
Abstract
Hakes of the genus Merluccius include 11 valid species as well a number of rare morphotypes suspected to be “cryptic species”. Concatenated nucDNA ITS1-rDNA and mtDNA cyt b sequences plus nested ITS1Nes sequences allowed to ascribe 14 specimens of nine rare morphotypes from the South Pacific and the South Atlantic to the phylogenetic backbone of this genus. Bayesian analyses pointed to M. bilinearis and M. albidus as the oldest species of the genus and the New World cluster, respectively. The phylogenetic status of M. angustimanus from the upper Gulf of California suggests its hybrid origin between M. gayi and M. productus from about 0.25 MYA, although an ever since confinement of a subset of those species cannot be ruled out. The molecular phylodiagnostic test suggests a common origin of all rare morphotypes and the absence of cryptic hake species in the Southern Cone. The molecular background of the morphotypes distributed between the Western Pacific South of New Zealand and the western Atlantic South of Argentina is compatible with their hybrid origin between M. gayi and both, M. australis or M. hubbsi, respectively.
Similar content being viewed by others


Introduction
The genus Merluccius comprises 11 valid species that occur on most temperate and tropical continental shelves except the Asian shores of the Pacific Ocean1. Hakes show an anti-tropical distribution in the Atlantic Ocean and the Eastern Pacific and a latitudinal bathymetric overlap between isotherms 7 °C and 23 °C2,3,4. Based on osteological data5,6 it is believed that genus Palaeogadus as ancestor of genus Merluccius, arose near Greenland in the early Eocene (ca. 50 MYA)7, dispersed southwards along the North American and Eurasian shelves and entered the Pacific8. The earliest known merluccid fossils date back to the Middle Oligocene (ca. 27–33 MYA) in a large inland sea that covered much of central Europe and connected to a temperate Arctic Ocean9. It is believed that either an ancestral species of Merluccius or the extant M. bilinearis experienced an evolutionary radiation in two superclusters, i.e. Old World hakes (the Euro-African supercluster) and New World hakes (the American supercluster)5,6,7,8. Also, it is hypothesized that a widening rift between Europe and North America plates prompted vicariant speciation and that recurrent dispersal events and adaptation to temperature regimes also played a role in the speciation of this genus10. Subsequent geological events such as the closure of the Panama Seaway over 3.5 MYA acted as a geographical barrier between Atlantic and Pacific lineages8,11. Successive population fragmentation and expansion due to climatic oscillations during Pleistocene glaciations allowing founder phenomena cannot be ruled out12. Such origin and dispersal hypotheses are congruent with the actual phylogeny of the genus worked out after parasite data8 morphology3,5,6,13 and genetic data14,15,16.
The Old World supercluster comprises five well-defined species and with occasional hybrids between sympatric species, e.g. M. capensis x M. paradoxus17. The New World supercluster comprises three clusters of two species each, an Atlantic north cluster that comprises M. bilinearis and M. albidus, a southern cluster that comprises M. hubbsi and M. australis, and a Pacific cluster that groups M. gayi and M. productus. Molecular systematics of Merluccius generally distinguishes those 11 species18, however there are still knowledge gaps in hake taxonomy as some specimens found in regions of species overlap show significant morphological divergence from extant species13. For instance, the phylogenetic relationships within the New World supercluster have been repeatedly interrogated due to the uncertain taxonomic status of some pairs of morphotypes such as M. gayi gayi19 vs. M. gayi peruanus20, M. angustimanus21 vs. M. hernandezi22, M. hubbsi vs. M. patagonicus23 or M. australis vs. M. polylepis or M. tasmanicus24. Several of those morphotypes have been recently taken as synonymous with extant species. For instance, the three stocks of M. productus believed to exist in the northeast Pacific corridor from Washington State to Costa Rica25,26 likely belong to a single hake species27,28. The high variability and overlap of meristic traits between M. hernandezi and M. angustimanus suggests they are synonymous forms13 and that M. angustimanus is a subpopulation of M. productus confined to the northern Gulf of California27. Also, meristic (see Fig. 1 from23) and molecular analyses29,30 showed that M. patagonicus could be synonymous with M. hubbsi and that M. tasmanicus could be synonymous with M. australis31. Taxonomic uncertainties are expected in closely-related species that show extensive overlap in morphological and meristic traits. Resolution of those questions has been complicated by the small number of morphotype samples available, the lack of a systematic sampling plan over a reasonable number of localities and the choice of the most appropriate phylogenetic reconstruction algorithm.

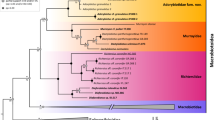
BEAST v1.8.485 phylogenetic reconstruction of genus Merluccius spp. using Bayesian inference on both, (a) the substitution model HKY85 + I + G on ITS1 variants and (b) the substitution model GTR + G on cyt b haplotypes. Percentage of trees over 5000 bootstrap replicates are shown above branches. OTU Gmor corresponds to the outgroup Gadus morhua. Scale bar indicates the No. of nucleotide substitutions per 100 DNA residues. Sample codes are given in Table 1 and are followed either by the number of ITS1 variants per species (Fig. 1a and Supp. Table S3) or by the number of cyt b haplotypes per species (Fig. 1b and Supp. Table S4).
We address those issues with the most comprehensive sample collection yet made in this genus, which comprises all 11 valid species13 as well as M. angustimanus from the Gulf of California21 and 14 specimens of nine rare morphotypes classified as M. tasmanicus from New Zealand waters23, M. polylepis from the Pacific coast of Chile32, M. patagonicus from the Atlantic South23, uncatalogued specimens of M. hubbsi from the Beagle Channel and Puerto Madryn in Argentina23, and rare specimens of M. australis from Chile23 and New Zealand24. An integrative multimarker phylogenetic reconstruction of genus Merluccius is enforced to determine both, the phylogenetic congruence and synergy between mtDNA (cyt b) and nucDNA (ITS1) sequences for inference of phylogenetic relationships in this genus and the genetic prospecting for cryptic species within the New World supercluster, as suspected on the rare morphotypes described so far.
Results
Description of samples and sequences
We examined 1205 specimens from 11 valid hake species and M. angustimanus (Table 1) as well as nine morphotypes (Table 2). The aligned ITS1 region had a length of 692 bp and comprised the ITS1-rDNA sequence, 53 bp from the 3′-end of the 18S-rDNA gene and 20 bp from the 5′-end of the 5.8S-rDNA gene33. A total of 254 variable sites were found among 85 specimens from 12 hake species (including M. angustimanus as putative species) (Supp. Table S1). The ITS1 sequences showed similar %GC content between species and a general low transversion rate (Supp. Table S2). The full dataset of 85 ITS1 sequences comprised 19 variants (Supp. Table S3). The aligned cyt b region had a length of 465 bp and comprised 428 bp from the 5′-end of the cyt b and 37 bp from the 3′-end of the mitochondrial DNA gene tRNA-Glu. A total of 129 variable sites were found among 66 specimens from 12 hake species as including M. angustimanus (Supp. Table S1). The 66 cyt b sequences comprised 29 haplotypes as one to four per species (Supp. Table S4). M. angustimanus showed a single haplotype (HakeCytb.18) that was molecularly close to those of M. productus, M. gayi and M. albidus. Concatenated sequence information from both DNA regions was available on 42 specimens, comprised 1158 bp in length with 381 variable sites (Supp. Table S1) and identified 25 variants (Supp. Figure S4). The aligned ITS1Nes sequences from 39 specimens of reference and 46 clones from 14 specimens of nine morphotypes had a length of 66 bp and 13 variable sites (Supp. Table S1). Those 85 ITS1Nes sequences comprised 11 variants (HakeITS1Nes.1–11) (Table 3). HakeITS1Nes.2 was shared among the Pacific species and four morphotypes, HakeITS1Nes.5 was shared between the Atlantic North species, HakeITS1Nes.6 was shared among all morphotypes and HakeITS1Nes.9 was shared between two morphotypes (Table 3). All the morphotypes shared at least one ITS1Nes variant among each other or with known species, but also exhibited specific variants, e.g. HakeITS1Nes.7-8-10-11. The recombination parameter (R) detected three pairs of sites in the ITS1Nes region with at least one recombination event34, i.e. between sites 5–8, 8–14 and 14–56.
Nucleotide divergence and genetic distance
The lowest average number of nucleotide substitutions per site among ITS1 sequences (Dxy = 0.004–0.009) as well as among cyt b sequences (Dxy = 0.007–0.014) were observed in pairwise comparisons of M. angustimanus, M. productus and M. gayi (Supp. Table S5; Supp. Table S6). M. gayi showed a similar evolutionary divergence of ITS1 (d = 0.004) and cyt b (d = 0.008–0.010) from both, M. angustimanus and M. productus, respectively. Those latter species showed the lowest pairwise divergence in the dataset, i.e. for ITS1 (d = 0.001) (Supp. Table S5) as well as for cyt b (d = 0.005) (Supp. Table S6). The divergence pattern after ITS1Nes sequences was less than that observed on full sequences of ITS1 or cyt b because of the lower number of variable sites in the former, and M. angustimanus-M. productus-M. gayi did not diverge among each other (Supp. Table S7). The average number of nucleotide substitutions per site (Dxy) (Table 4a, Supp. Table S8a) and the net evolutionary divergence between species (Table 4b, Supp. Table S8b) from ITS1Nes showed that the morphotype-specific sequences of M. patagonicus (PATA), M. tasmanicus (TASM), M. polylepis (POLY), M. hubbsi (TASM) and M. australis (AUST) were more similar to Pacific hakes than to any other species in the New World supercluster (see also ITS1Nes variation in Table 3).
Clustering methods
The major groups of species as inferred from the Euclidean divergence of the PCoA correctly identified a) the two major complexes of hakes, Old World versus New World hakes (Supp. Figure S1a, b) Atlantic versus Pacific New World hakes (Supp. Figure S1b, and c) the closeness of hake morphotypes to both, the Pacific group and the Austral group (Supp. Figure S1c). The AMOVA agreed with the partition among the groups identified by PCoA using ITS1 variants, i.e. Old World/ New World and Atlantic New World /Pacific New World /Austral New World (Table 5). Noteworthy, the joint analysis including the morphotypes in the Pacific cluster produced the largest within cluster variation as compared to any other hierarchical level.
Phylogenetic reconstruction
Evaluation of the relationship between the best phylogenetic signal and the most plausible phylogenetic scenario upon previous knowledge on this genus showed that Bootstrap support from Bayesian inference was maximum on concatenated ITS1—cyt b sequences (Table 6). The most congruent topology from likelihood-based trees was issued from concatenated ITS1—cyt b sequences (Fig. 2a; Table 6).

Phylogeny of genus Merluccius spp. comprising 11 valid species and M. angustimanus as built with concatenated sequences from ITS1-rDNA (HKY85 + I + G substitution model) and cyt b (GTR + G substitution model), (a) Maximum likelihood tree (-lnL = -4244.841) from IQ-TREE87; supporting values are written on branches (SH-aLRT (%)/ultrafast bootstrap (%)). (b) Bayesian tree from MRBAYES v3.2.686; branches are annotated with bootstrap values, resp. percent posterior probabilities. OTU Gmor corresponds to the outgroup Gadus morhua. Sample codes are given in Table 1 and are followed by the alphanumeric entry code for each specimen in the authors laboratory, e.g. aust_ma01_2 is the specimen No. 2 of sample ma01 from M. australis (aust). The maps have been modified after13.
ITS1-based phylogeny
The phylogenetic reconstruction of ML (-lnL = 1285.511) performed with PAUP on ITS1 variants recovered a correct supercluster split, a large polytomy within superclusters and an unclear age status of extant species (Supp. Figure S2a). The Bayesian reconstruction from MRBAYES on ITS1 variants recovered a single polytomy comprising all the well-recognized species clusters (Supp. Figure S2b). The Bayesian reconstruction of BEAST on ITS1 variants supported a correct supercluster split, the absence of polytomies and the odd basal placement of M. hubbsi in the New World supercluster (Fig. 1a).
Cyt b-based phylogeny
Likelihood computation of dN and dS using HyPhy package35 on cyt b sequences showed that six out of 142 codons contained nonsynonymous substitutions. Provided that p-values were not significant, the null hypothesis of neutral evolution was accepted. The best –lnL value and bootstrap support were observed on cyt b haplotypes but its topological features were unsatisfactory due to multiple polytomies (Supp. Fig S3a; Table 6). The ML phylogenetic reconstruction on cyt b haplotypes (-lnL = 1137.282) worked out with PAUP showed a poor definition of the Old World supercluster with a high impact of polytomy (Supp. Figure S3a). The best topology among Bayesian-based supported trees was obtained upon inference on cyt b haplotypes (Fig. 1b). The Bayesian phylogenetic reconstruction from MRBAYES on cyt b haplotypes recovered a strong supercluster split and a polytomy within the New World supercluster to which M. bilinearis was ancestor (Supp. Figure S3b). The Bayesian phylogenetic reconstruction of BEAST on cyt b haplotypes recovered a well-supported both, supercluster split and cladogenesis within superclusters, where M. bilinearis was basal to New World hakes (Fig. 1b).
Concatenated ITS1—cyt b phylogeny
The phylogenetic reconstruction using concatenated sequences of both genes showed bootstrap values > 90% for species and clusters. ML (Fig. 2a) and Bayesian (Fig. 2b) methods recovered two superclusters (New World vs. Old World), each of which contained two clusters (Pacific North + Pacific South, and North Euro-Africa + South Africa, respectively), the ancient status of M. bilinearis to both superclusters and that of M. albidus to the New World one. Consistently across reconstructions, M. angustimanus branched between M. productus and M. gayi. The Bayesian reconstruction placed M. angustimanus at the base of the sister taxons of M. productus and M. gayi (Supp. Figure S4). The phylogenies inferred from the methods ML and Bayesian were topologically similar to each other (Fig. 3). The Bayesian reconstruction failed to resolve the phylogenetic status of M. bilinearis regarding the two superclusters and exhibited a large polytomy within the New World supercluster.

Comparison of reconstruction methods from a synthetic phylogeny of genus Merluccius spp. on concatenated sequences from ITS1-rDNA (HKY85 + I + G substitution model) and cyt b (GTR + G substitution model). Left: Phylogeny of genus Merluccius spp. using Maximum likelihood (-lnL = − 4244.841) after IQ-TREE87. Right: Bayesian tree after MRBAYES v3.2.686. Circled nodes indicate topology differences between both reconstruction methods. Gadus morhua was used as outgroup.
Coalescence
The relative coalescence times inferred from a typical 2% mutation rate of cyt b in fish was ~ 9 MYA between Merluccius and the codfish genus Gadus, ~ 3.5 MYA between the New World and the Old World superclusters, and ~ 2.3 MYA between M. bilinearis and other New World hakes (Fig. 4). Atlantic and Pacific New World hakes would have diverged some ~ 1.4 MYA.

Bayesian reconstruction of genus Merluccius spp. on sequence variation of cyt b majority haplotypes (GTR + G substitution model) using BEAST v1.8.485. Divergence time is given in MYA on nodes as calculated upon a 2% mutation rate averaged among 26 pairs of major intraspecific phylogroups of fishes40. Gmor corresponds to the outgroup Gadus morhua. Scale bar indicates the No. of nucleotide substitutions per 100 DNA residues. Sample codes are given in Table 1.
ITS1Nes-based phylodiagnosis
The Neighbor-Joining reconstruction performed with the ITS1Nes fragment included six valid species and M. angustimanus, and comformed the basal phylogenetic backbone of this genus in the New World against which new phylogenetic hypotheses can be tested. The basal support afforded from the ITS1Nes fragment was quite close to that obtained on the full ITS1 sequence (Fig. 1a) but including a polytomy for M. gayi–M. productus (Fig. 5). Re-runs of morphotypes against the basal tree assigned five morphotype clones of M. hubbsi to the valid M. hubbsi cluster. The remaining 41 morphotype clones (M. polylepis, M. tasmanicus, M. patagonicus, M. australis, M. hubbsi) were grouped in a large intermediate and weakly supported cluster placed between the Pacific cluster (M. gayi–M. productus–M. angustimanus) and the Austral cluster (M. australis–M. hubbsi) (Fig. 6). Two additional minor clusters of morphotypes closely branched either to the Pacific cluster or to the Austral cluster.

Basal NJ tree on ITS1Nes sequences after PHYLIPv3.684 built to reconstruct the phylogenetic backbone of genus Merluccius spp. from the New World. The European hake (merl) was used as outgroup. Gray rectangles delineate a single cluster for each valid species. The percentage of trees over 1000 bootstrap replicates are shown above branches. Codes for valid species are given in Table 1 and are followed by the alphanumeric entry code for each specimen in the authors laboratory, e.g. aust_ma01_3 is the specimen No. 3 of sample ma01 from M. australis (aust).

Phylodiagnostic NJ tree (sensu18) on ITS1Nes sequences after PHYLIPv3.684 built to allocate morphotype clones (red branches and codes in uppercase) to the phylogenetic backbone of genus Merluccius spp. from the New World (clusters in grey rectangles, black branches and codes in lowercase). The percentage of trees over 1000 bootstrap replicates are shown above branches. Codes for valid species are given in Table 1 and are followed by the alphanumeric entry code for each specimen in the authors laboratory, e.g. aust_ma01_3 is the specimen No. 3 of sample ma01 from M. australis (aust). Codes for morphotypes are given in Table 2 and are followed by an alphanumeric entry from the authors laboratories (Supp. Table S9), e.g. TASM_M3_1 is a ITS1Nes sequence from muscle tissue (1) of specimen M3 (M3) of morphotype M. tasmanicus (TASM).
Discussion
Genetic homogeneity is the null hypothesis in widely-distributed marine taxa with large population size and sometimes morphological divergence may be a first clue to hypothesize on the existence of genetically divergent units13. Demonstration of intraspecific genetic differentiation requires comprehensive spatio-temporal sample designs, the identification of suitable genetic markers at the resolution level concerned, and the choice of appropriate phylogenetic algorithms18. The nuclear ITS1-rDNA region has been successfully applied in the identification of closely related taxa36, in fish phylogeography37, in phylogenetic inference38 as well as in forensic authentication of species33. Also, the cyt b gene is a well-known mtDNA gene in structure and function39 and is useful in phylogenetic reconstruction at many taxonomic levels, including congeneric species and confamiliar genera40 but see exceptions41. The synergy afforded from concatenated analyses of the nuclear ITS1-rDNA and the mitochondrial DNA cyt b allows comparison of interspecific levels of divergence and to achieving phylogenetic scenarios unaffordable from single-markers approaches42. The higher interspecific variation observed between New World and Old World superclusters (e.g. ITS1, Dxy ≈ 0.090) than within superclusters (e.g. ITS1, Dxy ≈ 0.040) is expected if previous calibrations were robust33. In consequence a higher net evolutionary divergence (d) existed among Old World species (d ≈ 0.080) than among New World species (d ≈ 0.030). In contrast, the minimum interspecific divergences (d and Dxy) observed among Pacific New World hakes (M. gayi–M. productus–M. angustimanus) represented the most recent evolutionary scenario in this genus, as reported from morphometric data5, allozyme data43 and mtDNA plus microsatellite data27.
The PCoA performed on ITS1 sequences agreed with AMOVA partitions that separated New World hakes into Atlantic, Austral, and Pacific groups. Those exploratory analyses also allocated morphotypes to either the Pacific or the Austral groups. The phylogenetic support for those groups tested between methods (Bayesian inference vs. ML) and markers (ITS1, cyt b) with non-parametric bootstrapping produced the best-supported Bayesian-based reconstruction on cyt b haplotypes. Such reconstruction was characterized by a well-defined supercluster split and the placement of M. bilinearis at the base of the New World supercluster (BEAST, Fig. 1b). However, the best Bayesian tree and the best ML tree were not fully congruent with previous studies, a handicap if congruence among marker reconstructions is a relevant asset, e.g. the better ML-value on cyt b does not grant any better species tree than that from ML-ITS1 because the topology of the latter is more congruent with previous studies. Such scenario highlights the insufficiency of the ML method on cyt b variation as compared to the Bayesian approach (but see44). The Bayesian phylogeny performed on both genes with BEAST was the unique method recovering both, a well-supported supercluster split and a full intracluster resolution, including the full definition of the Austral cluster. Discrepancies between markers using BEAST consisted on both, the branching of M. hubbsi as the oldest taxon among New World hakes with ITS1 and the branching of M. albidus within the Austral cluster with cyt b. Nonetheless, caution is needed on rejecting those odd positions regarding previous studies, since they could result from differential evolutionary rates among the markers applied.
It is expected that deep phylogenetic rooting of ancient hake lineages could be better afforded from conservative mtDNA haplotypes45. However, more recent evolutionary processes such as hybridization and drift could be better unveiled by highly recombinant nuclear DNA markers. Therefore, speciation histories based on mtDNA alone can be extensively misleading and large phylogenetic discrepancies have been reported between nuclear DNA and mitochondrial DNA46. Successful reconstructions have been achieved on concatenated sequences of the ITS region and COI47 or on nuclear genes48. Also, current data showed that concatenated data from ITS1-rDNA and cyt b at reconstructing hakes phylogeny using Bayesian inference have dramatically improved topology and support regarding individual markers49. Letting alone the well-supported supercluster split and the absence of polytomy, three topological novelties consisted on the placement of M. bilinearis as the oldest species of the genus, the basal branching of M. albidus to all New World hakes, and the intermediate branching of M. angustimanus between M. gayi and M. productus (see IQ-TREE, Fig. 2a). Those unexpected scenarios in view of previous studies can be explained as a synergical advantage of data concatenation as genes add up to produce a more balanced signal of the interspecific evolutionary divergence than that afforded from single genes47. Since each gene responds to distinct evolutionary dynamics it is expected that concatenated gene reconstructions approach the average evolutionary signal of their common history50.
The major phylogenetic split in Merluccius comprises two monophyletic superclusters, the Old World one comprises five species and the New World one comprises six species, as shown with parasites5,8,51, morphology and meristic traits6, allozymes14,52 and nucDNA33. This genus is believed to emerge in the Cretaceous after the opening of the southern Atlantic Ocean basin between South America and Africa7. However, its evolutionary bifurcation into New World and Old World superclusters is believed to have begun in the Oligocene53 when the northern margins of those continents begun to diverge (~ 30 MYA54). Application of an average 2% evolutionary clock to the cyt b as calibrated on 23 fishes40,55 suggests that genus Merluccius and genus Gadus diverged some 9 MYA and the supercluster split would have taken place some 3.4 MYA, what is congruent with estimates from the evolutionary rate of four mtDNA genes56 which dated back the supercluster split to some 3.0–4.2 MYA57. Such major evolutionary split seems to have been determined by a rapid vicariant-subsequent speciation in both continents in the early Pleistocene (ca. 2.58–0.77 MYA) with separation of Old World hakes into two clusters around 1.9 MYA, i.e. much later than reported after allozyme data (4.2–3 MYA52). The actual speciation scenario was not accomplished until ~ 0.3 MYA with the divergence within the species pairs M. merluccius–M. senegalensis and M. productus–M. gayi. Nevertheles, such divergence times as computed on a cyt b—based molecular clock should be taken as relative inferences because the evolutionary scenario from concatenated-based trees differs substantially from any other single-marker reconstruction. The Old World supercluster is believed to have experienced strong bottlenecks after the early divergence of M. polli and M. paradoxus, followed by a later speciation of M. merluccius, M. capensis and M. senegalensis52. A consensus exists on the monophyly of those two clusters8,14,15,52. Such phylogenetic partition is biogeographically counterintuitive since sympatric M. capensis and M. paradoxus in Atlantic South Africa are believed to emerge from two independent dispersions of North Atlantic taxa along the west coast of Africa58. Moreover, provided that M. polli and M. paradoxus are two benthonic species closer to the New World supercluster, they have been proposed both, either to descend from an early Old World Merluccius cluster52 or to represent a distinct speciation process in the Eastern Atlantic59.
The variance partition, the multivariate approach, and the phylogenetic inference, all them support three clusters within the New World supercluster, namely the Atlantic, the Austral and the Pacific. In the Atlantic cluster, the evolutionary status of M. bilinearis is disaggregated in classical hake phylogenies. Generally, this species has been placed as ancient to New World species with various methods and markers, including likelihood52 and parsimony14 on allozymes; NJ, ML, UPGMA on the D-loop15 and current Bayesian analyses on cyt b haplotypes. However, new scenarios appear after concatenated ITS1—cyt b sequences which indefectibly place M. bilinearis as the oldest among extant hake species (see Fig. 2).
Most previous studies agree on that hake originated in the North Atlantic and entered the Pacific, but disagree on the origin of M. hubbsi, i.e. as diverted either from an eastern south Pacific stock6,60 or from a western north Atlantic stock5,8. A novelty in the current concatenated approach is a well-supported Austral cluster that is congruent with parasite studies in which M. hubbsi and M. australis are closely related taxa of Austral origin6. The concatenated ML tree also shows the basal status of M. albidus to all New World hakes except M. bilinearis, what agrees with previous morphological and meristic studies5. Moreover, its broad western Atlantic distribution suggests that M. albidus is the colonizer of de Southern Cone and the primeval species of the Austral cluster. The phylogenetic proximity of M. hubbsi and M. australis suggests that M. hubbsi would have speciated after M. albidus some 1.5 MYA and would have reached its present distribution by a dispersal route along the Atlantic coast5. Noteworthy, other marker types offer the different scenario of M. hubbsi speciation prior to M. albidus (see also14).
Pacific New World hakes are believed originated upon the early migration of North Atlantic New World hakes into the Pacific8. Combined allozyme data and mtDNA14,52 and hake otoliths61,62 support the hypothesis of the separation between M. bilinearis and both, M. productus–M. gayi and M. albidus before the closure of the Panama Isthmus (3.5 MYA63) and advanced the divergence of M. bilinearis to the Miocene some 12 MYA52. However, present data suggest that M. albidus diverged first from M. bilinearis and that the divergence of actual north Pacific species from Atlantic species begun with the rise of the Panama Isthmus. A more recent speciation in the Pacific seems to have occurred between M. productus and M. gayi some 0.5 MYA. Such temporal estimate is congruent with the origin of M. angustimanus as a result of a post-speciation confinement of one of those species or their hybrids in the northern Gulf of California, some 0.25 MYA. Those hypothetical timeframes should be further investigated in relation to reported population fragmentations and expansions due to climatic oscillations that took place during Pleistocene glaciations in that area12.
Several species have been proposed to occur in the Pacific New World cluster such as M. productus, M. angustimanus, M. hernandezi, M. gayi gayi, and M. gayi peruanus22,64,65 as well as morphs (dwarf, normal, etc.)27,66,67 all with close morphological and meristic similarity among each other5,6 as well as in protein-coding loci43. Recently, it has been suggested that M. gayi68 could be the single pan-Pacific species28 although the North-Eastern Pacific hake has to be named Merluccius productus by priority28. Despite being considered by FAO as a variant of M. angustimanus3 and reproductively isolated from M. productus and M. angustimanus14, Merluccius hernandezi22 from Sinaloa state would likely be a dwarf morphotype of M. productus28. After its first description as a “tropical deep-water species off the Pacific coast of Central America from Mexico to Panama which is sandwiched between M. productus to the north and M. gayi to the south”14, M. angustimanus has been a more recognized taxon than M. hernandezi69. In the first genetic description of M. angustimanus33 it was named M. hernandezi provided its northern California origin. Following recovering of its trawling data from the seventies70 as well as the examination of its large scales (Mathews, personal communication) such sample was properly renamed as M. angustimanus18. That genetic analysis of M. angustimanus showed that the PCR–RFLP pattern on the ITS1 spacer was very similar to those of M. productus and M. gayi18,33. Also, current data showed that M. angustimanus shared its two unique ITS1 variants with M. productus. That result is congruent with the within species concerted evolution of the ITS1-rDNA gene family37 and also with the recently proposed confinement and drift of M. productus in the Gulf of California giving rise to its present divergent population27,27. Present data also suggest the putative hybrid origin of M. angustimanus by means of a trans-equatorial incursion of M. gayi5,6,14,22 in the territories of M. productus in the Gulf of California (see the bootstrap weakness within the Pacific cluster in all concatenated reconstructions, Fig. 2). Moreover, the six specimens of M. angustimanus examined had no evolutionary novelties in their cyt b region but rather were a chimeric genome from extant neighboring species plus an A-139 residue shared with M. albidus (see Supp. Table S4). In summary, M. angustimanus seems to be a hake population trapped in the northern Gulf of California27 whose origin dates back some 0.25 MYA either from hybridization between M. productus and M. gayi or from a confinement of a subset of those species therein. Such hypotheses would explain the weak morphological and molecular differentiation of M. angustimanus from its neighboring hake species22. Whether that confined population can be self-sustainable as a relic of an ancient hybridization or if its viability depends on ongoing genetic contribution from the surrounding M. productus, are unexplored questions.
The morphotypes under test other than M. angustimanus were analyzed with the nested fragment ITS1Nes which offered a more conservative view of the interspecific diversity than the full ITS1 sequence, i.e. 11 HakeITS1Nes variants versus 19 HakeITS1 variants. In addition to morphotype-specific variants, all morphotypes shared the specific HakeITS1Nes.2 sequence from Pacific hakes, what suggests their common origin. Such commonality does not preclude the presence of specific sequences within the ITS1 spacer family that trace back the origin of their carriers. For instance, although some ITS1Nes variants of a given morphotype fully grouped into the M. hubbsi clade (e.g. HUBBM131) other intraindividual variants branched intermediately between the Pacific cluster and the Austral cluster. That is a typical scenario for interspecific hybrids which can be ascertained by inspection of the dramatic decrease of the basal tree bootstrap support of the Austral and the Pacific clusters in the phylogenetic backbone (see Figs. 5 and 6). The significant No. of recombination events detected in those sequences also comes to support the working hypothesis of a hybrid origin for those morphotypes. The small molecular differentiation observed among morphotypes does not support either their distinct origin nor the existence of cryptic species around the Southern Cone. For instance, the six clones of the M. hubbsi paratype from the Beagle Channel (HUBBM91 and HUBBM92) shared ITS1Nes variants with all other morphotypes, including M. patagonicus (PATAM101 and PATAM111). After the description of the M. patagonicus23 some additional analyses suggested its synonymy with M. hubbsi29,31. However, the three paratypes of M. patagonicus did not share any sequence variant with true specimens of M. hubbsi (e.g. hubb153.2) but branched between the clusters of Austral hakes and Pacific hakes. Likewise, M. tasmanicus24 and M. polylepis from the Southern Ocean32 had been proposed as synonymous of M. australis6,29,30. However, their phylogenetic status was much like the rest of morphotypes, e.g. the paratypes of M. australis from Chile and from New Zealand. It is probable that those rare morphotypes resemble extant species when analyzed with more conservative markers such as the mtDNA Cytochrome c oxidase subunit I (COI)29. However, less conservative regions such as the ITS1-rDNA suggest that the morphotypes examined are the result of recurrent hybridization between adjacent M. gayi and M. australis or M. hubbsi in the Southern Cone. The wide distribution range of those parent species would make probable to find hybrids commonplace from the western Atlantic South in Argentine to the Western Pacific South in New Zealand.
Methods
Sample collection
This study was conducted in accordance with the European directive 2010/63/EU and the Spanish legislation on animal welfare (RD53/2013 and RD1386/2018). All the experiments were approved by the local ethics committee of Centro Oceanográfico de Vigo. Samples were taken from fishing or formalin-preserved corps; therefore no restrictions apply on taking tissue samples for species identification. Eleven valid hake species were sampled in their natural ranges (Table 1) together with M. angustimanus from the northern Gulf of California and nine morphotypes as three from the western Atlantic South, three from the eastern Pacific South and three from the south Western Pacific (Table 2, Supp. Table S9). The Atlantic cod (Gadus morhua) was used as a phylogenetic outgroup. The world-wide sampling was performed on board of factory ships and research vessels between 2000 and 200333 when 9–277 specimens per species were preserved in 95% ethanol or frozen upon collection and their GPS recorded on board (Table 1). All specimens were identified using species-specific morphological traits such as otoliths, shape of abdominal vertebrae (parapophysis), cranial shape and pectoral fin length, following classification criteria previously established6. Whole specimens were boiled to recover those structures and to facilitate bone cleaning. Six specimens of M. angustimanus were sampled as preserved frozen from a bottom trawl campaign (80–523 m in depth) carried out in 1973 by Instituto Nacional de la Pesca, Mexico70. Fourteen additional specimens as holotypes, paratypes and rare morphotypes were either sampled in situ by the authors or taken from museum specimens as preserved in formalin. Two specimens of M. tasmanicus (DM5566 and DM3963) and four specimens of M. patagonicus were sampled and described in23,24, respectively. The paratype USNM 157765 of M. polylepis from Puerto Montt (Chile) was described by23 and the M. polylepis holotype off Chiloe (Chilean Pacific) was described by32. Three rare morphotypes of M. hubbsi from the Beagle Channel (IIPB92/1987) and from Puerto Madryn were described by23. Finally, two rare morphotypes of M. australis from New Zealand (MOVI 27492–27493, formerly NMNZ P.13122) were described by24 and two additional ones from the Chilean Pacific (off the Aysén Region) were described in23.
Molecular data
Genomic DNA was extracted with FENOSALT71 including a preliminary 24 h hydration step for formalin-fixed samples. Two synergic tools for hakes identification were applied, one based on the ITS1-rDNA spacer33 and another based on the mtDNA cyt b gene18. Those targets are suitable DNA regions for the taxonomic classification of closely related taxa and their combinatory power has proven to be a robust approach to reduce the authentication error from hake-based commercial products72. Amplification conditions for the ITS1-rDNA spacer followed previous developments in this genus33,73. Electropherograms were revised with Chromas software (Technelysium, Tewantin, Australia). A total of 85 specimens were sequenced as averaging ~ 6 specimens per species (Supp. Table S1). Formalin-preserved samples produced 46 ITS1 nested sequences (ITS1Nes) of 66 bp in length using the new primer pair, PARIB152 (5′-GTTTCGCTGACCCCGTTGG-3′) and PARIB197 (5′-CCGCACTCTCCCTCGTACCTC-3′). The PCR reactions contained 1 µl DNA template, 20 pmol of each primer, 200 µM of each dNTP, 1.6 mM MgCl2, 5 µl of 5X Colorless GoTaq Flexi Buffer and 1.5U GoTaq Flexi DNA Polymerase (Promega) in a total volume of 25µL. Amplification was performed in an Eppendorf Mastercycler Gradient under the following conditions: one cycle at 95ºC for 5 min, 30 cycles of 1 min at 95 °C, 1 min at 57 °C and 1 min at 72 °C and a final extension at 72 °C for 10 min. Amplicons were cleaned using the Wizard SV Gel and PCR Clean-Up System (Promega) according to the manufacturer’s protocol. Clean PCR products were cloned into the pGEM-T Easy Vector System II (Promega) following the manufacturer protocol using a 3:1 insert:vector ratio. Clones were lysed and their plasmid purified using the NucleoSpin Extract (Mackerey -Nagel). Sequencing was performed on both strands in an ABI Prism 3100 Sequencer (Applied Biosystems) using T7 and SP6 primers. Several ITS1Nes sequences were obtained from multiple cloning per specimen of each morphotype (Supp. Table S1, Supp. Table S9). The 3′-end of the cyt b gene was obtained as described for this genus18,74. Sixty-six specimens were sequenced for cyt b as 5.42 ± 2.47 specimens per species in addition to the outgroup Gadus morhua.
Sequence alignment
The 3′ end of the 18S gene and the 5′-end of the 5.8S gene were used to align the 85 ITS1 sequences obtained from 11 hake species using the program SeqLab from the GCG software package (Genetics Computer Group, Madison, Wisconsin)75. The alignment of 66 sequences from the 464–465 bp cyt b region was performed from the 3′-end of the mitochondrial tRNA-Glu gene. The molecular properties of those sequences per species were calculated using MEGA v7.0.2076. Nucleotide diversity (Pi) was computed using DnaSP v5.10.177. The average number of nucleotide substitutions per site (Dxy) and estimates of the Net Evolutionary Divergence between species (d) were compared among taxa using the confidence interval calculated as CI = \(\stackrel{-}{x}\)±1.96*(S.D./\(\sqrt{n}\)). The Principal Coordinates Analysis (PCoA) built with GenAlEx v6.50378 on the molecular variation of ITS1 variants was used to check for the correct divergence signal between superclusters (New World and Old World) as well as between clusters and between species. Genetic variation within and between clusters were contrasted with an Analysis of Molecular Variance (AMOVA) as implemented in Arlequin v3.5.2.279.
Comparative phylogeny and synthetic phylogeny
The phylogenetic reconstructions using the ITS1 region, the ITS1Nes fragment and the cyt b region included reference samples used at establishing the phylogenetic backbone of the genus18,33. The phylogenetic test enforced to allocate morphotypes was based on the phylogenetic backbone of New World hakes built with the ITS1Nes fragment. The most suitable nucleotide substitution models were selected using jModeltest v2.1.1080. The HKY85 + I + G model81 was the best fitted to the ITS1-rDNA variation and the GTR + G model82 was the best fitted to that of the cyt b gene. Likelihood computations of nonsynonymous (dN) and synonymous (dS) substitution rates were conducted using HyPhy software package35. The phylogenetic reconstruction was carried out using full sequences (ITS1, ITS1Nes and cyt b) as well as sequence variants (ITS1 and ITS1Nes) and cyt b haplotypes, as obtained with program DnaSP v5.10.177. That software was also used to estimate the Recombination parameter (R)34 as (R = 4Nr) where N is the population size and r is the recombination rate per sequence. The rate R allowed to assess whether recombination fingerprints can be detected among ITS1Nes sequences as expected in highly recombinant nuclear DNA markers37.
Character-based phylogenies were first constructed using Maximum Likelihood (ML) hypothesis testing after PAUP v4.083, a gamma distribution coefficient (gamma-ITS1 or gamma-ITS1Nes = 0.269; gamma—cyt b = 0.420), a transition/transversion rate (Ti/Tv-ITS1 and Ti/Tv-ITS1Nes = 1.761; Ti/Tv—cyt b = 7.143) and the nucleotide substitution rate matrix observed for each marker. An algorithmic Neighbor-joining tree was built on ITS1Nes using PHYLIP v3.6 upon 5000 bootstrap replicates84. Bayesian phylogenetic hypotheses on ITS1 variants as well as on cyt b haplotypes were tested using the programs BEAST v1.8.485 and MRBAYES v3.2.686 and taking into account the best fitted evolutionary models. The comparative phylogeny consisted on contrasting tree support values and topological consistence per marker type (ITS1 and cyt b) among reconstruction methods (ML and Bayesian). The synthetic phylogeny of the genus consisted on a ML reconstruction made upon concatenated data sets of both genes using IQ-TREE (http://iqtree.cibiv.univie.ac.at/87) which allows partitioning the analysis to simultaneously conjugate two evolutionary models and also enforces the Shimodaira–Hasegawa test (SH) for testing gene trees. The null hypothesis (H0) stated that all the trees tested would be equally performant at explaining the observed data, while the alternate hypothesis (H1) stated that only one among several trees was a better proxy to real data. The Bayesian models implemented in MRBAYES and BEAST were employed in the phylogenetic reconstruction of concatenated sequences. The Net Evolutionary Divergence (d) among sequences (ITS1 variants, ITS1Nes variants, cyt b haplotypes) was calculated upon divergence between OTUs (species, clusters and superclusters) as (PAB) and corrected for within-OTU variation (PA and PB) as:
The net divergence values among cyt b sequences were converted to proxy estimates of coalescence times using a standard mtDNA clock for fishes, i.e. 2% sequence divergence per million years between two lineages55.
References
Rafinesque, C. S. Caratteri di alcuni nuovi generi e nuove specie di animali e piante della Sicilia: con varie osservazioni sopra i medesimi. Per le stampe di Sanfilippo 105 (1810). https://doi.org/10.5962/bhl.title.104418.
Alheit, J. & Pitcher, T. J. Hake: Biology, fisheries and markets. Fish. Res. 15, 478. https://doi.org/10.11646/zootaxa.3956.1.2 (1995).
Cohen, D. M., Inada, T., Iwamoto, T. & Scialabba, N. FAO Catalogue of Species Vol.10. FAO species Cat. Vol. 10 Gadiform Fishes world (Order Gadiformes). FAO Fish. Synop. 125(10), 1–442 (1990). http://www.fao.org/tempref/docrep/fao/009/t0243e/t0243e00.pdf.
Jones, B. W. World resources of hakes of the genus Merluccius, in: Sea Fisheries Research (ed. F.R. Harden Jones) 139–166 (London, 1974).
Ho, S. J. Phylogeny and biogeography of hakes (Merluccius; Teleostei): a cladistic analysis. Fish. Bull. 88, 95–104 (1990). https://ci.nii.ac.jp/naid/10017483742/.
Inada, T. Studies on the merlucciid fishes. Res. Lab. Shimizu, Japan. Bull. Far Seas Fish. 18, 1–172 (1981).
Fedotov, V. F. & Bannikov, A. F. On phylogenetic relationships of fossil Gadidae. Pap. Syst. Gadiform Fishes. 32, 187–196 (1989). http://ci.nii.ac.jp/naid/10016455908/en/.
Kabata, Z. & Ho, J. The origin and dispersal of hake (Genus Merluccius: Pisces: Teleostei) as indicated by its copepod parasites. Oceanogr. Mar. Biol. Annu. Rev. 19, 381–404 (1981). http://www.vliz.be/en/imis?refid=97503.
Hallam, A. Phanerozoic Sea-Level Changes (Columbia University Press, New York, 1992).
Savin, S. M., Douglas, R. G. & Stehli, F. G. Tertiary marine paleotemperatures. Bull. Geol. Soc. Am. 86(11), 1499–1510. https://doi.org/10.1130/0016-7606(1975)86%3c1499:TMP%3e2.0.CO;2 (1975).
Knowlton, N., Weigt, L. A., Solórzano, L. A., Mills, D. E. K. & Bermingham, E. Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science 260, 1629–1632. https://doi.org/10.1126/science.8503007 (1993).
Savage, J. M. Evolution of a peninsular herpetofauna. Syst. Zool. 9(3/4), 184–212. https://doi.org/10.2307/2411967 (1960).
Lloris, D., Matallanas, J. & Oliver, P. Hakes of the world (Family Merlucciidae). An annotated and illustrated catalogue of hake species known to date, in: FAO Species Catalogue for Fishery Purposes. No. 2. (FAO) 57 (Rome, 2005). https://digital.csic.es/bitstream/10261/32166/9/Lloris_et_al_2005.pdf.
Grant, W. S. & Leslie, R. W. Inter-ocean dispersal is an important mechanism in the zoogeography of hakes (Pisces: Merluccius spp.). J. Biogeogr. 28, 699–772. https://doi.org/10.1046/j.1365-2699.2001.00585.x (2001).
Quinteiro, J., Vidal, R. & Rey-Méndez, M. Phylogeny and biogeographic history of hake (genus Merluccius), inferred from mitochondrial DNA control-region sequences. Mar. Biol. 136, 163–174. https://doi.org/10.1007/s002270050019 (2000).
Roldán, M. I., García-Marín, J. L., Utter, F. M. & Pla, C. Population genetic structure of European hake, Merluccius merluccius. Heredity 81(3), 327–334. https://doi.org/10.1046/j.1365-2540.1998.00383.x (1998).
Henriques, R., Von der Heyden, S. & Matthee, C. A. When homoplasy mimics hybridization: a case study of Cape hakes (Merluccius capensis and M. paradoxus). PeerJ 4, e1827 (2016). https://doi.org/10.7717/peerj.1827.
Pérez, M. & Presa, P. Validation of a tRNA-Glu-cytochrome b key for the molecular identification of 12 hake species (Merluccius spp.) and Atlantic cod (Gadus morhua) using PCR-RFLPs, FINS, and BLAST. J. Agric. Food Chem. 56(22), 10865–10871. https://doi.org/10.1021/jf801700x (2008).
Gatica, C., Neira, S., Arancibia, H. & Vásquez, S. The biology, fishery and market of Chilean hake (Merluccius gayi gayi) in the South eastern Pacific Ocean, in: Hakes: Biology and Exploitation (ed. Wiley Blackwell) 126–153 (Oxford, 2015). https://doi.org/10.1002/9781118568262.ch5.
Espino, M., Castillo, R. & Fernández, F. Biology and fisheries of Peruvian hake (M. gayi peruanus), in: Hake (ed. Springer Netherlands) 339–363 (Dordrecht, 1995). https://doi.org/10.1007/978-94-011-1300-7_12.
Garman, S. Results of an exploration of the west coasts of México, Central and South America, and off the Galapagos Island. Fish. Mem. Mus. Comp. Zool. 26, 183–185 (1899).
Mathews, C. P. Meristic studies of the Gulf of California species of Merluccius, with a description of a new species. J. Nat. Hist. 19, 697–718. https://doi.org/10.1080/00222938500770421 (1985).
Lloris, D. & Matallanas, J. Description of a new species of hake: Merluccius patagonicus sp. Nov. (Gadiformes: Merlucciidae) from the waters of Argentina. Sci. Mar. 67, 323–326. https://doi.org/10.3989/scimar.2003.67n3323 (2003).
Matallanas, J. & Lloris, D. Description of Merluccius tasmanicus sp. Nov. and redescription of Merluccius australis (Pisces: Merlucciidae). J. Mar. Biol. Assoc. U. K. 86(1), 193–199. https://doi.org/10.1017/S0025315406013038 (2006).
Iwamoto, E. M., Elz, A. E., García-De León, F. J., Silva-Segundo, C. A., Ford, M. J., Palsson, W. A. & Gustafson, R. G. Microsatellite DNA analysis of Pacific hake Merluccius productus population structure in the Salish Sea. ICES J. Mar. Sci. 72(9), 2720–2731 (2015). https://doi.org/10.1093/icesjms/fsv146.
Iwamoto, E., Ford, M. J. & Gustafson, R. G. Genetic population structure of Pacific hake, Merluccius productus, in: The Pacific Northwest, in: Genetics of Subpolar Fish and Invertebrates (ed. Springer) 187–199 (Dordrecht, 2004). https://doi.org/10.1023/B:EBFI.0000022895.10683.c5.
García De León, F. J., Galván-Tirado, C., Velasco, L. S., Silva-Segundo, C. A., Hernández-Guzmán, R., Barriga-Sosa, I. D. L. A., Jaimes, P. D., Canino, M. & Cruz-Hernández, P. Role of oceanography in shaping the genetic structure in the North Pacific hake Merluccius productus. PLoS ONE 13(3), e0194646 (2018). https://doi.org/10.1371/journal.pone.0194646.
Silva-Segundo, C. A. et al. Clarifying the taxonomic status of Merluccius spp in the northeastern Pacific: A combined morphological and molecular approach Rev. Fish Biol. Fish. 21(2), 259–282. https://doi.org/10.1007/s11160-010-9166-6 (2011).
Deli Antoni, M. Y., González-Castro, M. & Díaz de Astarloa, J. M. New tools (DNA barcoding), old hypothesis: the case of the taxonomic identity of the Argentine hakes (Actinopterygii: Merluccius). J Fish Biol. 87(3), 783–793. https://doi.org/10.1111/jfb.12745 (2015).
Díaz de Astarloa, J. M. et al. Morphological, morphometric, meristic and osteological evidence for two species of hake (Actinopterygii: Gadiformes: Merluccius) in Argentinean waters. J. Fish Biol. 78(5), 1336–1358. https://doi.org/10.1111/j.1095-8649.2011.02937.x (2011).
Antoni, M. Y. D., Delpiani, S. M., Stewart, A. L., González-Castro & M., Díaz De Astarloa, J. M. Merluccius tasmanicus Matallanas & Lloris 2006 is a junior synonym of M. australis (Hutton 1872) (Gadiformes: Merluciidae) based on morphological and molecular data. Zootaxa 3956(1), 029–055 (2015). https://doi.org/10.11646/zootaxa.3956.1.2.
Ginsburg, I. Whitings on the coasts of the American continents. U.S. Fish Wildl. Serv., Fish Bull. 56, 187–208 (1954). https://www.st.nmfs.noaa.gov/spo/FishBull/56-1/ginsburg.pdf.
Pérez, M., Vieites, J. M. & Presa, P. ITS1-rDNA-based methodology to identify world-wide hake species of the genus Merluccius. J. Agric. Food Chem. 53(13), 5239–5247. https://doi.org/10.1021/jf048012h (2005).
Hudson, R. R. & Kaplan, N. L. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111(1), 147–164 (1985).
Kosakovsky Pond, S. L., Frost, S. D. W. & Muse, S. V. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. https://doi.org/10.1007/0-387-27733-1_6 (2005).
Pleyte, K. A., Duncan, S. D. & Phillips, R. B. Evolutionary relationships of the salmonid fish genus Salvelinus inferred from DNA sequences of the first Internal Transcribed Spacer (ITS 1) of ribosomal DNA. Mol. Phylogenet. Evol. 1(3), 223–230. https://doi.org/10.1016/1055-7903(92)90019-D (1992).
Presa, P., Pardo, B. G., Martínez, P. & Bernatchez, L. Phylogeographic congruence between mtDNA and rDNA its markers in brown trout. Mol. Biol. Evol. 19, 2161–2175. https://doi.org/10.1093/oxfordjournals.molbev.a004041 (2002).
Baldwin, B. G. et al. The ITS Region of Nuclear Ribosomal DNA: a valuable source of evidence on angiosperm phylogeny. Ann. Missouri Bot. Gard. 82(2), 247–277. https://doi.org/10.2307/2399880 (1995).
Esposti, M. D. et al. Mitochondrial cytochrome b: evolution and structure of the protein. Biochim. Biophys. Acta. Bioenergy 1143(3), 243–271. https://doi.org/10.1016/0005-2728(93)90197-N (1993).
Johns, G. C. & Avise, J. C. A comparative summary of genetic distances in the vertebrates from the mitochondrial cytochrome b gene. Mol. Biol. Evol. 15(11), 1481–1490. https://doi.org/10.1093/oxfordjournals.molbev.a025875 (1998).
Farias, I. P., Ortí, G., Sampaio, I., Schneider, H. & Meyer, A. The cytochrome b gene as a phylogenetic marker: The limits of resolution for analyzing relationships among cichlid fishes. J. Mol. Evol. 53(2), 89–103. https://doi.org/10.1007/s002390010197 (2001).
Cuéllar-Pinzón, J., Presa, P., Hawkins, S. J. & Pita, A. Genetic markers in marine fisheries: Types, tasks and trends. Fish. Res. 173, 194–205. https://doi.org/10.1016/j.fishres.2015.10.019 (2016).
Stepien, C. A. & Rosenblatt, R. H. Genetic Divergence in Antitropical Pelagic Marine Fishes (Trachurus, Merluccius, and Scomber) between North and South America. Copeia https://doi.org/10.2307/1447522 (1996).
Erixon, P., Svennblad, B., Britton, T. & Oxelman, B. Reliability of bayesian posterior probabilities and bootstrap frequencies in phylogenetics. Syst. Biol. 52(5), 665–673. https://doi.org/10.1080/10635150390235485 (2003).
Moore, W. S. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution 49(4), 718–726. https://doi.org/10.2307/2410325 (1995).
Shaw, K. L. Conflict between nuclear and mitochondrial DNA phylogenies of a recent species radiation: What mtDNA reveals and conceals about modes of speciation in Hawaiian crickets. Proc. Natl. Acad. Sci. 99(25), 16122–16127. https://doi.org/10.1073/pnas.242585899 (2002).
Woodyard, E. T., Rosser, T. G. & Rush, S. A. Alligator wrestling: morphological, molecular, and phylogenetic data on Odhneriotrema incommodum (Leidy, 1856) (Digenea: Clinostomidae) from Alligator mississippiensis Daudin, 1801 in Mississippi, USA. Parasitol. Res. 116(11), 2981–2993. https://doi.org/10.1007/s00436-017-5607-7 (2017).
Kocsubé, S. et al. Aspergillus is monophyletic: evidence from multiple gene phylogenies and extrolites profiles. Stud. Mycol. 85, 199–213. https://doi.org/10.1016/j.simyco.2016.11.006 (2016).
Hedges, S. B. et al. Phylogenetic relationships of the hoatzin, an enigmatic South American bird. Proc. Natl. Acad. Sci. USA 92(25), 11662–11665. https://doi.org/10.1073/pnas.92.25.11662 (1995).
Hughes, J. M. & Baker, A. J. Phylogenetic relationships of the enigmatic hoatzin (Opisthocomus hoazin) resolved using mitochondrial and nuclear gene sequences. Mol. Biol. Evol. 16, 1300–1307. https://doi.org/10.1093/oxfordjournals.molbev.a026220 (1999).
Soliman, I. A. Variations of fishes of genus Merluccius in Atlantic Ocean and Mediterranean Sea. Acta Ichthyol Piscat. 3, 29–64 (1973).
Roldán, M. I., García-Marín, J. L., Utter, F. M. & Pla, C. Genetic relationships among Merluccius species. Heredity 83, 79–86. https://doi.org/10.1038/sj.hdy.6885300 (1999).
Van Andel, T. H. An eclectic overview of plate tectonics, paleogeography, and paleoceanography, in: Historical Biogeography, Plate Tectonics and the Changing Environment. Proc. (ed. 37th Ann. Biol. Colloquium and selected Papers) 9–25 (Oregon State University Corvallis, 1979).
Van Syoc, R. J. Barnacle mitochondrial DNA: determining genetic relationships among species of Pollicipes. Crustacean issues 10, 269–296 (1995).
Avise, J. C., Walker, D. & Johns, G. C. Speciation durations and Pleistocene effects on vertebrate phylogeography. Proc. R. Soc. Biol. Sci. 265(1407), 1707–1712. https://doi.org/10.1098/rspb.1998.0492 (1998).
McCune, A. R. How fast is speciation? Molecular, geological, and phylogenetic evidence from adaptive radiations of fishes, in: Molecular evolution and adaptive radiation (ed. T. J. Givnish & K. J. Sytsma) 585–610 (Cambridge University Press, 1997).
Campo, D., Machado-Schiaffino, G., Pérez, J. & García-Vázquez, E. Phylogeny of the genus Merluccius based on mitochondrial and nuclear genes. Gene 406, 171–179. https://doi.org/10.1016/j.gene.2007.09.008 (2007).
Grant, W. S., Becker, I. I. & Leslie, R. W. Evolutionary divergence between sympatric species of southern African hakes, Merluccius capensis and M. paradoxus I: electrophoretic analysis of proteins. Heredity 61, 13–20. https://doi.org/10.1038/hdy.1988.86 (1988).
Becker, I. I., Grant, W. S., Kirby, R. & Robb, F. T. Evolutionary divergence between sympatric species of southern African hakes, Merluccius capensis and M. paradoxus II: restriction enzyme analysis of mitochondrial DNA. Heredity 61(2), l–30 (1988). https://doi.org/10.1038/hdy.1988.87.
Szidat, L. La fauna de parásitos de Merluccius hubbsi como carácter auxiliar para la solución de problemas sistemáticos y zoogeográficos del género Merluccius L. Com. Inst. Nac. Cienc. Nat., Bs. As. 3(1), 1–54 (1955).
Fitch, J. E. Fossil records of certain schooling fishes of the California current system. Rapp. CALCOFI 13, 71–80 (1969).
Zinsmeister, W. J. A late Pliocene macrofossil fauna of Newport Beach, Orange County. Bull. S. Calif. Acad. Sci. 69, 121–125 (1970). https://scholar.oxy.edu/handle/20.500.12711/11488.
Vawter, A. T., Rosenblatt, R. & Gorman, G. C. Genetic divergence among fishes of the Eastern Pacific and the Caribbean: Support for the molecular clock. Evolution 34(4), 705–711. https://doi.org/10.1111/j.1558-5646.1980.tb04009.x (1980).
Hernández, C., Galleguillos, R. & Oyarzun, C. Diferenciación genética de Merluccius gayi gayi y Merluccius gayi peruanus (Pisces, Merlucciidae) y antecedentes paleogeográficos de su área de distribución. Rev. Chil. Hist. Nat. 73(1), 23–29. https://doi.org/10.4067/S0716-078X2000000100003 (2000).
Mathews, C. P. Some observations on the ecology and the population dynamics of Merluccius angustimanus in the South Gulf of California. J. Fish Biol. 7(1), 83–94. https://doi.org/10.1111/j.1095-8649.1975.tb04578.x (1975).
Balart-Páez, E. F. Biología y Ecología de la merluza Baja Californiana Merluccius angustimanus Garman, 1899, en la costa occidental de Baja California Sur, México. Doctoral Thesis, Universidad Autónoma de Nuevo León, Monterrey (2005). http://eprints.uanl.mx/id/eprint/20622.
Zamora-García, O. G. et al. Catch rate, length, and sex ratio of Pacific hake (Merluccius productus) in the northern Gulf of California Issue. Fish. ull. 118(4), 365–379. https://doi.org/10.7755/FB.118.4.6 (2020).
Berry, F. H. & Perkins, H. C. Survey of pelagic fishes of the California Current area. Fish. Bull. US. 65, 625–682 (1965).
Pitcher, T. J. & Alheit, J. What makes a hake? A review of the critical biological features that sustain global hake fisheries, in: Hake (ed. Springer Netherlands) 1–14 (Netherlands, 1995). https://doi.org/10.1007/978-94-011-1300-7_1.
De la Rosa, M. & Pedrin, O. Informe de crucero realizado a bordo del barco "Antonio Alzate" entre Punta Piaxtla, Sin. y Bahía Kino, Son. del 17 de octubre al 3 de noviembre de 1973 (AA-73–10). Secretaria de Industria y Comercio, Subsecretaria de Pesca, Instituto Nacional de Pesca, 13 (1974).
Pérez, M. & Presa, P. FENOSALT: Un método sintético para la extracción de ADN de peces y moluscos, in: Métodos y Técnicas de Investigación Marina (ed. García-Estévez, J. M., et al. Editorial Tecnos) 81–89 (Madrid, 2011). https://agris.fao.org/agris-search/search.do?recordID=ES2015B00724.
Pérez, M., Santafé-Muñoz, A. M., Balado, M. & Presa, P. Methodological evaluation of DNA-based molecular keys to identify categories of mislabelling in commercial products from genus Merluccius spp. Food Chem. 239, 640–648. https://doi.org/10.1016/j.foodchem.2017.06.138 (2018).
Pérez, M., Cabado, A. G., Vieites, J. M. & Presa, P. Experimental Assessment of a New rDNA-based Method for the Identification of Merluccius capensis and Merluccius paradoxus in Commercial Products. J. Aquat. Food Prod. Technol. 13(2), 49–57. https://doi.org/10.1300/J030v13n02_05 (2004).
Pérez, M. et al. Identification of south Atlantic hakes (Merluccius australis and Merluccius hubbsi) in processed foods by PCR-RFLPs of Cytochrome b gene. J. Aquat. Food Prod. Technol. 13(2), 59–67. https://doi.org/10.1300/J030v13n02_06 (2004).
Edelman, I., Olsen, S. & Devereux, J. Program Manual for the Wisconsin Package, version 1.0. Genetics Computer Group (GCG) (Madison, 1996).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33(7), 1870–1874. https://doi.org/10.1093/molbev/msw054 (2016).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Peakall, R. & Smouse, P. E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Res. 6(1), 288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x (2006).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. JModelTest 2: More models, new heuristics and parallel computing. Nat. Methods. 9(8), 772–772. https://doi.org/10.1038/nmeth.2109 (2012).
Hasegawa, M., Kishino, H. & Yano, T. A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22(2), 160–174. https://doi.org/10.1007/BF02101694 (1985).
Rodríguez, F., Oliver, J. L., Marín, A. & Medina, J. R. The general stochastic model of nucleotide substitution. J. Theor. Biol. 142, 485–501. https://doi.org/10.1016/S0022-5193(05)80104-3 (1990).
Swofford, D. L. P. A. U. P. Phylogenetic analysis using parsimony. Options 42, 294–307. https://doi.org/10.1007/BF02198856 (2002).
Felsenstein, J. PHYLIP (phylogeny inference package) version 3.695. Distributed by the author. Department of Genome Sciences, University of Washington, Seattle, WA (2013). https://ci.nii.ac.jp/naid/10027221536/.
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. https://doi.org/10.1093/molbev/mss075 (2012).
Huelsenbeck, J. P. & Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17(8), 754–755. https://doi.org/10.1093/bioinformatics/17.8.754 (2001).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1), W232–W235. https://doi.org/10.1093/nar/gkw256 (2016).
Acknowledgements
The authors are grateful to all fishing organizations that assisted in collection of hakes in their native habitats, particularly to O. Pedrin who provided samples of M. angustimanus and to C. P. Mathews for his expert taxonomic assistance on this species. The authors are indebted to four anonymous reviewers for their critical inputs on a previous draft of this manuscript.
Author information
Authors and Affiliations
Contributions
All the authors analyzed the data, discussed results and implications, commented on the manuscript at all stages and approved the final article for submission. More specific tasks were the responsibility of P.P. and M.P (conceptualization and sampling), J.M. and D.L. (acquisition of morphotypes and morphometric data), M. P. and M.F-M (acquisition of molecular data), M. F-M., P. P., and M. P. (Data analysis and manuscript drafting). The Regional Government of Xunta de Galicia supported the M. F-M.’s Ph. D. grant (ED481A-2017/362). This research was partially supported with the project LETSHAKE (AGL2013-4846-R) co-funded by MINECO (Ministerio Español de Economía y Competitividad) and EU-FEDER to M.P. as well as with grant (IN607B 2018/14) to M. P. from Xunta de Galicia-Axencia Galega de Innovación. This work was also partly funded with grants from “Consellería de Educación e Ordenación Universitaria Xunta de Galicia (Galician Regional Government) cofunding from the European Regional Development Fund (ERDF) in the framework of the Operational Program Galicia 2014–2020 (CIM-UVIGO), “A way to build Europe”.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pérez, M., Fernández-Míguez, M., Matallanas, J. et al. Phylogenetic prospecting for cryptic species of the genus Merluccius (Actinopterygii: Merlucciidae). Sci Rep 11, 5929 (2021). https://doi.org/10.1038/s41598-021-85008-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-85008-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
