector with electrospray ionization (Agilent Technologies). Spectral data for compounds are presented in Supplementary Methods Section.
References
Francklyn, C. S. & Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 294(14), 5365–5385 (2019).
Ho, J. M., Bakkalbasi, E., Söll, D. & Miller, C. A. Drugging tRNA aminoacylation. RNA Biol. 15(4–5), 667–677 (2018).
Kwon, N. H., Fox, P. L. & Kim, S. Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug Discovery 18(8), 629–650 (2019).
Rajendran, V., Kalita, P., Shukla, H., Kumar, A. & Tripathi, T. Aminoacyl-tRNA synthetases: structure, function, and drug discovery. Int. J. Biol. Macromol. 111, 400–414 (2018).
Gudzera, O. I. et al. Discovery of potent anti-tuberculosis agents targeting leucyl-tRNA synthetase. Bioorg. Med. Chem. 24(5), 1023–1031 (2016).
Li, X. et al. Discovery of a Potent and Specific M tuberculosis Leucyl-tRNA Synthetase Inhibitor:(S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy) benzo [c][1, 2] oxaborol-1 (3 H)-ol (GSK656). J. Med. Chem. 60(19), 8011–8026 (2017).
Green, L. S. et al. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob. Agents Chemother. 53(1), 86–94 (2009).
Barros-Alvarez, X. et al. The crystal structure of the drug target Mycobacterium tuberculosis methionyl-tRNA synthetase in complex with a catalytic intermediate. Acta Crystallographica Section F: Struct. Biol. Commun. 74(4), 245–254 (2018).
Wang, W. et al. Structural characterization of free-state and product-state Mycobacterium tuberculosis methionyl-tRNA synthetase reveals an induced-fit ligand-recognition mechanism. IUCrJ 5(4), 1 (2018).
Eissa, A. G. et al. Targeting methionyl tRNA synthetase: design, synthesis and antibacterial activity against Clostridium difficile of novel 3-biaryl-N-benzylpropan-1-amine derivatives. J. Enzyme Inhib. Med. Chem. 31(6), 1694–1697 (2016).
Faghih, O. et al. Development of methionyl-tRNA synthetase inhibitors as antibiotics for Gram-positive bacterial infections. Antimicrob. Agents Chemother. 61(11), 1 (2017).
Huang, W. et al. Optimization of a binding fragment targeting the “enlarged methionine pocket” leads to potent Trypanosoma brucei methionyl-tRNA synthetase inhibitors. Bioorg. Med. Chem. Lett. 27(12), 2702–2707 (2017).
Hussain, T., Yogavel, M. & Sharma, A. Inhibition of protein synthesis and malaria parasite development by drug targeting of methionyl-tRNA synthetases. Antimicrob. Agents Chemother. 59(4), 1856–1867 (2015).
Jarvest, R. L. et al. Nanomolar inhibitors of staphylococcus a ureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J. Med. Chem. 45(10), 1959–1962 (2002).
Kumari, M., Chandra, S., Tiwari, N. & Subbarao, N. High throughput virtual screening to identify novel natural product inhibitors for methionyltRNA-synthetase of brucella melitensis. Bioinformation 13(1), 8 (2017).
Lee, J. et al. Methionyl adenylate analogues as inhibitors of methionyl-tRNA synthetase. Bioorg. Med. Chem. Lett. 9(10), 1365–1370 (1999).
Nayak, S. U. et al. (2017) Safety, tolerability, systemic exposure, and metabolism of CRS3123, a methionyl-tRNA synthetase inhibitor developed for treatment of Clostridium difficile, in a phase 1 study. Antimicrob. Agents Chemother. 61(8), 1 (2017).
Ranade, R. M. et al. Inhibitors of methionyl-tRNA synthetase have potent activity against Giardia intestinalis trophozoites. Antimicrob. Agents Chemother. 59(11), 7128–7131 (2015).
Robles, S., Hu, Y., Resto, T., Dean, F. & Bullard, J. M. Identification and characterization of a chemical compound that inhibits methionyl-tRNA synthetase from Pseudomonas aeruginosa. Curr. Drug Discov. Technol. 14(3), 156–168 (2017).
Torrie, L. S. et al. Chemical validation of methionyl-tRNA synthetase as a druggable target in Leishmania donovani. ACS Infect. Dis. 3(10), 718–727 (2017).
Kovalenko, O. P. et al. Dual-target inhibitors of mycobacterial aminoacyl-tRNA synthetases among N-benzylidene-N′-thiazol-2-yl-hydrazines. MedChemComm 10(12), 2161–2169 (2019).
Volynets, G. P. et al. Dual-targeted hit identification using pharmacophore screening. J. Comput. Aided Mol. Des. 33(11), 955–964 (2019).
Yesylevskyy, S. O., Ramseyer, C., Savenko, M., Mura, S. & Couvreur, P. Low-density lipoproteins and human serum albumin as carriers of squalenoylated drugs: insights from molecular simulations. Mol. Pharm. 15(2), 585–591 (2018).
Marrink, S. J., Lindahl, E., Edholm, O. & Mark, A. E. Simulation of the Spontaneous Aggregation of Phospholipids into Bilayers. J. Am. Chem. Soc. 123(35), 8638–8639 (2001).
Bodian, D. L. et al. Inhibition of the fusion-inducing conformational change of influenza hemagglutinin by benzoquinones and hydroquinones. Biochemistry 32(12), 2967–2978 (1993).
Ewing, T. J., Makino, S., Skillman, A. G. & Kuntz, I. D. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J. Comput.-Aided Mol. Des. 15(5), 411–428 (2001).
Ring, C. S. et al. Structure-based inhibitor design by using protein models for the development of antiparasitic agents. Proc. Natl. Acad. Sci. 90(8), 3583–3587 (1993).
Shoichet, B. K., Stroud, R. M., Santi, D. V., Kuntz, I. D. & Perry, K. M. Structure-based discovery of inhibitors of thymidylate synthase. Science 259(5100), 1445–1450 (1993).
Yakovenko, O. Y., Oliferenko, A., Golub, A., Bdzhola, V. & Yarmoluk, S. The new method of distribution integrals evaluations for high throughput virtual screening. Ukr. Bioorg. Acta 1, 52–62 (2007).
Yakovenko, O., Oliferenko, A. A., Bdzhola, V. G., Palyulin, V. A. & Zefirov, N. S. Kirchhoff atomic charges fitted to multipole moments: implementation for a virtual screening system. J. Comput. Chem. 29(8), 1332–1343 (2008).
Discovery Studio Visualizer 4.0.
Berendsen, H. J., van der Spoel, D. & van Drunen, R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91(1–3), 43–56 (1995).
Hess, B., Kutzner, C., Van Der Spoel, D. & Lindahl, E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4(3), 435–447 (2008).
Van Der Spoel, D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26(16), 1701–1718 (2005).
Hub, J. S., De Groot, B. L. & Van Der Spoel, D. g_wham - A Free Weighted Histogram Analysis Implementation Including Robust Error and Autocorrelation Estimates. J. Chem. Theory Comput. 6(12), 3713–3720 (2010).
Automated Topology Builder.
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65(1–2), 55–63 (1983).
Volynets, G. et al. Identification of protein kinase fibroblast growth factor receptor 1 (FGFR1) inhibitors among the derivatives of 5-(5, 6-dimethoxybenzimidazol-1-yl)-3-hydroxythiophene-2-carboxylic acid. Monatshefte für Chemie-Chemical Monthly 150(10), 1801–1808 (2019).
Naro, Y. et al. Small molecule inhibition of MicroRNA miR-21 rescues chemosensitivity of renal-cell carcinoma to topotecan. J. Med. Chem. 61(14), 5900–5909 (2018).
Pokhodylo, N. T. & Matiychuk, V. S. Synthesis of new 1, 2, 3-triazolo [1, 5-a] quinazolinones. J. Heterocycl. Chem. 47(2), 415–420 (2010).
Obushak, N., Pokhodylo, N., Pidlypnyi, N. & Matiichuk, V. Synthesis of 1, 2, 4-and 1, 3, 4-oxadiazoles from 1-aryl-5-methyl-1H-1, 2, 3-triazole-4-carbonyl chlorides. Russ. J. Org. Chem. 44(10), 1522 (2008).
Acknowledgments
This work was supported by the NAS of Ukraine grant for young scientists (Project № 0119U001863), NRFU Grant “Leading and Young Scientists Research Support” (Project № 0120U104850), and also by the National Institutes of Allergy and Infectious Diseases Preclinical Service Contract 75N93019D00005. The authors are thankful to Dr. Stephen Cusack and Dr. Andres Palencia (EMBL Grenoble Outstation, France) for the gift of plasmid encoding M. tuberculosis LeuRS. We are grateful to Dr. Clifton Barry III (National Institute of Allergy and Infectious Disease, National Institute of Health, Maryland, USA) for his interest and support in our study. Publication was financially supported by the US-Ukraine Foundation Biotech Initiative.
Author information
Authors and Affiliations
Contributions
M.Y.R. performed enzymes purification, in vitro assays with LeuRS and MetRS, formal analysis of the primary data and validated the data, drafted a manuscript; A.O.B. performed chemical synthesis, 1H NMR/13C NMR/LC–MS spectral analysis for compounds, analyzed data, drafted a manuscript; A.P.Y. performed MTT assays, validated the data; I.M.K. performed 1H NMR spectral analysis for compounds, analyzed the data; S.A.S. and V.G.B. selected compounds for screening, analyzed and validated the data; L.L.L. validated the data; S.M.Y. validated the data; M.Y.R., S.M.Y. and G.P.V. obtained funding; M.A.T. analyzed the data, drafted the manuscript; G.P.V. performed umbrella sampling and selected compounds for screening, supervised the project, validated the data, drafted the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rybak, M.Y., Balanda, A.O., Yatsyshyna, A.P. et al. Discovery of novel antituberculosis agents among 3-phenyl-5-(1-phenyl-1H-[1,2,3]triazol-4-yl)-[1,2,4]oxadiazole derivatives targeting aminoacyl-tRNA synthetases. Sci Rep 11, 7162 (2021). https://doi.org/10.1038/s41598-021-86562-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-86562-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
