
Abstract
Deinococcus bacteria are extremely resistant to radiation and other DNA damage- and oxidative stress-generating conditions. An efficient SOS-independent response mechanism inducing expression of several DNA repair genes is essential for this resistance, and is controlled by metalloprotease IrrE that cleaves and inactivates transcriptional repressor DdrO. Here, we identify the molecular signaling mechanism that triggers DdrO cleavage. We show that reactive oxygen species (ROS) stimulate the zinc-dependent metalloprotease activity of IrrE in Deinococcus. Sudden exposure of Deinococcus to zinc excess also rapidly induces DdrO cleavage, but is not accompanied by ROS production and DNA damage. Further, oxidative treatment leads to an increase of intracellular free zinc, indicating that IrrE activity is very likely stimulated directly by elevated levels of available zinc ions. We conclude that radiation and oxidative stress induce changes in redox homeostasis that result in IrrE activation by zinc in Deinococcus. We propose that a part of the zinc pool coordinated with cysteine thiolates is released due to their oxidation. Predicted regulation systems involving IrrE- and DdrO-like proteins are present in many bacteria, including pathogens, suggesting that such a redox signaling pathway including zinc as a second messenger is widespread and participates in various stress responses.
Similar content being viewed by others

Introduction
For all organisms it is important to respond efficiently to changes in environmental constraints leading to stress conditions within cells. In case of DNA damage-generating stress, many bacteria use the SOS response to induce expression of DNA repair genes. The molecular signal inducing the SOS response is the presence of single-stranded DNA1. RecA filaments formed on single-stranded DNA stimulate self-cleavage and inactivation of LexA, the repressor of the SOS response. Not all bacteria use the SOS response to induce DNA repair genes2. In Deinococcus bacteria, induction of recA and other DNA repair genes occurs in a RecA/LexA-independent manner and in the present work we aim to identify the intracellular molecular signal that induces this SOS-independent response.
Deinococcus species are famous for their extreme tolerance to DNA damage- and oxidative stress-generating conditions such as radiation and desiccation, and for their capacity to repair massive DNA damage3,4,5,6. This extreme tolerance results from a combination of multiple factors and mechanisms7, including limitation of oxidative protein damage8,9,10 that has been correlated with a high intracellular Mn2+/Fe2+ ratio (e.g. 0.24 in Deinococcus radiodurans versus < 0.01 in radiation-sensitive Escherichia coli)11 and the accumulation of small antioxidant Mn2+-containing complexes (with intracellular Mn2+ concentrations ranging from 0.2 to 2 mM, 70% of which not bound to proteins)12. An efficient radiation/desiccation response (RDR) mechanism for induced expression of a set of genes forming the RDR regulon is also essential for radiation tolerance13,14,15,16. The predicted RDR regulon in different Deinococcus species consists of about 20 genes, including classical DNA repair genes (e.g. recA, uvrB) and novel genes more specific to Deinococcus (e.g. ddrB, ddrO)17. We previously demonstrated that RDR regulon expression is controlled by two proteins highly conserved in Deinococcus species: IrrE and DdrO14.
IrrE, also called PprI, is a COG2856 domain-containing metalloprotease that induces expression of the RDR genes by cleaving and inactivating DdrO, the transcriptional repressor of the RDR regulon14,17,18,19,20. DdrO has a helix-turn-helix (HTH) motif-containing DNA-binding domain of the XRE family19. The HTH_XRE-type domain is present in many transcriptional regulators and has been named after the PBSX repressor protein Xre from the defective Bacillus prophage PBSX21. DdrO cleavage has been demonstrated when IrrE and DdrO are co-expressed from plasmids in E. coli without exogenous stress, and in vitro with both proteins purified from E. coli. In Deinococcus itself, however, the cleavage is somehow induced when bacteria are exposed to radiation14. The N-terminal domain of IrrE is structurally similar to zinc metalloprotease thermolysin22, and includes the active site/metal ion binding motif HEXXH that is essential for protease activity14. No metal ion was observed in the crystal structure of IrrE, but briefly soaking apo-IrrE crystals in a Zn2+-containing solution resulted in binding of Zn2+ in the expected site, accompanied by re-orientation of the side chains of two active site residues towards the Zn2+ ion22. Addition of Zn2+ to metal-free IrrE was sufficient to restore DdrO cleavage in vitro17. The in vitro protease activity of IrrE could also be restored after addition of Mn2+ or Fe2+ ions to metal-free enzyme17. Such in vitro re-activation not only by Zn2+ but also by some other metals is commonly found for zinc metalloproteases, including thermolysin23,24. Besides its protease domain, the crystal structure of IrrE revealed an additional domain with structural similarity to GAF domains22, which may have a role in signaling or in protein–protein interactions25,26. Interestingly, predicted but largely uncharacterized COG2856/XRE protein pairs have been identified in many other bacterial genera, including pathogens and bacteria used in biotechnological industry14,19,27,28,29, suggesting that stress response mechanisms involving such protein pairs are more widespread than currently recognized.
The molecular signaling mechanism by which radiation triggers cleavage of DdrO by the constitutively expressed IrrE in Deinococcus is unknown. Exposure of the cells to radiation or desiccation causes redox imbalance through formation of reactive oxygen species (ROS)6. ROS can react with and damage any molecule in the cell, including DNA. In turn, DNA damage can trigger ROS production30. Besides causing damage, ROS and other reactive species may also activate proteins through oxidative post-translational modification of cysteine residues, as exemplified by chaperone Hsp33 and transcription factor OxyR in E. coli and several other bacteria31,32. Here, we investigated if ROS are involved in IrrE activation, either by direct redox modification of IrrE or indirectly by generating another molecular signal acting downstream the stress-induced ROS and redox imbalance. We show that IrrE is a zinc-dependent enzyme in vivo in Deinococcus, that induction of IrrE protease activity still occurs when IrrE lacks its GAF-like domain, and that IrrE is activated directly by increased intracellular levels of free zinc ions following a zinc shock. Moreover, an increase in free zinc and IrrE protease activation is also observed after oxidative treatment of the cells. We propose that induction of the RDR regulon by radiation and oxidative stress involves a zinc signal generated by impaired redox homeostasis. Such role for zinc as a second messenger in regulation of cellular enzyme activities has been described previously for eukaryotes33,34,35, but to our knowledge this is the first time for prokaryotes.
Results
DdrO cleavage in Deinococcus is stimulated by oxidative treatments
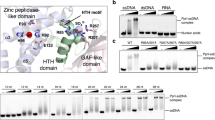
Cleavage of RDR regulon repressor DdrO (129 aa) by metalloprotease IrrE (281 aa) in D. deserti has been observed after exposure of the cells to gamma or UV radiation14. The cleavage occurs between L106 and R107 of DdrO. Here we show that this cleavage is also induced after a short exposure to mitomycin C (MMC) (Fig. 1; Supplementary Fig. S1). MMC is known in particular as an alkylating agent that generates interstrand DNA cross-links, but the metabolism of this quinone-containing antibiotic in the cell also results in oxidative damage to other macromolecules via the generation of various ROS6,36,37,38. The antioxidant compound thiourea scavenges hydroxyl and superoxide radicals and hydrogen peroxide (H2O2)39,40, and has been found to inhibit the effects of the MMC-induced ROS production in E. coli36. Here we observed that the MMC-induced DdrO cleavage in D. deserti was decreased in the presence of thiourea (Fig. 1; Supplementary Fig. S1), suggesting the involvement of MMC-induced ROS in DdrO cleavage stimulation. Another antioxidant, TEMPOL, which catalyzes superoxide dismutation, facilitates catalase-like metabolism of H2O2 and limits hydroxyl radical formation41,42,43, also inhibited DdrO cleavage in cells exposed to MMC (Fig. 1). When the cells were directly exposed to H2O2 for 10 min, DdrO cleavage was also efficiently induced, which was inhibited by thiourea and TEMPOL (Fig. 1), further confirming the participation of redox signaling in IrrE activation.

Mitomycin C and H2O2 induce DdrO cleavage in D. deserti. Wild-type cells were exposed to MMC (0.5 μg/ml) or H2O2 (10 mM) for the indicated time in the absence or presence of the antioxidants thiourea (150 mM) or TEMPOL (10 mM), and DdrO cleavage was analyzed by immunoblotting. P, 20 ng purified DdrO (129 aa, 14.7 kDa). Each cropped blot corresponds to a single independent blot; the four uncropped blots are shown in Supplementary Fig. S9.
We first presumed that stress-generated ROS directly oxidize IrrE. The sulfur-containing amino acids methionine (Met) and cysteine (Cys) are particularly sensitive to oxidation, and fulfill a key role in redox signaling. Thus for some proteins oxidation of these residues leads to their activation (e.g. HypT, OxyR, Hsp33)44. Met and Cys residues highly conserved in deinococcal IrrE proteins were mutated in D. deserti IrrE. None of the mutations affected radiation resistance and DdrO cleavage in D. deserti (Supplementary Fig. S2), excluding a role of these residues in IrrE activation upon stress exposure. This is in line with the previously reported in vitro cleavage of DdrO by IrrE, using purified proteins for which any modification (e.g. oxidized residues) was not detected by mass spectrometry14,17. Together, these data suggest that ROS stimulate IrrE activity in an indirect manner.
IrrE is a zinc-dependent metalloprotease in Deinococcus
DdrO cleavage by IrrE is not only stimulated by radiation and other stresses that induce ROS production, but also rapidly (within 5–10 min) and efficiently after exposure of Deinococcus cultures to an excess of zinc ions, but not of other metal ions including Mn2+ and Fe2+17, as confirmed here (Supplementary Fig. S1). The stimulation of IrrE protease activity by the zinc shock might be direct, if IrrE activity indeed requires Zn2+ as co-factor in vivo, or indirect, if the zinc shock, in common with radiation, causes ROS formation and DNA damage. To better understand the zinc shock-induced activation mechanism, we first investigated the metal dependency of the IrrE activity in vivo by applying different treatments in the presence of TPEN or dipyridyl, which are membrane-permeable chelators with high affinity for zinc (Zn2+) and iron (Fe2+) ions, respectively. Dipyridyl (DIP) was included in these tests because of oxidative stress through ROS formation often involves Fe2+. DIP (500 μM) inhibited DdrO cleavage only in H2O2-exposed cells (Fig. 2; Supplementary Fig. S3), showing that the observed cleavage stimulated by H2O2, but not by UV or MMC, requires Fe2+, most likely via the Fenton reaction where H2O2 reacts with Fe2+ generating oxygen-radical species. TPEN (25 to 50 μM) totally blocked DdrO cleavage in D. deserti and D. radiodurans exposed to each of the applied treatments (Fig. 2; Supplementary Fig. S3). Additional experiments showed that adding 10 μM TPEN to the culture was sufficient to inhibit stress-induced DdrO cleavage (Supplementary Fig. S3). It has been suggested that Mn2+ might be the co-factor required for IrrE activity in vivo in Deinococcus20. However, the applied TPEN concentrations are low compared to the near-millimolar concentration of Mn2+ in Deinococcus, and the affinity of TPEN for Zn2+ (2.6 × 10−16 M) is much higher than for Mn2+ (5.4 × 10−11 M)45. Together, the data strongly support that the in vivo protease activity of IrrE is dependent on zinc ions.

Stress-induced DdrO cleavage in D. deserti requires zinc ions. D. deserti strain RD19 (wild type) was exposed or not to UV (250 J/m2), H2O2 (10 mM for 10 min) or MMC (1 μg/ml for 10 min) in the presence of the Zn2+ chelator TPEN (25 or 50 μM) or the Fe2+ chelator DIP (500 μM), and DdrO cleavage was analyzed by immunoblotting. Because TPEN was dissolved in ethanol (EtOH), a control with EtOH (0.5% final concentration) was included. C, control without metal chelator. P, 20 ng purified DdrO. Each cropped blot corresponds to a single independent blot; the three uncropped blots are shown in Supplementary Fig. S9.
The zinc shock-induced cleavage is not accompanied by ROS production and DNA damage
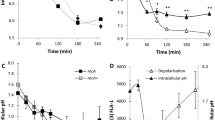
Next, we analyzed whether the zinc shock-induced cleavage occurs indirectly via generation of ROS and/or DNA damage. We have previously shown that exposure of D. deserti to radiation results in an increase of point mutations as detected by an increased number of rifampicin resistant mutants46. Such mutations can arise through direct DNA damage or after incorporation of oxidized nucleotides. Efficient cleavage of DdrO was observed when D. deserti was exposed to either zinc shock or H2O2 treatment. However, in contrast to H2O2 exposure, zinc shock was not accompanied by induced mutagenesis (Fig. 3a). In addition, ROS were not detected following the zinc shock when a fluorescent ROS detector was used (Supplementary Fig. S4). These results indicate that DdrO cleavage by IrrE after the zinc shock is induced directly by the sudden increase of the Zn2+ concentration, and not indirectly via the generation of ROS and DNA damage. An increase in intracellular free Zn2+ following zinc shock was indeed observed using Fluozin-3 AM, a membrane-permeable molecule that after de-esterification in the cell can detect Zn2+ (Supplementary Fig. S5). The direct activation by Zn2+ is further supported by the observation that the antioxidants thiourea and TEMPOL did not inhibit the zinc-induced DdrO cleavage (Fig. 3b). Moreover, also dipyridyl did not inhibit the zinc-induced cleavage, showing that this induction did not occur indirectly via a step that involves Fe2+ (Fig. 3b).

The zinc shock activates IrrE without generating ROS and DNA damage. (a) Induced mutagenesis experiments showing that the zinc shock-induced DdrO cleavage does not occur indirectly via DNA damage/oxidative stress. In contrast to the H2O2 exposure (10 mM for 10 min), the zinc shock (250 μM for 10 min) does not lead to an increase in DNA mutations, whereas both conditions induce DdrO cleavage (inlay). C, untreated control cells. A two-sided Mann–Whitney U test was performed on the data, and an asterisk indicates a p value < 0.05. NS, not significant. In box plots: center line, median; box limits, upper and lower quartiles; whiskers, 1.5 × interquartile range. (b) Western blots showing that DdrO cleavage induced by the zinc shock (250 μM for 10 min) is neither inhibited by antioxidants thiourea (150 mM) and TEMPOL (10 mM) nor by the Fe2+ chelator DIP (500 μM). The Zn2+ chelator TPEN (50 μM) partially inhibited the zinc shock-induced cleavage. C, control cells without metal chelator. P, 20 ng purified DdrO. Each cropped blot corresponds to a single independent blot; the four uncropped blots are shown in Supplementary Fig. S9.
The direct stimulation of IrrE-mediated DdrO cleavage by zinc ions suggests that radiation/redox-induced cleavage may also involve an increase in free zinc ions. A ROS-induced increase in intracellular free zinc ions is strongly supported by several previous studies. Indeed, many oxidants and other reactive species can cause rapid release of zinc ions from redox-sensitive cysteine-containing zinc sites such as zinc fingers and related structures in proteins, which allows novel interactions between zinc ions and other proteins31,33,34,47,48,49. Therefore, we hypothesized that oxidative treatments leading to DdrO cleavage, such as exposure to H2O2, could modify the free zinc ion content in Deinococcus. Using Fluozin-3 AM we showed that H2O2 concentrations in the range of 10 to 30 mM indeed lead to substantial increases in the level of free zinc ions (Fig. 4). These data strongly support a role for zinc in oxidative-stress-induced activation of IrrE.

Detection of free intracellular zinc in D. deserti after H2O2 exposure. After de-esterification of FluoZin-3 AM in the cells, increase of free intracellular zinc was measured after addition of H2O2. The values are corrected for the fluorescence observed after the incubation for 60 min with FluoZin-3 AM but before H2O2 treatment. This observed fluorescence indicates that FluoZin-3 chelates intracellular Zn2+ already before treatment. Error bars are standard deviation of three independent experiments (biological replicates). no FZ, cells incubated without FluoZin-3 AM.
Identification of Deinococcus deserti proteins with zinc/cysteine sites
This section describes possible sources of ROS-induced zinc mobilization in D. deserti. In addition to zinc/cysteine (Zn/Cys) sites in proteins, zinc ions can be bound by bacillithiol (BSH), a low-molecular-weight thiol produced by Deinococcus and several other bacteria, with oxidation of BSH (to BSSB) causing release of the zinc50. The zinc bound to BSH could thus be an important source for IrrE in oxidative conditions. However, with respect to DdrO cleavage upon stress and DdrO re-accumulation during recovery, no difference was observed between wild-type and BSH biosynthesis deficient mutant strains of D. deserti (Supplementary Fig. S6), showing that BSH is not crucial for regulating IrrE activity.
Release of zinc ions from Zn/Cys sites after oxidation or other modifications of the Zn2+-coordinating thiolates has been reported for several bacterial proteins49,51. Numerous proteins that bind zinc through Cys residues, or through a combination of Cys and histidine (His) residues, have been described or predicted. The zinc-binding residues are generally present as two pairs of closely spaced Cys/His (e.g. the most frequently found CXXC motif, but also others such as CXC, CXXXC and CXXH) separated by a longer spacer within the protein sequence35,52. To find D. deserti proteins that could be potential sources of oxidative stress-induced increase of intracellular free zinc, we searched its genome for proteins containing Zn/Cys sites. We found that it encodes homologs of at least 62 proteins harboring Zn/Cys sites (Supplementary Table S1), with a large fraction (27%) consisting of proteins involved in DNA replication and repair (Table 1). Similar Cys motifs and thus potential Zn/Cys sites are present in more than 30 additional D. deserti proteins, including uncharacterized small proteins containing four or more cysteine residues (e.g. the radiation-induced 74-residue-long DdrS with two CXXC motifs) (Supplementary Table S1). In a ddrS mutant, no difference was observed for DdrO cleavage induction upon stress when compared to the wild-type strain (Supplementary Fig. S6). Taking into consideration all these data, we presume that zinc ions are released from various different proteins following disturbed redox homeostasis. Indeed, the list of identified Zn/Cys site-containing D. deserti proteins (Supplementary Table S1) contains homologs of at least 10 proteins known to release zinc in stress conditions: Hsp33, Trx2, FUR family proteins, DnaG, PriA, QueC, RpoC, ThrS, AlaS, ClpX49,51.
The GAF-like domain of IrrE is not essential to induce DdrO cleavage
The results described above indicate that IrrE protease activity is stimulated by increased availability of zinc ions following exposure to radiation and other agents that cause ROS production and redox imbalance. Then what could be the role of the C-terminal GAF-like domain of IrrE? GAF domains may bind small signaling molecules or may have a role in protein–protein interactions25,26. IrrE/DdrO-related systems are found in other bacteria, but a GAF-like domain is not present in all IrrE-like proteins19. Therefore, it is interesting to investigate whether or not this domain plays a role in relation with zinc signaling. When a truncated IrrE lacking this domain was co-expressed with DdrO in E. coli, DdrO cleavage was observed (Fig. 5a), showing that the GAF-like domain is not strictly necessary for the cleavage by the N-terminal zinc peptidase domain. Nevertheless, the cleavage was clearly less efficient than with entire IrrE. The truncated IrrE could not be purified from E. coli, indicating that the C-terminal domain has at least a structural role important for protein stability, and possibly contributes in the interaction with DdrO, as suggested by modeling of the IrrE-DdrO complex (Supplementary Fig. S7)19. IrrE lacking its C-terminal domain was also expressed, under control of the irrE promoter, in a D. deserti irrE deletion mutant. Strikingly, some DdrO cleavage was observed after exposure of the cells to zinc shock, showing that the GAF-like domain is not absolutely required for the induction of IrrE protease activity in D. deserti (Fig. 5b; Supplementary Fig. S8).

DdrO cleavage by a truncated IrrE lacking its GAF-like domain. (a) SDS-PAGE showing cleavage of DdrO when co-expressed with IrrE or IrrE lacking its GAF-like domain (IrrEΔGAF) in E. coli (lanes 4 to 9). The same gel contains purified DdrO (lane 1), IrrE (lane 2) and the products of in vitro cleavage (10 min 37 °C; lane 3). IrrE and IrrEΔGAF have an N-terminal His-tag, DdrO a C-terminal His-tag. M, molecular weight marker proteins (masses in kDa). (b) Western blots showing induced cleavage of DdrO by IrrEΔGAF in D. deserti. Strain RD42 (ΔirrE) was transformed with a derivative of plasmid pI3 containing the indicated cloned gene, and DdrO cleavage was analyzed after zinc shock (250 μM for 10 min). In IrrEΔGAF-SPA, the GAF-like domain is replaced by a SPA-tag. Except for the strain without cloned gene, induced DdrO cleavage is (faintly) visible in each strain (appearance of cleavage product and decrease of intact DdrO). The cropped blots (1 and 2) are from two independent series of experiments (uncropped blots are shown in Supplementary Fig. S9). A third independent experiment is shown in Supplementary Fig. S8. P, 20 ng purified DdrO.
Discussion
The deinococcal irrE and ddrO genes have been described for the first time more than 15 years ago, with irrE being required for radiation resistance and radiation-induced expression of recA53, and ddrO as one of the genes highly induced after radiation or desiccation16. The precise function of IrrE and DdrO remained unknown for many years until we showed that IrrE and DdrO function together, which led to the description of a novel stress response mechanism where metalloprotease IrrE cleaves and inactivates transcriptional repressor DdrO when the cells are exposed to radiation14, thereby inducing expression of RDR regulon genes (e.g. recA and other DNA repair genes)17. Very recently we showed that DdrO consists of two domains: the predicted N-terminal DNA-binding domain, and a C-terminal dimerization domain showing a new fold19. Cleavage of this C-terminal domain by IrrE abolishes dimerization and DNA binding of DdrO. Insight in the protease activity of IrrE in Deinococcus, and its stimulation, was obtained in the present study, offering a more complete view of the regulatory mechanism (Fig. 6). We showed that DdrO cleavage by IrrE in Deinococcus cells is dependent on zinc ions, and, after having investigated several possible activation mechanisms, that this in vivo activity is stimulated directly by an increase in available zinc ions very likely resulting from modification in the cell redox status.

Proposed model of the IrrE/DdrO-controlled stress response mechanism. (a) Under standard conditions, repressor DdrO exist as a dynamic equilibrium between monomers and free and DNA-bound dimers. DdrO dimerization is mediated by its C-terminal domain. Binding of DdrO dimer to a conserved target DNA motif located in the promoter region of IrrE/DdrO-regulated genes represses their transcription. The majority of metalloprotease IrrE is inactive. (b) Exposure to conditions such as radiation generates oxidative stress through formation of ROS (step 1), which can cause oxidation of cysteine residues of zinc/cysteine sites in proteins (step 2), concomitantly causing release of zinc ions from these sites and a transient increase in the intracellular concentration of free, available zinc ions (step 3). The released zinc functions as second messenger, and increases the amount of active zinc-bound IrrE that cleaves the C-terminal domain of monomeric DdrO (step 4) abolishing its dimerization and shifting the DdrO equilibrium toward cleavable monomers. The diminished amount of DdrO leads to induced expression of the IrrE/DdrO-regulated genes (including several DNA repair genes and ddrO itself) (step 5). A zinc shock directly augments the intracellular free zinc ion concentration. Figure based on data obtained here and previously14,17,19.
IrrE-mediated DdrO cleavage in Deinococcus is induced after exposure to radiation, desiccation, mitomycin C, H2O2 and zinc shock, indicating that these conditions share a common intracellular signal stimulating the cleavage. Radiation, desiccation, mitomycin C and H2O2 have in common that they induce DNA damage and oxidative stress6. The latter may involve formation of oxygen-radical species through the Fe2+-catalyzed Fenton reaction. However, the zinc shock-induced DdrO cleavage was not accompanied by DNA damage or ROS production and was not inhibited by an Fe2+ chelator.
Many proteins and enzymes bind Zn2+, this ion usually having a catalytic or structural role. At catalytic sites, such as in IrrE, the zinc ion is often bound to histidine, glutamic acid and aspartic acid residues. At structural sites, the Zn2+ is often bound to the thiolates of four cysteines (e.g. two pairs of the CXXC motif) or to a combination of cysteine and histidine residues35,52. Such zinc-binding residue pairs may also be part of a protein dimer interface34, as proposed, for example, regarding dimerization of the DNA repair protein SbcC through a zinc-bridge coordinated by CXXC motifs of two monomers49. Impaired redox homeostasis and increased ROS content can result in oxidation of the thiolates that coordinate Zn2+ and destroy the Zn2+-binding site, resulting in rapid release of zinc ions that can subsequently bind to other proteins31,33,34. Oxidation or other modifications of the zinc-bound thiolate may occur directly by various ROS or other reactive species33,34,47,48, or indirectly by upstream redox sensors that transmit oxidizing equivalents54,55,56,57,58. Hsp33, Trx2, FurS, RsrA, RslA and DksA are examples of bacterial proteins for which oxidative stress-induced zinc release has been reported49,56. These proteins have been studied because of their role in sensing and responding to oxidative stress. Their Zn/Cys centers function as redox switches, and oxidation/reduction regulates the activity of these proteins. For example, under non-stress condition the four conserved Cys residues of Hsp33 bind a zinc ion and Hsp33 is inactive. Oxidants such as H2O2 and hydroxyl radicals oxidize the Zn/Cys center, inducing zinc release and intramolecular disulfide bond formation, and Hsp33 can acquire its chaperone activity after conformational rearrangement31,59. Besides the redox-induced modification of the activity of these proteins, the released zinc may participate in other pathways. Of note, Hsp33 proteins are highly conserved in Deinococcus7. Although there are lower levels of Fe2+-catalyzed oxidative protein damage in Deinococcus compared to radiation-sensitive species9, it can be assumed that proteins like Hsp33 function in the same way in Deinococcus as in other bacteria, that is, involving oxidation of Cys residues (with concomitant release of zinc ions). Many other Deinococcus proteins, such as FUR family members and Trx2 (see Results), may release zinc upon oxidation of their Zn/Cys sites.
Zinc shock and oxidative stress have thus in common that they transiently augment the intracellular concentration of free zinc ions (i.e. not bound to proteins). We propose that released zinc acts as a second messenger that signals redox imbalance and, by becoming more available to IrrE, increases the amount of active zinc-bound IrrE to induce the radiation response (Fig. 6). Under standard conditions, bacterial cells maintain an extremely low concentration of free zinc (less than one atom per cell), and most zinc ions are bound to proteins or other molecules60. Even a small increase in free zinc can thus be a very potent signal, which can be sensed by zinc-responsive proteins (e.g. IrrE in Deinococcus). A role of released zinc as second messenger in regulation of enzyme activities has been described for eukaryotic cells33,34,35, but such a role in induction of an oxidative stress/DNA damage response in bacteria has, to our knowledge, not been described previously. Remarkably, many DNA replication and repair proteins have Zn/Cys sites (Table 1). It is tempting to speculate that destruction of their zinc-binding sites inhibits their function, while the mobilized zinc stimulates IrrE activity and expression of DNA repair genes.
Genes encoding IrrE/DdrO-like protein pairs consisting of a putative metalloprotease (COG2856; ImmA/IrrE family) and a transcriptional regulator (XRE family) have been identified in many bacteria and bacteriophages14,27,29. Only a few have been studied experimentally, which showed a stress-induced inactivation of the XRE family repressor by the COG2856 domain-containing protein, resulting in induction of prophage or transposon excision27,28,61,62. In each case, however, it is unclear how stress activates the COG2856 protein. Of note, these other studied COG2856 domain proteins are smaller than IrrE and, unlike IrrE, do not contain an additional GAF-like domain19. As we observed protease activation of truncated IrrE lacking the GAF-like domain, this supports the proposed mechanism of IrrE activation and suggests that oxidative stress-induced release of zinc may also be involved in activation of GAF-lacking IrrE-like proteins. One of these metalloproteases, MpaR in Listeria monocytogenes, was found to be activated during intracellular growth of this pathogen in macrophages61, during which bacteria experience oxidative and nitrosative stress. Activated MpaR triggers excision of a prophage, thereby promoting virulence of L. monocytogenes61. The potential role of zinc signaling in MpaR activation is supported by another recent study that showed that nitric oxide produced by macrophages causes release of zinc from Zn/Cys sites of at least 15 proteins in the pathogen Salmonella enteritica51, including homologs of eight Zn/Cys site-containing proteins conserved in Deinococcus. The COG2856 protein Rir in Streptococcus thermophilus, a species used dairy industry, is involved in prophage induction causing cell lysis by interacting with and preventing oligomerization of repressor Crh without proteolytic cleavage of the latter, despite the presence of a predicted HEXXH zinc-binding active site in Rir28. In this case, it can be hypothesized that zinc binding stabilizes Rir or the Rir/Crh interaction. Taken together, a role for zinc as second messenger following stress exposure of bacteria may be more universal than currently recognized.
Methods
Growth media and culture conditions
The strains used in this study are listed in Supplementary Table S2. E. coli was grown in Luria–Bertani (LB) at 37 °C. D. deserti and D. radiodurans were grown at 30 °C in tenfold diluted Tryptic Soy Broth (Fluka, Sigma-Aldrich, T8907) (TSB/10) supplemented with trace elements22 or on agar (1.6%) plates containing the same growth medium. Antibiotics were used at the following concentrations for D. deserti: streptomycin, 10 µg ml−1; kanamycin, 10 µg ml−1; chloramphenicol, 2 µg ml−1. For E. coli, the antibiotics concentrations were: kanamycin, 50 µg ml−1; ampicillin, 100 µg ml−1.
Plasmids and DNA manipulations
Plasmids used are listed in Supplementary Table S3. Standard molecular biology techniques were used to construct plasmids. PCR products obtained with primers that included restriction sites were cloned in pCR4Blunt-TOPO prior to recloning in the desired vector. All cloned PCR fragments and site-directed mutations were analyzed by DNA sequencing to verify absence of potential PCR errors. Primer sequences are listed in Supplementary Table S4. D. deserti genes cloned in the E. coli expression vectors pET-TEV and pET22b are under control of the T7 promoter. Point mutations in IrrE (M18V, M243L and C116A) were introduced in pRD48, which contains an XbaI-HindIII fragment with irrE and its promoter region, using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) and appropriate primers. Then the XbaI-HindIII fragments with the mutations were recloned into pI3. The cloned XbaI-HindIII fragment in pI3-irrEΔGAF contains the promoter of irrE and the region encoding IrrE lacking its C-terminal 114 residues corresponding to the GAF-like domain. A similar DNA fragment is present in pI3-irrEΔGAF-SPA, except that it encodes an IrrE protein with its GAF-like domain replaced by a SPA-tag (69 aa) to render the truncated IrrE potentially more stable. The fusion of IrrEΔGAF with the SPA-tag was obtained with fusion PCR. The different pI3 plasmids were used to transform D. deserti RD42 as described previously46.
Exposure to UV, H2O2, MMC or zinc shock
Deinococcus strains grown to OD600 0.4 were exposed to zinc shock, H2O2, MMC or UV radiation (gamma radiation facilities in our institute have been dismantled). For UV irradiation, portions of 5 ml of the culture were exposed (in a Petri dish without lid) to UV-C (254 nm) using a Bio-Link BLX (Vilber Lourmat). After UV irradiation, cells were incubated at 30 °C for 10 min (or other times if indicated in the figures). For other exposures, H2O2 (Riedel-de Haën, 18312), MMC (Sigma-Aldrich, M0440), ZnCl2 (Sigma-Aldrich, 229997) or MnCl2 (Prolabo, 25222.233) was added to 10 ml of culture and incubation was continued (final concentrations and incubation times as indicated in the figures).
Exposure to these agents was also performed in the presence of metal chelators (TPEN for Zn2+, or DIP for Fe2+) or antioxidants (thiourea or TEMPOL) that were added at final concentrations as used previously41,63,64. For this, bacteria were grown to OD600 0.4 and then either 10–50 µM TPEN (N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine; Sigma-Aldrich, P4413), 500 µM DIP (2,2′-dipyridyl; Sigma-Aldrich, D216305), 150 mM thiourea (Sigma-Aldrich, T7875), or 10 mM TEMPOL (4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl; Sigma-Aldrich, 581500) was added and incubation was continued for 30 min before the cells were exposed to zinc shock, UV, H2O2 or MMC.
After the incubations, cells were collected by centrifugation for 1 min at 10,000g and directly lysed by heating for 10 min at 95 °C in Novex NuPAGE LDS Sample Buffer supplemented with Novex NuPAGE Sample Reducing Agent (Invitrogen) (volume corresponding to 100 µl sample buffer for 1 ml of culture with an OD600 of 1) and by passing through a syringe needle, and then frozen at − 20 °C. Protein separation by SDS-PAGE (20 μl sample per lane) and immunoblotting to analyze DdrO cleavage were performed as described previously14. A lane with 20 ng DdrO, purified as described17, was included on the Western blots. The experiments were performed in triplicate (biological replicates), and representative results are shown.
Co-expression of IrrE or IrrEΔGAF with DdrO in E. coli
Two plasmids were used for co-expression in E. coli: a pET-TEV derivative encoding IrrE or IrrEΔGAF (both with N-terminal His-tag) and a pET22b derivative encoding DdrO (with C-terminal His-tag). E. coli BL21 (AI) cells freshly transformed with the two plasmids were grown at 37 °C overnight to saturation in 10 ml of LB medium containing kanamycin and ampicillin. This pre-culture was used to inoculate (start OD600 0.05) 100 ml of LB medium with the antibiotics and grown at 37 °C with aeration. At OD600 of 0.6–0.7, IPTG (0.1 mM final concentration) and L-arabinose (0.2% final concentration) were added and the cells were further incubated at 37 °C for 3 h. Five hundred µl of induced cells were taken, centrifuged (10,000g, 2 min, 4 °C) and re-suspended in Novex NuPAGE LDS Sample Buffer (100 µl of sample buffer for 1 ml of culture at OD600 1), and heated for 10 min at 95 °C. Samples (20 μl per lane) were loaded on SDS-PAGE gels (Novex NuPAGE 10% Bis–Tris Gels; Invitrogen), and migrated in 1X NuPAGE MES SDS Running Buffer (Invitrogen) for 1 h at 130 V. In vitro cleavage control was performed as described previously14. The proteins were visualized by staining with Imperial protein stain (Pierce).
Induced mutagenesis
D. deserti strains were grown to OD600 0.4 and then 250 µM ZnCl2 or 10 mM H2O2 or nothing (control) was added, and cultures were incubated further for 10 min at 30 °C. One part (1 ml) of the cultures was taken to prepare samples for immunoblotting to analyze DdrO cleavage as described above. The other part of the cultures was centrifuged for 5 min at 5000 g, and the cell pellets were re-suspended in fresh growth medium and incubated for 20 h at 30 °C with shaking to permit fixation of mutations. Then 1 ml of each culture were spread in duplicate on plates with 10 µg ml−1 rifampicin. In parallel, 0.1 ml of serial dilutions 10−6 and 10−7 were spread in duplicate on plates without rifampicin. The entire experiment was performed in triplicate (biological replicates). Colony-forming units (CFU) were determined after incubation for 3–4 days at 30 °C. The mutation frequency was calculated by the ratio "number of RifR CFU/ml" / "total number of CFU/ml".
Zinc measurements with fluorescent probe FluoZin-3
Experiments to detect free intracellular Zn2+ were based on a previously described protocol65. D. deserti was grown to OD600 0.4. Bacteria were resuspended in fresh TSB/10 and then incubated with 5 µM FluoZin‐3, AM, cell permeant (Thermo Fisher Scientific, F24195) for 60 min at room temperature in the dark to allow complete de‐esterification of intracellular acetoxymethyl (AM) esters. The bacteria were then washed three times in TSB/10. Fluorescence measurements were performed in a microplate at 28 °C and using a spectrofluorimeter (Infinite 200 Pro; Tecan). After excitation of the sample at 485 nm, fluorescence emission was recorded at 519 nm before and after addition of H2O2 or ZnCl2.
ROS measurements with fluorescent probe H2DCFDA
ROS detection using H2DCFDA (2′,7′-Dichlorodihydrofluorescein diacetate; Sigma-Aldrich, D6883) was performed after adaptation of a previously described method66. D. deserti was grown to OD600 0.4. Bacteria were washed once in 0.85% KCl and then incubated with 25 µM H2DCFDA for 60 min at room temperature in the dark, and then washed again in 0.85% KCl. Fluorescence measurements were performed in a microplate at 28 °C and using a spectrofluorimeter (Infinite 200 Pro; Tecan). After excitation of the sample at 480 nm, fluorescence emission was recorded at 526 nm before and after addition of H2O2 or ZnCl2.
Construction of D. deserti gene deletion mutants
D. deserti strains in which ddrS (Deide_04721), bshA (Deide_11680) or bshC (Deide_13730) are deleted and replaced by a kanamycin resistance gene, were constructed as described previously for other deletion mutants46. Briefly, DNA fragments corresponding to upstream and downstream regions of the gene to be deleted, were cloned upstream and downstream of a kanamycin resistance cassette in a pUC19 derivative. Resulting plasmids, which do not replicate in D. deserti, were introduced in D. deserti, and double homologous recombination events and complete deletion of the gene of interest were verified by diagnostic PCR.
Data availability
The IrrE and DdrO genes and proteins in this study correspond to SwissProt entries C1CZ84 and C1CYP4, respectively. All data that support the findings of this study are available from the corresponding author on request.
References
Baharoglu, Z. & Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 38, 1126–1145 (2014).
Kreuzer, K. N. DNA damage responses in prokaryotes: Regulating gene expression, modulating growth patterns, and manipulating replication forks. Cold Spring Harb. Perspect. Biol. 5, a012674 (2013).
Battista, J. R. Against all odds: The survival strategies of Deinococcus radiodurans. Annu. Rev. Microbiol. 51, 203–224 (1997).
Cox, M. M. & Battista, J. R. Deinococcus radiodurans—The consummate survivor. Nat. Rev. Microbiol. 3, 882–892 (2005).
de Groot, A. et al. Alliance of proteomics and genomics to unravel the specificities of Sahara bacterium Deinococcus deserti. PLoS Genet. 5, e1000434 (2009).
Slade, D. & Radman, M. Oxidative stress resistance in Deinococcus radiodurans. Microbiol. Mol. Biol. Rev. 75, 133–191 (2011).
Lim, S., Jung, J.-H., Blanchard, L. & de Groot, A. Conservation and diversity of radiation and oxidative stress resistance mechanisms in Deinococcus species. FEMS Microbiol. Rev. 43, 19–52 (2019).
Daly, M. J. Death by protein damage in irradiated cells. DNA Repair. 11, 12–21 (2012).
Daly, M. J. et al. Protein oxidation implicated as the primary determinant of bacterial radioresistance. PLoS Biol. 5, e92 (2007).
Krisko, A. & Radman, M. Protein damage and death by radiation in Escherichia coli and Deinococcus radiodurans. Proc. Natl. Acad. Sci. USA 107, 14373–14377 (2010).
Daly, M. J. et al. Accumulation of Mn(II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science 306, 1025–1028 (2004).
Daly, M. J. et al. Small-molecule antioxidant proteome-shields in Deinococcus radiodurans. PLoS ONE 5, e12570 (2010).
de Groot, A. et al. RNA sequencing and proteogenomics reveal the importance of leaderless mRNAs in the radiation-tolerant bacterium Deinococcus deserti. Genome Biol. Evol. 6, 932–948 (2014).
Ludanyi, M. et al. Radiation response in Deinococcus deserti: IrrE is a metalloprotease that cleaves repressor protein DdrO. Mol. Microbiol. 94, 434–449 (2014).
Makarova, K. S. et al. Deinococcus geothermalis: the pool of extreme radiation resistance genes shrinks. PLoS ONE 2, e955 (2007).
Tanaka, M. et al. Analysis of Deinococcus radiodurans’s transcriptional response to ionizing radiation and desiccation reveals novel proteins that contribute to extreme radioresistance. Genetics 168, 21–33 (2004).
Blanchard, L. et al. Conservation and diversity of the IrrE/DdrO-controlled radiation response in radiation-resistant Deinococcus bacteria. Microbiologyopen 6, e477 (2017).
Devigne, A. et al. DdrO is an essential protein that regulates the radiation desiccation response and the apoptotic-like cell death in the radioresistant Deinococcus radiodurans bacterium. Mol. Microbiol. 96, 1069–1084 (2015).
de Groot, A. et al. Crystal structure of the transcriptional repressor DdrO: Insight into the metalloprotease/repressor-controlled radiation response in Deinococcus. Nucleic Acids Res. 47, 11403–11417 (2019).
Wang, Y. et al. Protease activity of PprI facilitates DNA damage response: Mn2+-dependence and substrate sequence-specificity of the proteolytic reaction. PLoS ONE 10, e0122071 (2015).
Lewis, R. J., Brannigan, J. A., Offen, W. A., Smith, I. & Wilkinson, A. J. An evolutionary link between sporulation and prophage induction in the structure of a repressor:Anti-repressor complex. J. Mol. Biol. 283, 907–912 (1998).
Vujicic-Zagar, A. et al. Crystal structure of the IrrE protein, a central regulator of DNA damage repair in Deinococcaceae. J. Mol. Biol. 386, 704–716 (2009).
Fukasawa, K. M., Hata, T., Ono, Y. & Hirose, J. Metal preferences of zinc-binding motif on metalloproteases. J. Amino Acids 2011, 574816 (2011).
Holmquist, B. & Vallee, B. L. Metal substitutions and inhibition of thermolysin: spectra of the cobalt enzyme. J. Biol. Chem. 249, 4601–4607 (1974).
Shi, R., McDonald, L., Cygler, M. & Ekiel, I. Coiled-coil helix rotation selects repressing or activating state of transcriptional regulator DhaR. Structure 22, 478–487 (2014).
Zoraghi, R., Corbin, J. D. & Francis, S. H. Properties and functions of GAF domains in cyclic nucleotide phosphodiesterases and other proteins. Mol. Pharmacol. 65, 267–278 (2004).
Bose, B., Auchtung, J. M., Lee, C. A. & Grossman, A. D. A conserved anti-repressor controls horizontal gene transfer by proteolysis. Mol. Microbiol. 70, 570–582 (2008).
Koberg, S., Mohamed, M. D. A., Faulhaber, K., Neve, H. & Heller, K. J. Identification and characterization of cis- and trans-acting elements involved in prophage induction in Streptococcus thermophilus J34. Mol. Microbiol. 98, 535–552 (2015).
Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 4, 19 (2009).
Matic, I. The major contribution of the DNA damage-triggered reactive oxygen species production to cell death: Implications for antimicrobial and cancer therapy. Curr. Genet. 64, 567–569 (2018).
Ilbert, M., Graf, P. C. & Jakob, U. Zinc center as redox switch–new function for an old motif. Antioxid. Redox Signal. 8, 835–846 (2006).
Imlay, J. A. Transcription factors that defend bacteria against reactive oxygen species. Annu. Rev. Microbiol. 69, 93–108 (2015).
Kröncke, K. D. & Klotz, L. O. Zinc fingers as biologic redox switches?. Antioxid. Redox Signal. 11, 1015–1027 (2009).
Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox Signal. 8, 1419–1441 (2006).
Wouters, M. A., Fan, S. W. & Haworth, N. L. Disulfides as redox switches: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 12, 53–91 (2010).
Dapa, T., Fleurier, S., Bredeche, M.-F. & Matic, I. The SOS and RpoS regulons contribute to bacterial cell robustness to genotoxic stress by synergistically regulating DNA polymerase Pol II. Genetics 206, 1349–1360 (2017).
Polyakov, N. et al. Redox-active quinone chelators: Properties, mechanisms of action, cell delivery, and cell toxicity. Antioxid. Redox Signal. 28, 1394–1403 (2018).
Verweij, J. & Pinedo, H. M. Mitomycin C: Mechanism of action, usefulness and limitations. Anticancer Drugs 1, 5–13 (1990).
Kelner, M. J., Bagnell, R. & Welch, K. J. Thioureas react with superoxide radicals to yield a sulfhydryl compound. Explanation for protective effect against paraquat. J. Biol. Chem. 265, 1306–1311 (1990).
Kohanski, M. A., Dwyer, D. J., Hayete, B., Lawrence, C. A. & Collins, J. J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810 (2007).
Latifi, A., Jeanjean, R., Lemeille, S., Havaux, M. & Zhang, C.-C. Iron starvation leads to oxidative stress in Anabaena sp. strain PCC 7120. J. Bacteriol. 187, 6596–6598 (2005).
Wilcox, C. S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 126, 119–145 (2010).
Wilcox, C. S. & Pearlman, A. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol. Rev. 60, 418–469 (2008).
Ezraty, B., Gennaris, A., Barras, F. & Collet, J. F. Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 15, 385–396 (2017).
Shumaker, D. K., Vann, L. R., Goldberg, M. W., Allen, T. D. & Wilson, K. L. TPEN, a Zn2+/Fe2+ chelator with low affinity for Ca2+, inhibits lamin assembly, destabilizes nuclear architecture and may independently protect nuclei from apoptosis in vitro. Cell Calcium 23, 151–164 (1998).
Dulermo, R., Fochesato, S., Blanchard, L. & de Groot, A. Mutagenic lesion bypass and two functionally different RecA proteins in Deinococcus deserti. Mol. Microbiol. 74, 194–208 (2009).
Maret, W. Zinc in cellular regulation: The nature and significance of ‘zinc signals’. Int. J. Mol. Sci. 18, 2285 (2017).
Maret, W. The redox biology of redox-inert zinc ions. Free Radic. Biol. Med. 134, 311–326 (2019).
Ortiz de Orue Lucana, D., Wedderhoff, I. & Groves, M. R. ROS-mediated signalling in bacteria: zinc-containing Cys-X-X-Cys redox centres and iron-based oxidative stress. J. Signal Transduct. 2012, 605905 (2012).
Ma, Z. et al. Bacillithiol is a major buffer of the labile zinc pool in Bacillus subtilis. Mol. Microbiol. 94, 756–770 (2014).
Frawley, E. R. et al. Nitric oxide disrupts zinc homeostasis in Salmonella enterica serovar Typhimurium. mBio 9, 66 (2018).
Andreini, C., Banci, L., Bertini, I. & Rosato, A. Zinc through the three domains of life. J. Proteome Res. 5, 3173–3178 (2006).
Earl, A. M., Mohundro, M. M., Mian, I. S. & Battista, J. R. The IrrE protein of Deinococcus radiodurans R1 is a novel regulator of recA expression. J. Bacteriol. 184, 6216–6224 (2002).
Boronat, S. et al. Thiol-based H2O2 signalling in microbial systems. Redox Biol. 2, 395–399 (2014).
García-Santamarina, S. et al. Is oxidized thioredoxin a major trigger for cysteine oxidation? Clues from a redox proteomics approach. Antioxid. Redox Signal. 18, 1549–1556 (2013).
Kim, J.-S. et al. DksA-DnaJ redox interactions provide a signal for the activation of bacterial RNA polymerase. Proc. Natl. Acad. Sci. USA 115, E11780–E11789 (2018).
Stewart, E. J., Aslund, F. & Beckwith, J. Disulfide bond formation in the Escherichia coli cytoplasm: An in vivo role reversal for the thioredoxins. EMBO J. 17, 5543–5550 (1998).
Stöcker, S., Maurer, M., Ruppert, T. & Dick, T. P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 14, 148–155 (2018).
Ilbert, M. et al. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat. Struct. Mol. Biol. 14, 556–563 (2007).
Wang, D., Hosteen, O. & Fierke, C. A. ZntR-mediated transcription of zntA responds to nanomolar intracellular free zinc. J. Inorg. Biochem. 111, 173–181 (2012).
Argov, T. et al. Coordination of cohabiting phage elements supports bacteria-phage cooperation. Nat. Commun. 10, 5288 (2019).
Bose, B. & Grossman, A. D. Regulation of horizontal gene transfer in Bacillus subtilis by activation of a conserved site-specific protease. J. Bacteriol. 193, 22–29 (2011).
Dewachter, L., Herpels, P., Verstraeten, N., Fauvart, M. & Michiels, J. Reactive oxygen species do not contribute to ObgE*-mediated programmed cell death. Sci. Rep. 6, 33723 (2016).
Kashyap, D. R. et al. Peptidoglycan recognition proteins kill bacteria by inducing oxidative, thiol, and metal stress. PLoS Pathog. 10, e1004280 (2014).
Bayle, L. et al. Zinc uptake by Streptococcus pneumoniae depends on both AdcA and AdcAII and is essential for normal bacterial morphology and virulence. Mol. Microbiol. 82, 904–916 (2011).
Hakkila, K. et al. Oxidative stress and photoinhibition can be separated in the cyanobacterium Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 1837, 217–225 (2014).
Acknowledgements
We thank R. Dulermo, M. Ludanyi and B. Alonso for construction of some of the strains and plasmids, C. Brutesco for help with fluorescence spectroscopy, F. Confalonieri for helpful discussions, and D. Pignol and C. Carles for constant support. This work was funded by the Transverse Division n◦4 (Radiobiology) of the French Alternative Energies and Atomic Energy Commission (Programme n◦ 4 Radiobiologie) and by a grant from the Agence Nationale de la Recherche (ANR-19-CE12-0010). R.M. was supported by a Phare Ph.D. fellowship from the French Alternative Energies and Atomic Energy Commission.
Author information
Authors and Affiliations
Contributions
R.M. and A.d.G. designed and conceived the experiments. R.M. performed all experiments. L.B. and A.d.G. supervised the execution of the experiments. A.d.G. searched the D. deserti genome for Zn/Cys proteins. R.M., P.R., L.B. and A.d.G analyzed the data. A.d.G. wrote the paper with contributions from R.M., P.R. and L.B. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magerand, R., Rey, P., Blanchard, L. et al. Redox signaling through zinc activates the radiation response in Deinococcus bacteria. Sci Rep 11, 4528 (2021). https://doi.org/10.1038/s41598-021-84026-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-84026-x
This article is cited by
-
The Deinococcus protease PprI senses DNA damage by directly interacting with single-stranded DNA
Nature Communications (2024)
-
Engineered autonomous dynamic regulation of metabolic flux
Nature Reviews Bioengineering (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
