
Abstract
G protein-coupled receptors (GPCRs) are the largest class of transmembrane proteins, making them an important target for therapeutics. Activation of these receptors is modulated by orthosteric ligands, which stabilize one or several states within a complex conformational ensemble. The intra- and inter-state dynamics, however, is not well documented. Here, we used single-molecule fluorescence to measure ligand-modulated conformational dynamics of the adenosine A2A receptor (A2AR) on nanosecond to millisecond timescales. Experiments were performed on detergent-purified A2R in either the ligand-free (apo) state, or when bound to an inverse, partial or full agonist ligand. Single-molecule Förster resonance energy transfer (smFRET) was performed on detergent-solubilized A2AR to resolve active and inactive states via the separation between transmembrane (TM) helices 4 and 6. The ligand-dependent changes of the smFRET distributions are consistent with conformational selection and with inter-state exchange lifetimes ≥ 3 ms. Local conformational dynamics around residue 2296.31 on TM6 was measured using fluorescence correlation spectroscopy (FCS), which captures dynamic quenching due to photoinduced electron transfer (PET) between a covalently-attached dye and proximal aromatic residues. Global analysis of PET-FCS data revealed fast (150–350 ns), intermediate (50–60 μs) and slow (200–300 μs) conformational dynamics in A2AR, with lifetimes and amplitudes modulated by ligands and a G-protein mimetic (mini-Gs). Most notably, the agonist binding and the coupling to mini-Gs accelerates and increases the relative contribution of the sub-microsecond phase. Molecular dynamics simulations identified three tyrosine residues (Y112, Y2887.53, and Y2907.55) as being responsible for the dynamic quenching observed by PET-FCS and revealed associated helical motions around residue 2296.31 on TM6. This study provides a quantitative description of conformational dynamics in A2AR and supports the idea that ligands bias not only GPCR conformations but also the dynamics within and between distinct conformational states of the receptor.
Similar content being viewed by others
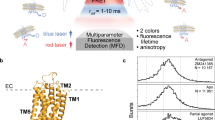


Introduction
G-protein-coupled receptors (GPCRs), also referred to as seven-transmembrane (7TM) helix domain receptors, are the largest protein family in humans and are responsible for a wide range of signaling processes such as sight, smell, inflammation, cell homeostasis, and neurotransmitter and hormone mediated responses1. In the human genome, GPCRs account for over 800 receptors, constituting nearly 4% of encoded proteins, and have important implications in the treatment of a wide range of diseases2. In particular, the adenosine A2A receptor (A2AR) is a class A GPCR that is important for treatment of sickle cell disease, cancer, inflammation, ischemia, neuronal disorders and various infectious diseases3. Deciphering the mechanism by which GPCRs are activated and the role of ligands in stabilizing specific conformational states is essential for rational drug design.
The promiscuous interactome of most GPCRs, in addition to their reputation for exhibiting a range of efficacies as a function of ligand, implies a complex conformational landscape as revealed by a majority of recent spectroscopic studies4,5,6,7. Studies of A2AR, rhodopsin, and a number of class A receptors have further identified a representative conformational ensemble in which ligands or interacting partners serve to stabilize specific functional states required for inactivation, G protein binding, and activation8,9. Thus, ligands bias the ensemble in a manner consistent with conformational selection and facilitate allosteric transitions between these states to achieve intracellular signaling via G protein or arrestin-mediated response7,10,11.
X-ray crystallography provides snapshots of GPCRs in active or inactive states through the use of thermostabilizing mutations, fusion-proteins, or high-affinity ligands12. Several crystal structures of A2AR have been resolved in various ligand-bound states, which show pronounced translation of the transmembrane 3 (TM3), inward shifts of TM7 and significant rotation of TM6 towards TM512,13,14,15. Similar ligand-induced TM movements have been reported across several other class A GPCRs, including light-activatable rhodopsin GPCRs8,9,16, and the β2-adrenergic receptor (β2AR)17. While X-ray crystallography does not fully capture the dynamic nature of these states, it provides a framework of key intermediates that can be used to interpret the results of techniques capable of measuring dynamics.
In recent years, several spectroscopic methods have been used to document the dynamic behavior of GPCRs, including electron paramagnetic resonance (EPR)11 and nuclear magnetic resonance (NMR)6. Advancements in pulsed-EPR techniques, such as double electron–electron resonance (DEER), provide an opportunity to refine GPCR structures obtained by X-ray crystallography or, more recently, cryo electron microscopy18, while simultaneously identifying distributions of conformations constituting the ensemble19,20. Moreover, this can be repeated under various conditions (i.e., pH, ligand, detergent, liposomes, nanodiscs etc.)21. At the same time, EPR measurements provide a measure of dynamics of specific states, particularly on nanosecond and microsecond timescales. A combination of DEER and fluorine NMR (19F-NMR) spectroscopy recently highlighted the key role of orthosteric ligands in altering the free energy landscape of A2AR , suggesting unique activation pathways for partial and full agonists4,5.
19F-NMR experiments using a label at the site V229C6.31 on TM6 suggest that ligand-free (apo) A2AR adopts two inactive states which undergo sub-millisecond exchange, resulting in a single (exchange-broadened) inactive state resonance (i.e. S1-2), in addition to two active-like states (S3 and S3′) which undergo slower (> 5 ms timescale) exchange with the inactive state4. The inverse agonist ZM241385 increases the population of the inactive states S1-2, the full agonist NECA preferentially stabilizes the S3′ activation intermediate, while the partial agonist LUF5834 increases the population of the S3 activation intermediate.
The binding of a peptide-mimetic of the C-terminal domain of the α subunit of G protein (Gαs 374–394) further stabilizes the S3′ state in presence of NECA, while no significant changes were observed for the LUF5834-saturated receptor4. The on–off flickering of the ionic lock between R1023.50 (TM3) and E2286.30 (TM6), which is thought to cause rapid transitions between the S1 and S2 state, was found to be suppressed in the agonist-bound state4,13,15. A2AR conformational dynamics on millisecond timescales was indirectly inferred from the line-broadening of the 19F-NMR spectra and from Carr-Purcell-Meiboom-Gill (CPMG) relaxation dispersion measurements4,22, as well as by 2D exchange spectroscopy (EXSY)23. However, a direct time-domain measurement of exchange rates between and within each of identified A2AR states is still lacking.
More recently, single-molecule fluorescence (SMF) techniques have been applied to GPCRs to measure their conformational heterogeneity and dynamics24,25. Single-molecule Förster Resonance Energy Transfer (smFRET) is an effective tool for measuring distance distributions/changes between fluorescent dyes attached to specific sites in GPCRs within the framework of conformational heterogeneity26,27,28. An smFRET study by Blanchard and coworkers on detergent solubilized β2AR showed evidence for ~ 14 Å outward movement of TM6 and for distance fluctuations occurring on a 1–10 ms timescale26. In addition, the FRET distributions were modulated by binding of orthosteric ligands and by the coupling to the Gs heterotrimer. Similarly, smFRET on the class C metamorphic glutamate receptor (mGluR)27 revealed both millisecond (> 4 ms) and sub-millisecond (~ 50–100 μs) conformational fluctuations28.
Ligand-free A2AR has been shown to be inherently dynamic, whereby the outward motion of TM6 is important for the binding of G protein4. However, the rates of these internal motions have not been directly measured. By avoiding ensemble averaging, SMF techniques have the unique ability to resolve distributions of GPCR conformers and their dynamics on a broad range of timescales25. Here, we used SMF methodologies that are sensitive to the movement of TM6 to measure intra- and inter-state exchange dynamics of different A2AR conformers. smFRET was used to measure the distribution of separation distances between TM4 and TM6 in detergent-purified A2AR samples labelled with a donor–acceptor dye pair at residues T119 and Q2266.28, respectively. The smFRET distributions exhibit ligand-dependent shifts of the mean transfer efficiency and significant changes in their widths, which are interpreted by invoking three distinct conformational states of the receptor.
In addition, we labelled TM6 at residue 2296.31 with a probe (BODIPY-FL) that is sensitive to quenching by aromatic amino acids at short-range distance (5–10 Å)29,30. This process is known as Photoinduced Electron Transfer (PET) and the resulting fluorescence intensity fluctuations report on local protein dynamics around the labelling site31. Fluorescence Correlation Spectroscopy (FCS) is widely used to analyze fluorescence intensity fluctuations on nanosecond-to-microsecond timescales32, including PET quenching processes (PET-FCS)30. PET-FCS was shown to capture the kinetics of conformational dynamics in proteins, such as ultrafast folding of small water-soluble proteins33 and movements of helical TM bundles in proton channels34.
In A2AR, our PET-FCS analysis resolved three lifetimes of protein-induced quenching processes in the proximity of residue 2296.31 on TM6: fast (150–350 ns), intermediate (50–60 μs), and slow (200–300 μs). The lifetimes and amplitudes of the three components are modulated by the binding of orthosteric ligands and coupling to mini-G protein (mini-Gs). A concomitant increase in amplitude and decrease in lifetime of the fast component is observed upon activation by agonist and mini-G, while the other two components show a moderate decrease in amplitude and lifetime.
Molecular dynamics (MD) simulations of the dye-labelled receptor identified three tyrosine residues as the being responsible for the (sub)microsecond PET quenching observed by FCS: Y112 on intracellular loop 2 (ICL2), and Y2887.53 and Y2907.55 on TM7. MD simulations also revealed novel helix-to-loop dynamics near the intracellular-end of TM3, which decreased significantly when the A2AR is prearranged to bind mini-Gs. This work shows that GPCR ligands modulate not only state populations and associated exchange dynamics of the ensemble, but also internal dynamics and energy barriers of these states (i.e., sub-states). SMF data provides a quantitative dynamic picture of GPCR activation, which encompasses structural changes on timescales from hundreds of nanoseconds to several milliseconds being controlled by interactions with ligands and intracellular effectors.
Results
smFRET reveals ligand-mediated displacement of TM6
smFRET experiments on a double-cysteine mutant (T119C on TM4 and Q2266.28 on TM6) labelled with AF488 (donor) and AF647 (acceptor) show the effect of saturating amounts of four orthosteric ligands on A2AR conformations. Observed changes in the FRET efficiency reflect displacements of TM6 relative to TM4, which remains relatively immobile (Supporting Information Sect. 1, Fig. S1). Solution smFRET experiments on detergent-reconstituted A2AR samples were performed on a custom-built single-molecule fluorescence microscope and the multiparameter photon data was analyzed using custom MATLAB scripts35,36. Labelled protein was diluted to 50 pM in a buffer solution (50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, 0.02% CHS, 10 mM cysteamine) to ensure a low probability (< 1%) of two independent receptors simultaneously occupying the confocal detection volume. Therefore, nearly all fluorescence intensity bursts originate from single, freely-diffusing, detergent-isolated A2AR molecules (Fig. 1a).

Ligand-induced changes in A2AR conformations measured by smFRET. (a) A double-cysteine mutant of A2AR (T119C Q226C) labelled with AF488 (donor) and AF647 (acceptor) was alternately excited with blue (473 nm) and red (635 nm) lasers; bursts of fluorescence of the donor (green) and the acceptor (red) were recorded from single receptors diffusing through the confocal detection volume. The FRET efficiency E for each burst was calculated using Eq. 1a and the values obtained from all bursts for each ligand condition were used to build smFRET histograms (c–f). (b) Width (ΔEFWHM) vs. mean (< E >) for the FRET efficiency distributions shown in (c–f); the error bars were estimated by statistical bootstrapping. Shown as reference are the regions of the shot-noise limit (blue) and of the quasi-static limit (dsDNA, pink) (see SI Sect. 1.2 for details). (c–f) smFRET distributions of A2AR in the (c) basal state (apo), and in the presence of (d) 100 μM ZM241385 (antagonist), (e) 100 μM LUF5834 (partial agonist), and (f) 100 μM NECA (full agonist), fitted to a sum of Gaussians (black line). Solid blue, green and red lines represent individual Gaussians and corresponding dashed vertical lines represent their peak center. The results of the fitting are shown in Table 1.
smFRET was measured for A2AR without ligands (apo) and in the presence of 100 μM ZM241385 (inverse agonist), 100 μM LUF5834 (partial agonist), and 100 μM NECA (full agonist). The corrected energy transfer efficiency E for each fluorescence burst was calculated using Eq. 1, and all E values for each sample were used to build FRET histograms (Fig. 1c–f). Without any ligands, as well as in the presence of inverse agonist ZM241385, the receptor exhibits relatively broad smFRET distributions centered around \(\left\langle E \right\rangle \)≈ 30%, with full-width-at-half-maximum (FWHM) values of ΔEFWHM ≈ 52% (Fig. 1b, Table 1). The partial agonist LUF5834 and the full agonist NECA shift the smFRET histogram to higher (\(\left\langle E \right\rangle \) = 38%) and lower (\(\left\langle E \right\rangle \) = 25%) average values, respectively, with concomitant narrowing (ΔEFWHM = 41% and 46%, respectively, Table 1).
To benchmark the conformational dynamics underlying the A2AR smFRET histograms, double-stranded DNA (dsDNA) control samples provided a quasi-rigid scaffold for the donor–acceptor pair (Supporting Information Sect. 1). As shown in Fig. 1b, the smFRET broadening observed for A2AR in all ligand conditions is significantly larger than both the theoretical shot-noise limit computed using the average total number of photons per burst and the quasi-static limit of dsDNA (Supporting Information, Sect. 1.2). While other factors may cause broadening of smFRET histograms (e.g., dye photophysics and orientational freedom), our observations are consistent with low-millisecond exchange between low-FRET and high-FRET states37. This range is similar, although somewhat faster than rate of conformational exchange estimated by 19F-NMR saturation transfer using different labelling sites4,23.
Fitting the smFRET histogram for the apo receptor requires two Gaussians, centered at Elow = 24 ± 1% (major) and at Ehigh = 52 ± 1% (minor) (Fig. 1c). This corresponds to two states, one where TM6 is relatively farther from TM4 (low FRET, open/active) and another where TM6 is relatively closer to TM4 (high FRET, closed/inactive). Similar states were resolved previously using smFRET for a different class A GPCR, the β2 adrenergic receptor26.
In the presence of the inverse agonist ZM241385, the smFRET histogram is described by the same two FRET states as in the apo form, however the weight of the inactive (high-FRET) state increases (by ~ 50%) at the expense of the active (low-FRET) state (Fig. 1d, Table 1). Binding of the partial agonist LUF5834 leads to a shift of the smFRET histogram to intermediate values between the low- and the high-FRET peaks and to significant narrowing (Fig. 1b). The histogram is satisfactorily fit with only one Gaussian centered at Emid = 37 ± 1% (Fig. 1e). This could originate from accelerated (μs-scale) transitions between the two FRET apo states27, or from a new (partially active) A2AR conformation with an intermediate TM6-TM4 separation4,23 (see also below).
With the full agonist (NECA) bound to receptor, the smFRET histogram exhibits a significant shift towards lower values, but only a small decrease in width compared to the apo state (Fig. 1b). The FRET distribution is best fitted by the sum of two Gaussians centered at Elow = 21 ± 3% and Emid = 36 ± 1%, having nearly identical weights, i.e., 0.51 ± 0.02 and 0.49 ± 0.02, respectively (Fig. 1f, Table 1). These states closely resemble the low-FRET (open/active) state found in the absence of ligands and the intermediate-FRET (partially active) state induced by the partial agonist, respectively. The results of fitting all smFRET histograms, including the error margins, are summarized in Table 1.
The Förster radius of the AF488-AF647 dye pair was estimated to be Ro = 50.1 ± 2.5 Å assuming full rotational freedom of the dyes attached to A2AR, although anisotropy effects introduce larger uncertainties (Supporting Information, Sect. 1.3). Using the Förster equation (Eq. S2), the donor–acceptor distances (RDA) were estimated to be 49.3 ± 1.4, 54.6 ± 1.6, and 60.6 ± 1.8 Å, for the high, intermediate, and low A2AR FRET states, respectively. This 5–10 Å outward movement/reorientation of TM6 as A2AR transitions from inactive, partially active, and active conformations closely resembles the effect of inverse, partial and full agonists on the TM6-TM4 separation distance reported by smFRET in β2 AR26. The increasing separation between TM4 and TM6 upon activation is also present in the crystal structures of A2AR in the ZM241385-bound state (3EML), the NECA-bound state (2YDO), and the mini-Gs coupled state (5G53) of A2AR (Fig. S1)14,15. The ligand-dependent shifts and broadening of smFRET distributions suggest that the exchange between active and inactive A2AR conformations occurs on timescales that are comparable to the average single-molecule burst duration (2.6 ± 0.5 ms), and much slower than the average inter-photon time in our experiments (~ 10 μs)28,37.
BODIPY-FL-labelled A2AR: rotational freedom and quenching
To dissect the dynamics of TM6, we labelled A2AR at residue V229C6.31 with an environment-sensitive dye (BODIPY-FL) with a short linker and performed fluorescence quenching experiments. In the crystal structure of A2AR (2YDO/4EIY), several aromatic residues (i.e., phenylalanine, histidine, and tyrosine) on ICL2, TM5 and TM7 are found in the proximity of residue 2296.31 and could act as BODIPY-FL quenchers (Fig. 2a). Thus, V229C6.31 is an adequate construct for monitoring local dynamics in A2AR via fluorescence quenching, assuming that quenching dynamics reflects the movement of the host (i.e., the receptor) and not the intrinsic movement of the dye or its linker34. The linker flexibility and the rotational freedom of BODIPY-FL attached to the apo receptor was measured by fluorescence anisotropy decay (FAD). The anisotropy decay curves were fit to Eq. 3b and show that, regardless of the ligand-condition (Fig. S2), the slow, global rotational dynamics of the receptor (ρslow = 52–60 ns) is dominant, while the fast, dye-linker rotational dynamics (ρfast = 0.28–0.42 ns) has a minor contribution (~ 10%) to the anisotropy decay (Fig. 2a–e, Table 2).

Rotational freedom and quenching of BODIPY-FL in A2AR. (a) Native aromatic residues (yellow hexagons) on TM3, TM5, and TM7 provide a dynamic quenching environment for BODIPY-FL (green) attached to residue 2296.31 in A2AR(V229C6.31). Fluorescence anisotropy (b) and lifetime (f) decay curves for apo A2AR (yellow) were fitted to multi-exponential functions, i.e. Eqs. 3b and 4, respectively (black). Similar data was acquired under excess (100 μM) of ligands, i.e. inverse agonist ZM241385, partial agonist LUF5834 and full agonist NECA, and was fitted to the same models. Slow and fast rotational correlation times for all ligand conditions shown as yellow and red bars in (c) and (d), respectively, and their corresponding amplitudes in (e). Unquenched and quenched fluorescence lifetimes shown as green and red bars in (g) and (h), respectively, and their corresponding amplitudes in (i). Standard deviations shown in (c–e) and (g–i) were estimated by parametric bootstrapping. The experiments were performed on 100 nM solutions of labelled receptors using pulsed 480-nm laser excitation. The fitting results are shown in Table 2 (anisotropy) and Table 3 (lifetime).
Fluorescence lifetime measurements describe the extent of quenching when BODIPY-FL is attached to residue 2296.31 in A2AR (Fig. 2f–i, Table 3). For the apo receptor (Fig. 2f), the experimental decay curve was best fitted to a sum of two exponential lifetimes, τq = 1.4 ± 0.1 ns and τnq = 5.6 ± 0.1 ns, with amplitudes Bq = 0.17 ± 0.01 and Bnq = 0.83 ± 0.01, respectively (Eq. 4). The long lifetime is similar to previous values reported for free BODIPY-FL in solution (i.e., 5.5 ns)38, while the short lifetime corresponds to the quenched state induced by the host protein. The two fluorescence lifetimes and amplitudes vary slightly across different ligand conditions, with most quenching occurring in the presence of partial and inverse agonists. Thus, the fluorescence of BODIPY-FL attached to TM6 at residue 2296.31 is partially quenched, and the quenching varies in response to different ligand-induced rearrangements of TM bundles or local helical movements.
BODIPY-FL as a probe for PET-FCS
To benchmark the intrinsic photoblinking of BODIPY-FL due to reversible transitions to its dark (triplet) state, FCS measurements of the free dye in solution were performed under the same buffer conditions as the receptor (Fig. S3a, Table S1), including photo-protectants (20 mM cysteamine and 2 mM Trolox) to enhance brightness and reduce photoblinking39. The measured autocorrelation curve is best fitted to a diffusion model with one triplet component (Eq. 5, with no quenching terms), and the triplet lifetime obtained (τt = 3 ± 1 μs) is in agreement with previous reports (2–8 μs)40. For the global analysis of FCS data of the receptor under different ligand conditions, the BODIPY-FL triplet lifetime was used as a shared fitting parameter (Eq. 5, see also below).
Of the several aromatic residues in A2AR near residue 2296.31, tyrosines are most likely to act as PET quenchers of BODIPY-FL fluorescence, due to their abundance and quenching power30. To verify the quenching of BODIPY-FL by tyrosine, FCS experiments of free dye (~ 10 nM) were performed in the presence of 1 mM and 3 mM tyrosine under similar buffer conditions as above (Fig. S3b,c). Data was fitted to Eq. 5 and the fitting parameters are summarized in Table S1. Fitting of the FCS curve obtained upon adding 1 mM tyrosine requires an additional lifetime (~ 40 μs) compared to the free dye in buffer solution. Upon increasing the tyrosine concentration to 3 mM, this decay component becomes more pronounced and also a faster decay component (~ 100 ns) appears.
Quenching collisions corresponding to the intermediate PET lifetime (~ 40 μs) occur with a frequency of ~ 25 ms−1, whereas those responsible for the fast PET lifetime (~ 100 ns) occur with a frequency of ~ 105 ms−1. These rates represent diffusion-limited quenching interactions between BODIPY-FL and tyrosine in an aqueous solution41. In A2AR, the effective concentration of tyrosine in the proximity of the dye is higher (~ 60 mM), which would normally lead to an increase of the quenching rates. However, the PET quenching rates in A2AR will be determined by both local helical dynamics and long-range TM movements, which can alter the proximity and the orientation of BODIPY-FL relative to native A2AR tyrosine residues34.
Direct measurements of A2AR conformational dynamics by PET-FCS
PET-FCS measurements were performed on the apo receptor, and upon addition of 100 μM of inverse agonist, partial agonist, or full agonist, as well as in the presence of 10 μM mini-Gs and 100 μM NECA, 2 mM Mg2+ and 50 μM GDP (Fig. 3). At the concentration of BODIPY labelled-A2AR used in FCS experiments (~ 10 nM) there is an approximately 104-fold excess of ligand and 103-fold excess of mini-Gs. For clarity, the fitted diffusion component was subtracted from each experimental FCS curve to obtain decay curves that included only contributions from photoblinking of the dye (i.e., triplet and PET quenching), as illustrated in Fig. 3a and described previously34.

FCS measurements of conformational dynamics around residue 2296.31 in A2AR. (a) FCS data (green line) of A2AR-V229C6.31-BODIPY-FL fitted to Eq. 5 (red line). The pure diffusion component in the fitted curve (black dashed line) can be subtracted from the data to highlight the decays due to excursions to the triplet state and to PET quenching. The inset shows the curves obtained after removing the diffusion term from the raw data (solid yellow) and the fitted curve (dashed blue). “Diffusion-free” FCS curves of the labelled receptor in the presence of (b) 100 μM ZM241385, (c) 100 μM LUF5834, (d) 100 μM NECA, and (e) 100 μM NECA, 2 mM MgCl2, 50 μM GDP and 10 μM mini G protein, respectively; raw data (solid yellow) and fitted curves to Eq. 5 (dashed blue). The fitting results for all sample conditions are summarized in Table 4.
The autocorrelation curves obtained for each sample condition were fitted to Eq. 5 and the results are shown in Fig. 4 and in Table 4. Using the diffusion times and the Stokes–Einstein equation (Eq. 6), the hydrodynamic radii were estimated to be RH = 6 ± 1 nm for all samples (slightly higher for apo, 7 ± 1 nm), which matches the size of the detergent micelle encapsulating the receptor. The data was fitted to a model including one triplet component and three PET quenching components. Global fitting of all FCS curves was performed, with the triplet lifetime shared and close to that of the free dye (ttrip = 5 ± 1 μs, Table 4), and with ligand-specific PET lifetimes and amplitudes.

PET quenching lifetimes and amplitudes in A2AR as modulated by ligands and a G-protein mimetic. PET lifetimes (tq,i) and corresponding amplitudes (Ki) obtained by fitting FCS data shown in Fig. 3 to a model described by Eq. 5. Three PET components were resolved for each ligand condition: fast (a,d) intermediate (b,e), and slow (c,f), with varying lifetimes and amplitudes. The error bars were derived by parametric bootstrapping.
The ligand-specific PET quenching lifetimes can be grouped into fast (tq, fast = 150–350 ns), intermediate (tq, int = 50–60 μs), and slow (tq, slow = 200–300 μs), with corresponding amplitudes (Ki) that vary as a function of ligand (Fig. 4a–f, Table 4). Most notably, the contribution of the fast kinetics increases in the fully active states (NECA- and mini-Gs-bound) relative to those from two slower kinetic components, as shown by the amplitudes Ki (Fig. 4d–f). The fast kinetics becomes even faster in the active states, with its lifetime decreasing from 340 ± 20 ns in the apo state to 180 ± 30 ns in the NECA-bound state and to 160 ± 10 ns in the mini-Gs-bound state (Fig. 4a). The intermediate lifetime slightly increases upon activation by NECA, i.e., from 52 ± 3 μs to 60 ± 3 μs, but it returns to the apo-state level (51 ± 1 μs) upon binding of mini-Gs (Fig. 4b). Similarly, the lifetime of the slow component is nearly unchanged between the apo and the partially/fully active states (245–265 μs), and it shows only a minor decrease in the inverse-agonist-bound state (225 ± 20 μs) (Fig. 4c).
The intermediate and the slow PET lifetimes (30–300 μs) reflect changes in the brightness of the dye that are likely driven by local helical movements and rearrangement of the TM bundles, as suggested previously34. These timescales of motion are on the same order of magnitude as those suggested by exchange broadening of the smFRET distributions (Fig. 1, see above). In particular, the sub-millisecond protein dynamics captured by PET-FCS could be associated to the on/off behavior of the ionic-lock for the salt-bridge between R1023.50 of TM3 and E2286.30 of TM64,40.
Molecular dynamics simulations of PET quenching in A2AR
MD simulations of A2AR were performed to identify the native aromatic residues and the protein motions responsible for the (sub)microsecond PET kinetics measured by FCS. To compute the spatial extension of the BODIPY-FL dye from its anchoring point on the receptor (Fig. S4a,b), we simulated an 11-residue segment of TM6 centered at residue 2296.31, with the dye attached at this residue (Fig. 5a). The dye linker relaxes rapidly, with an exponential distance autocorrelation lifetime of τacor = 0.8 ± 0.3 ns (Fig. S4c).

Molecular dynamics simulations of PET quenching dynamics in A2AR. (a) Simulations of the cloud of possible positions of a BODIPY-FL molecule (with boron replaced by carbon) attached to position 2296.31 within a small peptide segment of A2AR (224–234). (b) The simulated BODIPY-FL cloud from (a) overlaid with the PDB structure of A2AR, with tyrosine residues (green) in the proximity representing possible PET quenching candidates. Probability distributions for the distance between the beta-carbon of position 229 to the center of mass of the aromatic system of tyrosine Y112 on ICL2 (c), Y2887.53 (e) and Y2907.55 (g) on TM7: inverse agonist (red), full agonist (blue), full agonist + miniG (green). Inter-residue distance corresponding to the cut-off distance for PET quenching of BODIPY-FL by tyrosine, dq = 1.81 nm, is shown as a vertical dash line. The frequency of quenching/de-quenching transitions for Y112 (d), Y2887.53 (f) and Y2907.55 (h) as a function of cut-off distance in presence of inverse agonist (red squares), full agonist (blue circles), and full agonist + mini G (green triangles).Error bars are standard deviations from 6 replicate simulations. The shaded regions in (d,f,h) represent the most likely cut-off distance for PET quenching of BODIPY-FL by tyrosine, dq = 1.8 ± 0.1 nm. MD simulations were initiated using crystal structures in which the A2AR was crystalized bound to an inverse agonist, an agonist, or an agonist plus mini-G, as shown in Table S2.
This rapid relaxation permitted a simplifying approximation in which simulations of unmodified A2AR were post-processed to mimic the presence of a temporally averaged dye cloud (Fig. 5a,b). In order to quantify the dynamic quenching of BODIPY-FL by specific aromatic residues in the receptor, we defined the radius of a spherical surface surrounding the Cβ atom of A2AR residue V2296.31 as < l > = 0.81 ± 0.01 nm. This value represents the mean distance from the Cβ atom of V2296.31 to the center of geometry of non-hydrogen atoms in the dye's aromatic group obtained from three replicate simulations (Fig. S4). Since the typical quenching distance for BODIPY-FL is 1.0 nm or less29,42, we defined a quenching cutoff distance of dq = (1 + < l >) = 1.81 nm from the Cβ atom of V2296.31 to the center of mass of the side chain of each quenching candidate residue in A2AR (Fig. S5).
Simulations of A2AR in a hydrated lipid bilayer composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) were performed. An aggregate time of 210 μs was obtained via a total of 42 independent 5-μs simulations initiated from seven crystal structures containing three different types of ligands: inverse agonist, full agonist and mini G (see Table S2). Although ligands were not explicitly included in the simulations, the signatures of different starting structures remained mostly intact during the 5-μs MD trajectories (see below and Figure S8). We quantified quenching based on the distance, d, from the Cβ atom of V2296.31 to the center of mass of each aromatic side chain in the simulated region of the A2AR (S6 to V307). Given the “dye-cloud” approximation (Fig. 5a,b), we applied a cutoff distance of dq = 1.8 ± 0.1 nm, such that states with d < dq are defined to be “quenched” (dark) and states with d > dq are defined to be “unquenched” (fluorescent) (Fig. S5). The inter-residue distance distributions from the Cβ atom of V2296.31 to all possible quenching candidates (Trp, Tyr, Phe and His) show that 6 of the 42 residues monitored have overall populations that span across the dq cutoff (dashed red arrows, Figure S6): Y1033.51, Y112, Y1975.58, Y2887.53, Y2907.55, and F295 (see e.g., Fig. 5c,e,g).
To quantify reversible transitions between quenched and unquenched states, we analyzed the time trajectories of d in each simulation, separately for each of the 6 residues above. Statistical noise was reduced by smoothening the trajectories with a 20-ns running average and transitions between quenched and unquenched states were interpreted using a cut-off distance that was varied between dq = 1 nm and dq = 2 nm. By comparing the frequency of MD quenching/dequenching transitions to the experimental values, we propose that Y112 on ICL2, and Y2887.53 and Y2907.55 on TM7 are likely responsible for the fast PET-FCS lifetime. These tyrosines exhibit a maximum number of quenching-dequenching transitions at the expected cut-off distance of 1.8 ± 0.1 nm for each ligand condition (Fig. 5d,f,h). The MD estimates of the PET rates are an order of magnitude higher than the experimental values and should be viewed as an upper limit, as the relative orientation of the dye and the tyrosine, which is critical for electron transfer, has not been taken into account in the calculations.
The residue Y112 is on ICL2 and forms a helical bridge between TM3 and TM4, which makes frequent transitions (i.e., 4–10 μs−1) from an ordered helical configuration to a disordered loop-like configuration in the MD simulations (Fig. 5d). This transition to a loop-like configuration brings Y112 within quenching range of BODIPY-FL on residue V2296.31 (Movie S1, colored green and blue, respectively) and is more pronounced in the inactive state (red lines, Fig. 5c,d). Comparing the MD-estimated quenching/dequenching rate to that estimated by PET-FCS in the inverse-agonist-bound state (i.e., 1/ tq,fast ~ 3.7 μs−1), the fast PET lifetime can be assigned to quenching by Y112. As the receptor switches into more active states, such as those stabilized by full agonist and mini-G protein, however, the frequency of Y112 transitions decreases, implying stabilization of the helix (blue, and green lines, Fig. 5c,d). This decreasing trend in PET dynamics can be rationalized in terms of the crystal structure of the mini-G-bound receptor (5G53), which shows stabilizing interactions between Y112 on ICL2 of A2AR and H387 of the mini-G protein43 (see Supporting Information, Figure S7a,b).
The other two candidates, Y2887.53 and Y2907.55 on TM7 show rather opposing trends in MD simulations (Fig. 5e,g,f,h), such that the inverse-agonist bound state exhibits less frequent quenching transitions (~ 1.6 μs−1) when compared to the full-agonist-bound (~ 3.0 μs−1), or to the receptor bound to full-agonist and mini-G-protein (~ 1.9 μs−1) (Fig. 5f,g). The increasing number of transitions estimated by MD for Y2887.53 and Y2907.55 upon receptor activation suggests that there are larger fluctuations in the active states of A2AR that bring these two aromatic residues on TM7 within quenching range of BODIPY-FL attached to TM6. Another candidate quencher residue, Y1975.58 on TM5, shows little to no transitions in the presence of inverse and full agonist, and it is only significant in simulations initiated with a receptor that is pre-organized to bind the mini-G protein. This suggests a dynamic reversible rotation of TM5 relative to TM6 in a manner that increases the accessibility of Y1975.58 to residue V2296.31 (Figure S7c-e).
Discussion
This study combines fluorescence spectroscopy and molecular dynamics to examine the conformational dynamics of A2AR over a wide range of timescales. A2AR conformations were measured at the single-molecule level using FRET between specific sites in TM4 and TM6 that were labelled with a donor–acceptor dye pair. The smFRET data confirm previous 19F-NMR findings that active (i.e., S3 and S3′) and inactive (i.e., S1-2) states of the receptor co-exist and ligands alter their relative population4,5. Specifically, the broadening of the smFRET histogram for the apo receptor is consistent with the existence of different FRET sub-populations and millisecond exchange between high-FRET inactive (S1 and S2) and low-FRET active (S3 and S3′) A2AR conformations. In addition, using the quasi-static dsDNA reference, each FRET sub-population appears to be broadened (ΔEFWHM-low ~ 40% and ΔEFWHM-high ~ 50%, Fig. 1), suggesting millisecond-scale exchange between S3 and S3′, and between S1 and S2, respectively. Previous 19F-NMR studies have conflicting outcomes regarding the basal activity of A2AR, reporting either a high4 or a low23 fraction of active or activation intermediate states for the apo receptor. Our smFRET data confirms that the ligand-free A2AR is inherently dynamic, with a population bias towards active-like conformations that is consistent with significant basal activity44.
Binding of the inverse agonist ZM241385 did not change the overall broadening of the smFRET distribution, but led to ~ 50% increase in the fraction of inactive (high-FRET) sub-population (Table 1). This is consistent with a 19F-NMR study that showed a similar reequilibration of the apo conformational ensemble in the presence of inverse agonist23. An alternative interpretation would invoke the emergence of an intermediate state between the active and inactive states (dashed blue line between the green and red lines, Fig. 1d), corresponding to the S3 state that is enriched in the inverse-agonist-bound receptor4.
The partial agonist LUF5834 shifts the smFRET distribution to intermediate values between the low- and high-FRET sub-populations (Fig. 1e). It also considerably narrows the distribution to a level that is close to the quasi-static control (dsDNA) for smFRET. This is consistent with the partial agonist stabilizing a distinct A2AR conformation (blue line, Fig. 1e), which corresponds to the S3 state identified by 19FNMR4. Alternatively, the narrow smFRET peak could arise from a significantly increased exchange rate between the apo low- and the high-FRET states (~ 1–10 μs lifetime), although this appears to be at least 2 orders of magnitude faster than the exchange rates inferred by 2D [19F,19F]-EXSY23.
The full agonist NECA shifts the smFRET distribution to lower E while maintaining significant broadening (Fig. 1f). The data analysis reveals two nearly equal sub-populations that are similar to the low-FRET conformation of the apo receptor, and the mid-FRET conformation stabilized by partial agonist. smFRET confirms previous 19FNMR observations of two A2AR active states for the NECA-bound receptor4,23, with blue and green lines in Fig. 1f corresponding to S3 and S3′, respectively. Overall, the smFRET results provide new, independent evidence that inactive and active A2AR conformations co-exist in the absence of ligands, that transitions between these states occur on a millisecond timescale, and that ligand binding alters both the energetics and the dynamics of these states.
Since the smFRET measurements were performed in the presence of 100 mM NaCl, and sodium is a known allosteric modulator of A2AR45, the pronounced low-FRET population in both the apo and the inverse agonist bound receptor could also implicate the intermediate FRET state stabilized by the partial agonist (Fig. 1e). Similar effects were reported previously in A2AR at comparable concentrations of sodium5. The binding of sodium ions to A2AR may not only account for the low-FRET active-like state of the apo receptor (Fig. 1c,d), but also for the intermediate FRET state in the presence of the partial agonist LUF5834 or the full agonist NECA (Fig. 1e,f).
In previous 19FNMR studies, the point mutation V229C6.31 on TM6 provided good sensitivity to discriminate between active and inactive states, while still maintaining the overall integrity and function of A2AR4,5. These studies confirmed the co-existence of inactive and active states in the apo receptor; however, direct measurements of sub-millisecond conformational dynamics is still missing. To fill this gap, we performed fluorescence quenching experiments on A2AR-V229C6.31 labelled with BODIPY-FL in the absence and presence of ligands (Figs. 2, 3, 4). PET is a dynamic (collisional) quenching process, in which the formation of a weak complex between an organic dye and a quencher molecule results in the transfer of an excited electron from the donor (dye) to the acceptor (quencher) molecule causing a reduction in the excited-state lifetime of the dye31. Electron transfer is a short-range process occurring at dye-quencher distances less than ~ 10 Å and for favorable orientations between the two molecules. As A2AR has several aromatic residues in the vicinity of residue 2296.31, the displacement and orientation of TM6 will determine the proliferation of PET quenching interactions of the probe.
A short two-carbon linker between BODIPY-FL and the point of attachment on A2AR (V229C6.31) ensures that quenching of the dye is primarily due to the movement of TM6 (relative to tyrosines on ICL2, TM5, and TM7) and not due to the intrinsic movement of the dye/linker. Indeed, fluorescence lifetime and anisotropy decay analysis (Fig. 2, Table 2) reveal that quenching occurs upon labelling the receptor, and that the dye reorients slowly, on a timescale determined by the global rotation of the receptor. Additionally, the smFRET data (see above) shows binding of partial/full agonist displaces TM6 by ~ 5–10 Å relative to TM4, which is consistent with the reduction in distance required for quenching to occur between BODIPY-FL and tyrosine residues on TM7 (Y2887.53 and Y2907.55).
We made inferences on the sub-millisecond dynamics of A2AR based on PET quenching lifetimes (tq) measured by FCS. As such, fluorescence intensity fluctuations of BODIPY-FL-labelled A2AR were found to occur on fast (tq, fast = 150–350 ns), intermediate (tq, int = 50–60 μs) and slow (tq, slow = 200–300 μs) timescales, and they were modulated by orthosteric ligands (Fig. 4, Table 4). These lifetimes represent timescales of the local re-arrangement of ICL2, TM5, TM6, and TM7, which either proliferate or suppress quenching interactions between BODIPY-FL and tyrosine residues located on these TMs34.
Using MD simulations, we identified the residue Y112 on intracellular loop 2 (ICL2) between TM3 and TM4 as a promising candidate for dynamic quenching of BODIPY-FL on TM6 (Movie S1). Nevertheless, whereas PET-FCS indicates that addition of full agonist and mini-Gα significantly increases the frequency of reversible quenching (Fig. 4a,d), MD simulations predict fewer quenching transitions of Y112 as A2AR trends towards full activity (Fig. 5c,d). One possible explanation for this discrepancy could be the polarization bias in FCS measurements34. However, the FAD-measured rotational correlation time of A2AR is 50–60 ns (Fig. 2), i.e., 3–5 times faster than tq, fast in FCS. Thus, the fastest FCS decay predominantly reports on the dynamics of PET quenching, not depolarization. Corroborating this, FCS experiments on the free dye in a solution of 3 mM tyrosine reveal a similar quenching lifetime (tq ~ 100 ns, Fig. S3) as in A2AR, while the depolarization of the dye in solution is much faster (ρ ~ 0.3 ns, Table 2).
Therefore, we propose that conformational changes involved in receptor activation bring other aromatic quenchers into the proximity of BODIPY-FL. In MD simulations, residues Y2887.53 and Y2907.55 exhibit distance fluctuations to and from residue 2296.31 which significantly increase as the receptor trends toward full activity (Fig. 5e–h). These fluctuations bringing Y2887.53 and Y2907.55 on TM7 within close proximity to residue 2296.31 on TM6 may be responsible for the increase in the rate and the amplitude of fast PET-FCS component for the full agonist and mini-G protein bound case (Fig. 4a,d). Another residue on TM5, Y1975.58, may have a negligible contribution to the fast quenching phase in the absence of ligands, however it contributes significantly in the presence of mini-G (Figure S7d,e).
The sub-millisecond dynamics reveled by PET-FCS could also be related to the integrity of the ionic lock between R1023.50 (TM3) and E2286.30 (TM6), which was shown to be altered by different ligands4,5. 2D probability plots were generated to assess the integrity of the ionic lock in the inactive (inverse agonist), active (full agonist), and full agonist plus mini-G protein bound structures, respectively (Figure S8). As suggested previously, when the minimum distance between the two residues, \(d_{\text{ R97 - E223}}^{{\text{ N}}\eta - {\text{O}}\varepsilon }\), is less than 5 Å, the ionic lock remains intact due to formation of a salt bridge, whereas at longer distances the ionic lock is broken46,47.
During 5-μs MD simulations started from inverse agonist bound configurations the ionic lock remains mostly intact (Figure S8a), however, in simulations initiated from agonist (Figure S8b) or mini-G (Figure S8c) bound configurations, the ionic lock is predominantly open. For the active structures, two populations emerge, conformations where TM3 and TM6 are close to each other (~ 3–5 Å, ionic lock intact) or far apart (˃5 Å, ionic lock broken), with a significant population in the latter (Figure S8b,c). Ligand-induced breaking of this ionic lock may proliferate the movement of TM3 or TM6 in a manner that causes (sub)microsecond fluctuations in local helical regions or in the TM domains at large.
Conclusions
Single-molecule fluorescence techniques were applied to the A2A Adenosine Receptor and revealed new details about its structural plasticity and how this is modulated by ligands and a G-protein mimetic. smFRET analysis showed that orthosteric ligands alter the relative population of inactive and active conformations, with apo A2AR biased towards active states, and the inverse agonist increasing the fraction of inactive states. The partial agonist stabilizes an intermediate conformation with a TM6-TM4 separation distance that lies between the active (open/high-FRET) and inactive (closed/low-FRET) states. The full agonist promotes an active conformation of the receptor, which coexists with a nearly equally populated intermediate conformation. The broadening of smFRET distributions implies low-millisecond (> 3 ms) exchange between different conformations of A2AR.
The A2AR conformational dynamics was quantitatively analyzed using PET-FCS spectroscopy, by taking advantage of dynamic quenching of a dye attached to residue 2296.31 on TM6 by proximal tyrosines on ICL2, TM5, and TM7. PET-FCS analysis revealed fast (sub)microsecond scale protein motions with lifetimes of 150–350 ns, 50–60 μs, and 200–300 μs, respectively. This previously unresolved kinetics is modulated by the binding of orthosteric ligands and of a G-protein mimetic. Surprisingly, the precoupled state promoted by the full agonist shows increased flexibility. One could imagine a wide range of substates that the receptor can sample around this precoupled state, and we believe that this dynamics may facilitate the binding step with the G protein.
The dynamics resolved by FCS is not associated with exchange between active and inactive states of the receptor, but rather describes local fluctuations at the intracellular side of TM3, TM5, TM6 and TM7 in different ligand-biased conformations. These fast intra-state conformational fluctuations of the receptor may represent movement or rotation of helical domains in A2AR to facilitate the binding of the G protein or other intracellular effectors. The mini-G protein used here mimics the GTPase domain of the G protein and is specifically engineered to couple to the fully active receptor48. While this mimetic has no nucleotide dependence, the conformational flexibility of A2AR revealed by single-molecule fluorescence may be important for facilitating nucleotide exchange (i.e., GDP to GTP) in the receptor-coupled G protein. Understanding the rich multiscale dynamics of GPCRs will be key to develop a metric that distinguishes the effects of drugs, and their ramifications on the activation processes of G protein and their downstream signaling cascades.
Materials and methods
Plasmid construction and transformants screening of A2AR
The constructs of A2AR V229C6.31 for fluorescent experiments were same as previously published4,5. The construct, A2AR(T119C Q226C), was generated using QuikChange Site-Directed Mutagenesis Kits (Agilent Technologies). The mutant gene was verified by full-length DNA sequencing for target genes (The Center for Applied Genomics, Sick Kids Hospital, Toronto, Canada) with the AOX1 primer pair of PFAOX1 and PRAOX1 prior to expression transformant screening. Freshly prepared competent cells of a strain of Pichia pastoris SMD 1163 (Δhis4 Δpep4 Δprb1, Invitrogen) were electro-transformed with individually PmeI-HF (New England Biolabs) linearized plasmid using a Gene Pulse II (Bio-Rad). Clone selection was performed as previously described by an in-house two-step transformant screening strategy49 to maximize the expression of receptor for subsequent fluorescence labeling50.
A2AR expression, purification, and labeling
A2AR was expressed in yeast according to a previously published protocol49. Briefly, 60 h after inoculation, yeast cells were harvested and homogenized, and membrane fragments were solubilized in a detergent solution containing 1% MNG-3 (lauryl maltose neopentyl glycol) and 0.02% CHS (cholesteryl hemisuccinate). Solubilized membranes were incubated with Talon resin (Clontech) and then with 100 μM TCEP reducing agent to obtain A2AR-bound Talon resin suspended in 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, and 0.02% CHS51.
Apo A2AR was eluted from the Talon resin with 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, 0.02% CHS, and 250 mM imidazole. Sodium chloride and imidazole in the sample were removed by dialysis against 50 mM HEPES, pH 7.4, 0.1% MNG-3, 0.02% CHS for 3 h. A2AR was then incubated with BODIPY-FL-iodoacetamide (ThermoFisher Scientific, Cat. no. D6003) for 2 h for labelling at residue V229C6.31. The XAC-agarose gel, i.e., A2AR antagonist xanthine amine congener (XAC) conjugated to Affi-Gel 10 resin, and A2AR were subsequently incubated together for 2 h with gentle nutation. Non-functional A2AR and free unreacted BODIPY-FL- iodoacetamide was washed off with 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, and 0.02% CHS.
Functional A2AR labelled with BODIPY-FL was eluted with 50 mM HEPES, pH 7.4, 0.1% MNG-3, 0.02% CHS, 100 mM NaCl, and 20 mM theophylline. Talon resin was added to the eluted sample and incubated for another 2 h to bind functional A2AR-BODIPY-FL, and then was washed extensively with 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, 0.02% CHS, and 20 mM imidazole, to remove the theophylline. The functional apo A2AR was eluted with 50 mM HEPES, pH 7.4, 100 mM NaCl, 0.1% MNG-3, 0.02% CHS, and 250 mM imidazole, and the sample was dialyzed to remove imidazole for fluorescence experiments. A similar expression, purification, and dye labelling procedure as above was applied for A2AR(T119C Q226C), which was designed for smFRET.
Expression and purification of R373C-ΔCys-mini-Gs
The mini-Gs construct used in this study is based on the miniGs-393 construct described previously43. The miniGs-393 construct was modified to remove four cysteine residues (C237SG.H2.10, C359IG.S6.01, C365VG.s6h5.02, and C379VG.H5.11) producing a cysteine-less variant. This cysteine-less variant was developed to serve as a background for the introduction of cysteine point mutations for covalent chemical modification. One cysteine residue was introduced on helix 5 in the Ras domain at residue R373G.H5.05 to create the R373C-ΔCys-Mini-Gs construct used in the current study. Further details regarding the expression and purification can be found in the Supporting Information Sect. 2.
Single-molecule FRET
smFRET measurements were performed on a custom-built multiparameter fluorescence microscope with alternating-laser excitation (ALEX)35,36. The donor dye (AF488) was excited with a 473 nm diode-pumped solid-state laser (Cobolt Blue, Market Tech). The acceptor dye (AF647) was excited using a continuous wave 635-nm laser (World StarTech, TECRL-25GC-635-TTL-A). An acousto-optic modulator (Isomet, 1205C-2) switches between blue and red excitation with 50 μs duty cycle via a function generator (Tektronix, afg3022b). A multi-channel single-photon counting module (PicoHarp300, PicoQuant, Germany) collects digitally converted photon data from avalanche photodiodes. Green (donor) emission was separated from red (acceptor) emission using a dichroic mirror (FF640-Di01, Semrock) and band-pass filters (BP520/66 m and HQ685/80, Chroma), and broadband polarizing cubes further separate the two signals into two orthogonal polarization components.
The smFRET measurements were performed using a concentration of ~ 50 pM of sample in 20 mM HEPES (100 mM NaCl, pH 7.4) in the presence of 20 mM cysteamine and 0.01% Tween20 and using blue and red laser excitation with an intensity of ~ 25 kW/cm2 and ~ 12 kW/cm2, respectively, at the objective. A custom MATLAB script based on the ‘MLT’ algorithm52 was used to identify fluorescence bursts and sort them into donor-only, acceptor-only and dual-labelled (FRET) populations35. The energy transfer efficiency for each burst, E, was estimated from the number of photons detected in the donor, ID, and the acceptor, IA, channels as:
Here the gamma factor, γ, corrects for the difference in detection efficiencies between the donor and acceptor channels (ηDem and ηAem), and in the quantum yields of the two dyes (ΦD and ΦA)53:
The gamma factor was estimated to be γ = 0.56 ± 0.02 using standard FRET dsDNA samples (see Supporting Information Sect. 1), as described previously54.
The donor–acceptor stoichiometry, S, for the data was calculated as follows,
where, IDD, and IAA, represent the spectrally corrected intensities of the donor after donor excitation and of the acceptor after acceptor excitation, respectively54. Bursts with stoichiometry between 0.2 and 0.8 were used to build the smFRET histograms presented in this work.
Background and spectral-cross-talk corrections were applied using donor-only and acceptor-only bursts, as described previously53,55. The smFRET histograms were fitted to either one Gaussian distribution or to a sum of two Gaussian distributions. The choice of fitting models was informed by the minimization of reduced-χ2 and by the Akaike information criterion (AIC)56. The error margins were estimated using statistical bootstrapping57. Briefly, this was achieved via random sampling of n bursts from the original experimental pool containing, N, smFRET bursts, with replacement (i.e., some bursts are chosen multiple times, others not at all). As such, synthetic smFRET histograms are generated from each subset containing n bursts and each were fit to one or two Gaussians. This algorithm was repeated 2000 times. The standard deviation of the array of best fit parameters from the 2000 synthetic histograms were then taken as the error margin for reported parameter.
Fluorescence lifetime and anisotropy decay
Fluorescence lifetime and anisotropy decay measurements were performed using the custom-built multiparameter fluorescence microscope (see above) and the data was fit using custom MATLAB code36. BODIPY-FL was excited with 480-nm femtosecond pulses obtained by frequency-doubling the output of a tunable Ti:Sapphire laser (Tsunami HP, Spectra Physics, USA). Time- and polarization-resolved curves were obtained by binning the time delay between excitation and detection events for photons with distinct polarizations (parallel and perpendicular) compared to the polarization of the excitation laser. Measurements were performed with a concentration of ~ 100 nM receptor in 20 mM HEPES (100 mM NaCl, pH 7.4) for the ligand-free (apo) receptor, and in the presence of saturating amounts (100 μM) of ligands. All samples were excited at an intensity of 0.1 kW/cm2.
Fluorescence polarization anisotropy is defined using the parallel, I║, and perpendicular I┴, components of the detected fluorescence emission as:
where G is a factor that corrects for different sensitivities in detecting signals with different polarizations58.
Anisotropy decay time constants of the free and receptor-attached BODIPY-FL, i.e., rotational correlation times, were estimated by fitting the experimental anisotropy curve r (t) to a multi-exponential function:
where ro is the fundamental anisotropy of the fluorescence particle (~ 0.4) and Ai is the fraction of species with rotational correlation time ρi58.
The fluorescence lifetimes of the free and the receptor-attached BODIPY-FL were estimated by fitting the isotropic decay curve, i.e., the denominator in Eq. 3a, to a multi-exponential decay function:
where N is the total number of photon counts detected and Bi is the fraction of species with the lifetime τLi36. The quality of the fitting and the model validation were evaluated by minimization of χ2 and the Akaike information criterion (AIC)56. The standard deviation of the fitting parameters was estimated by parametric bootstrapping57. Briefly, this was achieved by using the best fit parameters from a single fit of a lifetime or an anisotropy decay curve and generating an idealized decay curve to which Poisson noise is added, which generated a synthetic curve, mimicking the original decay. Each synthetic curve was then fit using the above models (Eqs. 3–4) and was repeated 100 times. The standard deviation of the array of fitted parameters from the algorithm were then used as the reported error margins.
Fluorescence correlation spectroscopy
FCS measurements were performed on A2AR-V229C6.31-BODIPY-FL using a custom-built fluorescence confocal microscope, described in detail elsewhere59. For each experiment, a solution of 30 μL containing 10 nM receptor in 20 mM HEPES (100 mM NaCl, pH 7.4 supplemented with 20 mM cysteamine and 2 mM Trolox) was cast on a plasma-cleaned glass coverslip (VMR, cat. No. CA48366-249–1) and excited at 488 nm at ~ 0.2 kW/cm2 using a blue laser (TECBL-488 nm; WorldStarTech). FCS measurements were repeated under the same excitation/detection conditions for the ligand-free (apo) receptor, and in the presence of saturating amounts (100 μM) of an inverse agonist (ZM241385), a partial agonist (LUF5834), and a full agonist (NECA) w/wo a G-protein mimetic (mini-Gs). The resulting pseudo-autocorrelation curves were fitted to the following mathematical model33:
where \(\langle{N}\rangle\) is the average number of molecules in the detection volume (which is characterized by the width ωo ellipticity s, both measured using samples with known diffusion properties59), \({\tau }_{\mathrm{D}}\) is the average diffusion time, tt and Kt are the triplet-state lifetime and amplitude, respectively, and \({t}_{\mathrm{q},\mathrm{i}}\) and Ki are quenching lifetimes and corresponding amplitudes, respectively.
The hydrodynamic radius (RH) of the fluorescent molecules is estimated by using the FCS-derived diffusion time \({\tau }_{\mathrm{D}}\) using the Stokes–Einstein equation:
where ωo is the width of the confocal detection volume, kB is the Boltzmann constant, η is the viscosity of the solution, and T is the temperature. All FCS curves under different ligand-conditions were globally using custom MATLAB code, where the triplet lifetime of the BODIPY-FL was a shared parameter. The goodness of the fit and the number of free parameters in FCS fitting models were assessed using the minimization of residuals (χ2) and the AIC criterion56. For clarity, the diffusion component of the FCS curve was subtracted from the fitted curve and the raw FCS data, leaving only the triplet and quenching components, as shown in Fig. 3.
BODIPY-FL simulations
The initial structure for an 11-residue segment of A2AR (T224 to S234) was taken from PDB: 2YDO14. The V229C6.31 mutation and the conjugated dye were built with the molefacture plugin to VMD60. Parameters were constructed with the LigParGen web server61, supplemented with backbone parameters from cysteine and the C–C–S–C dihedral parameter from methionine. The dye's boron atom was replaced with a carbon atom to circumvent limitations of the LigParGen web server. No attempt was made to enforce boron-like geometry, though inaccuracies stemming from this simplification are likely minor due to regional rigidity. Three 2.5 μs simulations of this system were conducted in a cubic box with ~ 6000 water molecules and 100 mM excess KCl. To maintain helical structure throughout these simulations, residue restraints with force constants of 103 kJ/mol/nm2 were applied to all Cα atoms. To quantify the spatial separation of the dye from its protein attachment, distances, r, were measured from the modified cysteine Cβ atom to the center of geometry of non-hydrogen atoms in the dye's aromatic group (C1, C2, C4, C5, N1, C7, C8, N2, and B1, where B1 represents the boron that we modeled as carbon). Further details regarding these simulations can be found in the Supporting Information Sect. 3.
Molecular dynamics simulations of A2AR
We conducted six MD simulations for each of seven different initial receptor configurations, which were obtained from crystal structures containing either the inverse agonist ZMA (PDBs: 3PWH and 4EIY)45,62, the endogenous agonist adenosine (PDB: 2YDO)14, the synthetic agonist UK-432097 (PDB: 3QAK)13, or the agonist N-ethyl-5′-carboxamido adenosine (NECA) and an engineered G protein (PDBs: 5G53 and 6GDG)48,63. We used both chains A and B from the 5G53 structure independently. None of our simulations included ligand, G protein, or receptor modifications employed for crystallization. However, the MD trajectories retained the global features of the starting structures while capturing distinct local dynamics on a sub-microsecond timescale. The simulated sequence was S6 to V307 (i.e., ΔM1-G5 and ΔL308-S412). Residue identifiers always corresponded to the receptor's full-length wild-type sequence (UNIPROT ID: P29274). The receptor was not glycosylated, all titratable residues were in their standard states for pH 7, and the protein backbone termini were zwitterionic. Further details regarding the MD simulations can be found in the Supporting Information Sect. 3.
References
Fredriksson, R., Lagerström, M. C., Lundin, L.-G. & Schiöth, H. B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 63(6), 1256–1272 (2003).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there?. Nat. Rev. Drug Discov. 5(12), 993–996 (2006).
de Lera Ruiz, M., Lim, Y.-H. & Zheng, J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 57(9), 3623–3650 (2014).
Ye, L., Van Eps, N., Zimmer, M., Ernst, O. P. & Prosser, R. S. Activation of the A 2A adenosine G-protein-coupled receptor by conformational selection. Nature 533(7602), 265–268 (2016).
Ye, L. et al. Mechanistic insights into allosteric regulation of the A 2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 9(1), 1–13 (2018).
Nygaard, R. et al. The dynamic process of β2-adrenergic receptor activation. Cell 152(3), 532–542 (2013).
Weis, W. I. & Kobilka, B. K. The molecular basis of G protein–coupled receptor activation. Annu. Rev. Biochem. 87, 897–919 (2018).
Ernst, O. P. et al. Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms. Chem. Rev. 114(1), 126–163 (2014).
Hofmann, K. P. et al. AG protein-coupled receptor at work: The rhodopsin model. Trends Biochem. Sci. 34(11), 540–552 (2009).
Weikl, T. R. & Paul, F. Conformational selection in protein binding and function. Protein Sci. 23(11), 1508–1518 (2014).
Manglik, A. et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell 161(5), 1101–1111 (2015).
Manglik, A. & Kruse, A. C. Structural basis for G protein-coupled receptor activation. Biochemistry 56(42), 5628–5634 (2017).
Xu, F. et al. Structure of an agonist-bound human A2A adenosine receptor. Science 332(6027), 322–327 (2011).
Lebon, G. et al. Agonist-bound adenosine A 2A receptor structures reveal common features of GPCR activation. Nature 474(7352), 521–525 (2011).
Jaakola, V.-P. et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 322(5905), 1211–1217 (2008).
Choe, H.-W. et al. Crystal structure of metarhodopsin II. Nature 471(7340), 651–655 (2011).
Rasmussen, S. G. et al. Crystal structure of the β 2 adrenergic receptor–Gs protein complex. Nature 477(7366), 549–555 (2011).
García-Nafría, J. & Tate, C. G. Cryo-electron microscopy: Moving beyond X-ray crystal structures for drug receptors and drug development. Annu. Rev. Pharmacol. Toxicol. 60(1), 51–71 (2020).
Van Eps, N. et al. Gi- and Gs-coupled GPCRs show different modes of G-protein binding. Proc. Natl. Acad. Sci. 115(10), 2383–2388 (2018).
Wingler, L. M. et al. Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176(3), 468–478 (2019).
Van Eps, N., Caro, L. N., Morizumi, T. & Ernst, O. P. Characterizing rhodopsin signaling by EPR spectroscopy: from structure to dynamics. Photochem. Photobiol. Sci. 14(9), 1586–1597 (2015).
De Andrade, F. D., Netto, A. M. & Colnago, L. A. Qualitative analysis by online nuclear magnetic resonance using Carr–Purcell–Meiboom–Gill sequence with low refocusing flip angles. Talanta 84(1), 84–88 (2011).
Sušac, L., Eddy, M. T., Didenko, T., Stevens, R. C. & Wüthrich, K. A2A adenosine receptor functional states characterized by 19F-NMR. Proc. Natl. Acad. Sci. 115(50), 12733 (2018).
Tian, H., Fürstenberg, A. & Huber, T. Labeling and single-molecule methods to monitor G protein-coupled receptor dynamics. Chem. Rev. 117(1), 186–245 (2017).
Quast, R. B. & Margeat, E. Studying GPCR conformational dynamics by single molecule fluorescence. Mol. Cell. Endocrinol. 493, 110469 (2019).
Gregorio, G. G. et al. Single-molecule analysis of ligand efficacy in β2AR–G-protein activation. Nature 547(7661), 68–73 (2017).
Vafabakhsh, R., Levitz, J. & Isacoff, E. Y. Conformational dynamics of a class C G-protein-coupled receptor. Nature 524(7566), 497–501 (2015).
Olofsson, L. et al. Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. Nat. Commun. 5(1), 1–12 (2014).
Marmé, N., Knemeyer, J.-P., Sauer, M. & Wolfrum, J. Inter-and intramolecular fluorescence quenching of organic dyes by tryptophan. Bioconjug. Chem. 14(6), 1133–1139 (2003).
Doose, S., Neuweiler, H. & Sauer, M. Fluorescence quenching by photoinduced electron transfer: A reporter for conformational dynamics of macromolecules. ChemPhysChem 10(9–10), 1389–1398 (2009).
De Silva, A. P., Moody, T. S. & Wright, G. D. Fluorescent PET (photoinduced electron transfer) sensors as potent analytical tools. Analyst 134(12), 2385–2393 (2009).
Mazouchi, A., Bahram, A. & Gradinaru, C. C. Sub-diffusion decays in fluorescence correlation spectroscopy: Dye photophysics or protein dynamics?. J. Phys. Chem. B 117(38), 11100–11111 (2013).
Neuweiler, H., Johnson, C. M. & Fersht, A. R. Direct observation of ultrafast folding and denatured state dynamics in single protein molecules. Proc. Natl. Acad. Sci. 106(44), 18569–18574 (2009).
Rogers, J. M. et al. Photoinduced electron transfer and fluorophore motion as a probe of the conformational dynamics of membrane proteins: Application to the influenza a M2 proton channel. Langmuir 27(7), 3815–3821 (2011).
Gomes, G.-N. & Gradinaru, C. C. Insights into the conformations and dynamics of intrinsically disordered proteins using single-molecule fluorescence. Biochim. Biophys. Acta 11, 1696–1706 (2017).
Fernandes, D. D. et al. Characterization of fluorescein arsenical hairpin (FlAsH) as a probe for single-molecule fluorescence spectroscopy. Sci. Rep. 7(1), 1–15 (2017).
Gopich, I. V. & Szabo, A. FRET efficiency distributions of multistate single molecules. J. Phys. Chem. B 114(46), 15221–15226 (2010).
Mikhalyov, I., Gretskaya, N., Bergström, F. & Johansson, L. B. -Å. Electronic ground and excited state properties of dipyrrometheneboron difluoride (BODIPY): Dimers with application to biosciences. Phys. Chem. Chem. Phys. 4(22), 5663–5670 (2002).
Campos, L. A. et al. A photoprotection strategy for microsecond-resolution single-molecule fluorescence spectroscopy. Nat. Methods 8(2), 143–146 (2011).
Zhao, J. et al. Bodipy derivatives as triplet photosensitizers and the related intersystem crossing mechanisms. Front. Chem. 7, 821 (2019).
Doose, S., Neuweiler, H. & Sauer, M. A close look at fluorescence quenching of organic dyes by tryptophan. ChemPhysChem 6(11), 2277–2285 (2005).
Soranno, A. et al. Integrated view of internal friction in unfolded proteins from single-molecule FRET, contact quenching, theory, and simulations. Proc. Natl. Acad. Sci. 114(10), E1833–E1839 (2017).
Carpenter, B. & Tate, C. G. Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng. Des. Sel. 29(12), 583–594 (2016).
Bennett, K. A. et al. Pharmacology and structure of isolated conformations of the adenosine A2A receptor define ligand efficacy. Mol. Pharmacol. 83(5), 949–958 (2013).
Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337(6091), 232–236 (2012).
Neale, C., Herce, H. D., Pomès, R. & García, A. E. Can specific protein-lipid interactions stabilize an active state of the beta 2 adrenergic receptor?. Biophys. J . 109(8), 1652–1662 (2015).
Romo, T. D., Grossfield, A. & Pitman, M. C. Concerted interconversion between ionic lock substates of the β2 adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys. J . 98(1), 76–84 (2010).
Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. & Tate, C. G. Structure of the adenosine A 2A receptor bound to an engineered G protein. Nature 536(7614), 104–107 (2016).
Ye, L., Orazietti, A. P., Pandey, A. & Prosser, R. S. High-Efficiency Expression of Yeast-Derived G-Protein Coupled Receptors and 19 F Labeling for Dynamical Studies 407–421 (Springer, Berlin, 2018).
Scorer, C. A., Clare, J. J., McCombie, W. R., Romanos, M. A. & Sreekrishna, K. Rapid selection using G418 of high copy number transformants of Pichia pastoris for high–level foreign gene expression. Bio/Technology 12(2), 181–184 (1994).
Ye, L., Larda, S. T., Li, Y. F. F., Manglik, A. & Prosser, R. S. A comparison of chemical shift sensitivity of trifluoromethyl tags: Optimizing resolution in 19 F NMR studies of proteins. J. Biomol. NMR 62(1), 97–103 (2015).
Liu, B. et al. The effect of intrachain electrostatic repulsion on conformational disorder and dynamics of the Sic1 protein. J. Phys. Chem. B 118(15), 4088–4097 (2014).
Sisamakis, E., Valeri, A., Kalinin, S., Rothwell, P. J. & Seidel, C. A. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. Elsevier 475, 455–514 (2010).
Hellenkamp, B. et al. Precision and accuracy of single-molecule FRET measurements—a multi-laboratory benchmark study. Nat. Methods 15(9), 669–676 (2018).
Lee, N. K. et al. Accurate FRET measurements within single diffusing biomolecules using alternating-laser excitation. Biophys. J. 88(4), 2939–2953 (2005).
Akaike, H. A new look at the statistical model identification. IEEE Trans. Autom. Control 19(6), 716–723 (1974).
Efron, B. & Tibshirani, R. J. An introduction to the bootstrap (CRC Press, Boca Raton, 1994).
Gradinaru, C. C., Marushchak, D. O., Samim, M. & Krull, U. J. Fluorescence anisotropy: From single molecules to live cells. Analyst 135(3), 452–459 (2010).
Li, Y., Shivnaraine, R. V., Huang, F., Wells, J. W. & Gradinaru, C. C. Ligand-induced coupling between oligomers of the M2 receptor and the Gi1 protein in live cells. Biophys. J . 115(5), 881–895 (2018).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 14(1), 33–38 (1996).
Dodda, L. S., Cabeza de Vaca, I., Tirado-Rives, J. & Jorgensen, W. L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 45, W331–W336 (2017).
Doré, A. S. et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19(9), 1283–1293 (2011).
García-Nafría, J., Lee, Y., Bai, X., Carpenter, B. & Tate, C. G. Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife 7, e35946 (2018).
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Grant No. RGPIN 2017-06030 to C.C.G.) and by Canadian Institutes for Health Research (Grant No. MOP-43998 to R.S.P.) C.N. was funded by U.S. DOE Laboratory Directed Research and Development (LDRD) funds. Computations used resources provided by the Los Alamos National Lab Institutional Computing Program, which is supported by the U.S. DOE National Nuclear Security Administration under Contract No. DE-AC52-06NA25396.
Author information
Authors and Affiliations
Contributions
D.D.F. acquired and analyzed fluorescence data, prepared figures and wrote the manuscript; L.Y. designed the A2A cysteine mutants, L.Y. and A.P. purified and labelled them with fluorescent dyes; C.N. performed and analyzed molecular dynamics simulations; G.-N.W.G. wrote software for smFRET and FAD data analysis; Y.L. wrote software for PET-FCS data analysis; A.M. performed control FCS experiments of BODIPY-FL; A.P.O. and X.W. expressed and purified the mini-G protein; C.C.G. and R.S.P. guided the entire project, interpreted the data and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Video 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fernandes, D.D., Neale, C., Gomes, GN.W. et al. Ligand modulation of the conformational dynamics of the A2A adenosine receptor revealed by single-molecule fluorescence. Sci Rep 11, 5910 (2021). https://doi.org/10.1038/s41598-021-84069-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84069-0
This article is cited by
-
Exosomal secreted SCIMP regulates communication between macrophages and neutrophils in pneumonia
Nature Communications (2024)
-
Single-molecule visualization of human A2A adenosine receptor activation by a G protein and constitutively activating mutations
Communications Biology (2023)
-
Sub-millisecond conformational dynamics of the A2A adenosine receptor revealed by single-molecule FRET
Communications Biology (2023)
-
Probing Single-molecule Interfacial Electron Transfer Inside a Single Lipid Vesicle
Journal of Fluorescence (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
