
Abstract
In the ferruginous and anoxic early Earth oceans, photoferrotrophy drove most of the biological production before the advent of oxygenic photosynthesis, but its association with ferric iron (Fe3+) dependent anaerobic methane (CH4) oxidation (AOM) has been poorly investigated. We studied AOM in Kabuno Bay, a modern analogue to the Archean Ocean (anoxic bottom waters and dissolved Fe concentrations > 600 µmol L−1). Aerobic and anaerobic CH4 oxidation rates up to 0.12 ± 0.03 and 51 ± 1 µmol L−1 d−1, respectively, were put in evidence. In the Fe oxidation–reduction zone, we observed high concentration of Bacteriochlorophyll e (biomarker of the anoxygenic photoautotrophs), which co-occurred with the maximum CH4 oxidation peaks, and a high abundance of Candidatus Methanoperedens, which can couple AOM to Fe3+ reduction. In addition, comparison of measured CH4 oxidation rates with electron acceptor fluxes suggest that AOM could mainly rely on Fe3+ produced by photoferrotrophs. Further experiments specifically targeted to investigate the interactions between photoferrotrophs and AOM would be of considerable interest. Indeed, ferric Fe3+-driven AOM has been poorly envisaged as a possible metabolic process in the Archean ocean, but this can potentially change the conceptualization and modelling of metabolic and geochemical processes controlling climate conditions in the Early Earth.
Similar content being viewed by others

Introduction
Tropical inland waters and wetlands have been recognized as major sources of methane (CH4) to the atmosphere1. While progress has been made in refining the evaluation of the CH4 emission rates, less attention has been given to evaluate the underlying production and loss terms, i.e. methanogenesis and methane oxidation. In modern marine sediments where sulfate (SO42−) is more abundant by several orders of magnitude than any other electron acceptor, most of the CH4 removal is due to anaerobic CH4 oxidation (AOM) coupled to SO42− reduction2,3,4,5. However, SO42− abundance is typically much lower in freshwaters compared to marine ecosystems, so that CH4 oxidation in anoxic hypolimnion or sediments of lakes might be linked to the reduction of thermodynamically more favorable electron acceptors such as nitrite (NO2−)6, nitrate (NO3−)7, manganese IV (Mn4+) and ferric iron (Fe3+)8.
Kabuno Bay is a ferruginous, nearly isolated, sub-basin of Lake Kivu (RD Congo) with a marked and distinct physico-chemistry. Primarily due to high hydrothermal activity, a strong and stable stratification is established within Kabuno bay water column throughout the year, with waters being anoxic below ~ 11 m depth9,10. A consequence of this strong stratification is the occurrence of a particularly steep gradient in CH4 and iron (Fe2+/Fe3+) concentrations in the chemocline. Also, anoxic waters of Kabuno Bay are characterized by low sulfide (HS−) concentrations11. These combined features are rarely encountered in modern environments, Lake Matano (Indonesia) and Lake La Cruz (Spain) being one of the few others12, while they were widespread in the Archean ocean13. Llirós et al.11 reported the occurrence of a particularly active pelagic Fe cycle driven by photoferrotrophy in Kabuno Bay, with little net Fe oxidation, meaning that Fe reduction processes are tightly coupled to photoferrotrophic Fe oxidation. In the present study, we measured CH4 oxidation rates in the water column of Kabuno Bay, and investigated the potential importance of Fe3+ as a terminal electron acceptor for AOM. We hypothesized that Fe3+ could be the main electron acceptor for AOM given the high abundance of Fe species in the water column and the high in situ photoferrotrophic rates previously reported in Kabuno Bay11,14.
Material and methods
Description of the study site and the sampling device
Kabuno Bay (− 1.6216°N, 29.0497°E; Figure S1) was sampled in May 2013 (late rainy season), September 2013 (dry season) and August 2014 (dry season)15. Vertical profiles of temperature, conductivity, pH and oxygen were obtained with a Yellow Springs Instrument (YSI) 6600 V2 multiparameter probe, with a detection limit for dissolved oxygen of 0.01 mg L−1. High amounts of dissolved gases (in particular CO2) were present in superficial and deep waters of Kabuno Bay, causing losses of CH4 when samples were brought to the surface. To avoid that, a home-made sampler (Figure S2) was used; sealed N2-flushed 60 mL glass serum bottles (SUPELCO, Sigma Aldrich, 33109-U) were fixed on a two-meter high plate, every 0.25 m. Thin needles (0.6 × 25 mm) equipped with non-return valves (valves allowing the water to fill in the bottles but preventing gases to escape from the bottles) penetrated the grey butyl stoppers (WHEATON, USA). The non-return valves were sealed by a butyl stopper. A string was connected to stoppers in series (all stoppers were connected to the same string). The device was immersed at the sampling depth, and the stoppers were removed from the non-return valves by pulling the string, allowing water to enter the bottles through the needle. The system was left under water 10 min to fill the serum bottles. Once the sampling device was brought back to the surface, the needles were removed from the butyl stoppers and further processed as described below. Serum bottles were half-filled with water, and the other half was a N2 headspace.
The five main rivers of Kabuno Bay (Figure S1) were sampled every month from November 2013 to June 2014, by the mean of a Niskin bottle. Samples for total Fe and Mn concentrations determination were taken in plastic vials, stored at 4 °C and analyzed as described hereafter.
Chemical analyses
Samples for CH4 concentrations were collected in sealed (with butyl stoppers previously boiled in milli-Q water in the laboratory, and aluminium caps) N2-flushed 60 mL glass serum bottles, as described above. Two bottles were directly poisoned with 100 µL of HgCl2. CH4 concentrations were determined via the headspace equilibration technique and measured by gas chromatography (GC)16, as described by Borges et al.1. The precision of measurements was ± 3.9% and the detection limit of the method is 0.5 nmol L−1.
Samples for nutrient analyses were collected into 250 mL borosilicate bottles, using the same sampling device as for the gas sampling. Water was then collected from the bottles with a 50 mL-syringe, filtered through a 0.22 µm syringe filter (polyethylsulfone), preserved with 200 µL of H2SO4 5 N, and stored frozen. Nitrite (NO2−) and NO3− concentrations were measured by spectrophotometry, by the sulfanilamide method17 and the vanadium reduction to NO2− method18, respectively, while ammonia (NH4+) was determined with the dichloroisocyanurate–salicylate–nitroprussiate colorimetric method19. NO2− and NH4+ were quantified on a Thermo Spectronic Genesys 10vis spectrophotometer using a 5-cm light path, and NO3– was determined with a Multiskan Ascent Thermo Scientific multi-well plate reader. The detection limits for these methods were 0.03, 0.15 and 0.3 µmol L−1 for NO2−, NO3− and NH4+, respectively. The concentrations for NO3− and NO2− are reported here as NOx concentrations (NO3− + NO2−). Nutrients concentrations are not available in May 2013, due to a problem during samples preservation.
Samples for SO42− and sulfide (HS−) concentrations were collected in N2-flushed 60 mL serum bottles, by the same sampling method as described above. Water was rapidly filtered after collection through a 0.22 µm syringe filter, and collected in 5 mL Cryotube vials and 50 mL plastic vials for SO42− and HS−, respectively. Samples were preserved with 20 µL of 20% zinc acetate (ZnAc), for SO42− and 200 µL of ZnAc for HS−; both samples were then stored frozen. SO42− concentrations were quantified by ion chromatography (Dionex ICS-1500, with an autosampler Dionex AS50, a guard column Dionex AG22 and an analytical column Dionex IonPac AS22) and HS− concentrations were determined with a Thermo Spectronic Genesys 10vis spectrophotometer, using a 5-cm light path, according to the method described by Cline20. The detection limits were 0.5 and 0.25 µmol L−1 for SO42− and HS−, respectively.
Samples for Fe and Mn measurements were collected into sealed N2-flushed 60 mL glass serum bottles, with the sampler described above. Water was rapidly transferred from the bottles to the filtration set with a syringe equipped with a tube, and was passed through 25 mm glass fiber filters15. Filters were collected in 2 mL Eppendorf vials and preserved with 1 mL of a HNO3− 2% solution, while filtrates were collected into four 2 mL Eppendorf vials and preserved with 20 µL of a HNO3 65% solution. Particulate Fe and Mn concentrations were determined from the filters, which were digested with nitric acid in Teflon bombs in a microwave digestion apparatus (Ethos D, Milestone Inc.) and diluted with milli-Q water to a final volume of 50 mL. Dissolved Mn and Fe concentrations were determined from the filtrates, which were diluted with milli-Q water to a final volume of 50 mL. Fe and Mn concentrations were determined by inductively coupled plasma mass spectrometry (ICP-MS) using dynamic reaction cell (DRC) technology (ICP-MS SCIEX ELAN DRC II, PerkinElmer inc.). Analytical accuracy was verified by a certified reference material (BCR 715, Industrial Effluent Wastewater).
CH4 oxidation rate measurements
Samples for CH4 oxidation incubations were collected in N2-flushed 60 mL glass serum bottles sealed with butyl stoppers previously boiled in milli-Q water in the laboratory, and aluminum caps, using the sampling device described above. CH4 oxidation was determined following the methodology described by Roland et al.21. Briefly, two bottles were immediately poisoned with 100 µL of HgCl2 after collection (T0), five bottles received an inhibitor of sulfate-reducing bacteria (sodium molybdate, + Mo) and five other did not receive any amendment (− Mo). In May 2013, the molybdate (Mo) solution was prepared directly on the field with milli-Q water stored at ambient temperature and was not flushed, while it was prepared with fresh milli-Q water and was flushed before the transport in September 2013 and August 2014. The bottles of both treatments were incubated in the dark and at constant temperature close to in situ temperature (~ 23 °C). The biological activity in + Mo and – Mo bottles was stopped at ~ 12, 24, 48, 72 and 96 h by the addition of 100 µL of HgCl2. CH4 concentrations were determined via the headspace equilibration technique and measured by gas chromatography (GC)16, as described by Borges et al.1. The precision of measurements was ± 3.9% and the detection limit of the method is 0.5 nmol L−1. The precision was calculated based on the analysis of the two T0 bottles sampled in duplicate for each depth and then accounted for the variability induced by the handling of samples (samples collection, storage) and our analytical method.
CH4 oxidation rates were calculated as a linear regression of CH4 concentrations over time during the course of the incubation. Table S1 shows standard deviations, initial CH4 concentrations, percentage of CH4 consumed and the time lapse during which the CH4 oxidation rates were calculated for each depth15.
A correction of the CH4 oxidation rates has been applied taking into account the potential oxygen supply through the injection of the Mo solution. We considered that a maximum of 2.5 µmol L−1 of O2 were added to each bottle (250 µL of the solution were added to 30 mL of water). The calculations were made according to Roland et al.21. Only the time change in dissolved CH4 concentration and not the concomitant decrease in the concentration of the electron acceptors potentially involved in CH4 oxidation processes were monitored during the incubation. This is due to the interference caused by the HgCl2 poison addition with the analytical methods used to determine the electron acceptors concentrations.
Vertical flux calculations
The vertical fluxes (Fvertical) of NH4+, SO42−, HS−, Mn2+ and Fe2+ were calculated as described by Pasche et al.22 (Eq. 1):
where Dturbulent is the turbulent diffusion coefficient, Grad is the vertical concentration gradient of each element, C is the concentration of the element at a given depth, and Adv is the upwelling velocity. Vertical fluxes were computed by using a range of turbulent diffusion coefficient and of upwelling velocity of 1.4 × 10–7–1.0 × 10–6 m2 s−1 and 6.3 × 10–9–6.3 × 10–8 m s−1, respectively14.
Contribution to CH4 oxidation
Based on these vertical fluxes, the fraction of the integrated AOM rates potentially sustained by Fe reduction, NO3− reduction and SO42− reduction rates were calculated considering stoichiometry equivalences of 1:1 (SO42−:CH4), 8:5 (NO3−:CH4), 8:1 (Fe(OH)3:CH4) and 4:1 (MnO2:CH4)8,23, according to the following equations:
Bacteriochlorophyll pigments analyses
Samples for pigments analyses were collected every 0.25 m, from 9 to 13 m depth in September 2013, and from 8 to 12 m depth in August 2014. Water was collected with the sampler described above, and filtered through Whatman GF/F 47 mm diameter filters. The filtration volume depended on the depth sampled, but was on average 0.3 L. Filters were preserved in 5 mL Cryotube vials and stored frozen. The pigment extraction was made in 4 mL of 90% HPLC grade acetone. Two 15-min sonication steps separated by an overnight period at 4 °C in dark were applied, and extracts were stored in 2 mL-amber borosilicate vials. HPLC analyses were carried out as described by Sarmento et al.24.
Archaeal diversity
Samples for DNA analyses were collected as detailed by Inceoğlu et al.25. Genomic DNA was extracted as previously described26 and further subjected to FLX–titanium amplicon pyrosequencing27 from collected filters using ARCH 349F (5′-GYGCASCAGKCGMGAAW-3′) and ARCH 806R (5′-GGACTACVSGGGTATCTAAT-3′) as sequencing primers targeting the 16S rRNA V3–V4 region27. Reads used in the present study can be accessed through sequencing read archive (SRX349388). Archaeal 454-pyrosequencing sequences from Kabuno Bay water samples tentatively belonging to the Methanosarcinales order were subsequently analysed for further taxonomic refinement. All sequences were aligned using the SINA aligner28 and then imported into the latest SILVA 16S rRNA-ARB-compatible database (SSURef-132_NR_99_13_12_17_opt.arb; http://www.arb-silva.de) in ARB29. Two base frequency filters (“termini” and “ssuref:archaea”; positional variability by parsimony) were applied to exclude highly variable positions before adding sequences to the original database using the “parsimony quick add marked” tool from ARB.
Results and discussion
Environmental settings in Kabuno Bay
The water column of Kabuno Bay was sharply stratified and anoxic from 11.0 to 11.3 m depth during the three field campaigns (2013–2014; Fig. 1, Figure S3). Chemocline co-occurred with the oxycline. Methane was abundant in anoxic waters (up to ~ 200 µmol L−1 at 13 m) but its concentration decreased abruptly at the bottom of the chemocline to relatively modest values (0.1–1.1 µmol L−1), indicative of vigorous microbial CH4 oxidation. Ferruginous Kabuno Bay hypolimnion was characterized by high Mn2+ and Fe2+ concentrations (up to 55 and 600 µmol L−1, respectively; Figs. 1, 2). Dissolved Mn and Fe concentrations declined in the CH4 gradient, and were mirrored by an accumulation of particulate Mn and Fe species.

Physico-chemical conditions in Kabuno Bay are analogous to the Archean Ocean (a,b: May 2013; c,d: September 2013; e,f: August 2014). (a,c,e): bacteriochlorophyll contents (Bchle, µg L−1), dissolved oxygen (DO, µmol L−1) and CH4 concentrations (µmol L−1), specific conductivity (SPC, µS cm−1). (b,d,f): methane oxidation rates (µmol L−1 d−1) without molybdate added (− Mo) and with molybdate added (+ Mo).

Vertical profiles of electron acceptors potentially involved in AOM (a–c: May 2013; d–g: September 2013; h–k: August 2014). (d,h): NOx and NH4+ concentrations (µmol L−1). (a,e,i): SO42− and HS− concentrations (µmol L−1). (b,f,j(: particulate (MnO2) and dissolved (Mn2+) Mn concentrations (µmol L−1). (c,g,k(: particulate (Fe3+) and dissolved (Fe2+) Fe concentrations (µmol L−1).
Sulfate concentrations were relatively high in the chemocline and did not change substantially within the CH4 gradient, while HS− concentrations were always two orders of magnitude lower than SO42− (i.e., lower than 1.0 µmol L−1), with the exception of a maximum peak (10 µmol L−1) detected at 9.5 m depth in September 2013 (Fig. 2). The vertical fluxes of the potential electron acceptors for AOM and their reduced forms are shown in Table S2.
Five main rivers enter Kabuno Bay and are sources of Fe and Mn to the lake. Based on data gathered during a monthly monitoring of these rivers, we estimated that they supplied the lake with 3.3 mmol m−2 d−1 of Fe3+ oxide and 0.1 mmol m−2 d−1 of Mn4+ oxide.
Electron acceptors sustaining AOM in Kabuno Bay
Methane oxidation rates in oxic waters (maximum of 0.12 ± 0.03 µmol L−1 d−1 observed in May 2013) were one order of magnitude lower than those in anoxic waters with maximum rates of 51 ± 1 (at 11.5 m depth), 21 ± 4 (at 11.5 m) and 48 ± 7 (at 12.0 m) µmol L−1 d−1 in May 2013, September 2013 and August 2014, respectively (Fig. 1). Methane removal in anoxic waters by aerobic organisms has been found to be supported by oxygenic photosynthesis in the well-illuminated (10% of incident PAR) chemocline of Lake Cadagno30. Kabuno Bay chemocline is located below the photic zone so that light conditions (0.1–1% PAR) do not appear suitable to support significant phytoplankton activity. Pigments analysis carried out during our study revealed that the abundance of bacteriochlorophyll e (Bchl e), a pigment distinctive of low light adapted anoxygenic photoautotrophs Green Sulfur Bacteria (GSB)31,32, was at least one order of magnitude higher than chlorophyll a in the chemocline (Figs. 1 and S4). Also, Morana et al.14 showed that 74 ± 13% of particulate biomass in the chemocline derive from anoxygenic CO2 fixation by GSB. These multiple lines of evidence indicate that biological primary production in the chemocline is largely dominated by anoxygenic photoautotrophs so that a cryptic oxygen cycle sustained by oxygenic photosynthesis in Kabuno Bay anaerobic waters can be ruled out. Nevertheless, we cannot rule out the possibility of punctual and/or episodic oxygen incursions in the anoxic waters, which could explain the presence of NOx below the oxycline in September 2013. This oxygen could be rapidly used by aerobic CH4 oxidation, and the part of aerobic CH4 oxidation in the water column of Kabuno Bay might be more significant than reflected by the results of our in vitro incubations.
However, in the particular conditions of our in vitro incubations, we could estimate that 89–98% of the CH4 was oxidized under anoxic conditions, raising the question which electron acceptors supported AOM. The water column was particularly rich in SO42−, but SO42− concentrations did not decrease substantially with depth in the chemocline (Fig. 2), neither in anoxic waters. Indeed, SO42− reduction rates reported by Llirós et al.11 were relatively low compared to SO42− concentrations and were ca. 10 times lower than the AOM rates. Furthermore, except in May 2013 where AOM rates were lower with Mo added (what can be linked to the slightly different methodology used), our in situ incubation experiments revealed that AOM rates were up to 6 times higher (September 2013 and August 2014) in presence of Mo, an inhibitor of SO42− reduction (Fig. 1), contrary to what would have been expected if CH4 oxidation depended on SO42− reduction. All these multiple lines of evidence suggest that SO42− reduction did not sustain a significant part of the CH4 oxidation in the chemocline of Kabuno Bay. Similarly, it seems unlikely that Mn oxides and NO3− would fuel a substantial part of AOM. Their concentrations were always low, and the upward fluxes of Mn2+ and NH4+ could only have sustained at most 4% and 25% of the AOM rates, considering an extreme scenario where the totality of Mn2+ and NH4+ fluxes, once oxidized, would be exclusively involved in Mn oxides or NO3–dependent AOM, which is unlikely. Instead, it has been showed that denitrification and NO3− reduction to NH4+ (DNRA) linked to Fe oxidation occur in the water column of Kabuno Bay33. As both these processes are thermodynamically more favorable than NO3– driven AOM, we hypothesized that the NO3− formed in the water column of Kabuno Bay is rapidly consumed by DNRA and denitrification coupled to Fe oxidation rather than by NO3–driven AOM, which can also explain the low NO3− concentrations. Therefore, it is likely that AOM coupled to NO3− reduction did not occur at a significant extent in Kabuno Bay.
On the other hand, Llirós et al.11 showed that Fe3+ reduction in Kabuno Bay was ca. 24 times higher than SO42− reduction. This study also reported the existence of an important Fe-related bacterial community in the water column of Kabuno Bay, among which Chlorobium phaeoferrooxidans, a GSB capable of Fe oxidation34 was the dominant member. Here, we showed that Bchl e, a specific biomarker of the GSB, was mainly located in the Fe oxidation–reduction zone, which co-occurred with the maximum CH4 oxidation peaks (Fig. 1). 16S-RNA gene based pyrosequencing data from February 2012 showed the presence of an archaeal community related to Candidatus Methanoperedens nitroreducens (the most abundant retrieved OTUs showed 94.6 and 93.7% sequence similarity, respectively, against Candidatus Methanoperedens nitroreducens (JMIY01000002); Fig. 3 and Table S3). The peaks of abundance of these archaea co-occurred within the Fe oxidation/reduction zone and the maximum occurrence of photoferrotrophy previously reported by Llirós et al.11 (Fig. 4). Overall, putative AOM-related archaea in Kabuno bay represented ca. 16% of the whole community in February 2012. Recent studies have shown that microbes belonging to the Candidatus Methanoperedens archaeal group (Order Methanosarcinales) are particularly versatile, and can couple AOM with different electron acceptors, among which Fe, depending on environmental conditions35,36. Cai et al. recently demonstrated that these archaea were capable to catalyze Fe-linked AOM alone, without a bacterial partner, via the “reverse methanogenesis” pathway and possibly using a extracellular electron transport pathway37. Despite that these prokaryotes diversity data were acquired during a different sampling campaign25 than the AOM measurement reported here, the good agreement between (1) high Candidatus methanoperedens abundance and high Bchl e concentrations in the Fe oxidation–reduction zone in 2012, and (2) high AOM rates and high Bchl e concentrations in the Fe oxidation–reduction zone during this study, allow to hypothesize that Candidatus methanoperedens could represent the dominant microbes thriving AOM during the present study.

Presence of Candidatus Methanoperedens, capable of Fe-related AOM. 16S rRNA gene phylogenetic tree of the Candidatus Methanoperedens representative related OTUs (0.03 cut-off; only those OTUs containing more than 10 reads are shown) retrieved by pyrosequencing from Kabuno Bay water samples, the scale bar indicates 0.10 fixed point mutation per nucleotide position.

Co-occurence of Candidatus methanoperedens and ferrophototrophs in the chemocline of Kabuno Bay. (a) Vertical distribution of Candidatus methanoperedens and (b), Vertical profiles of particulate Fe concentrations (µmol L−1) and Bacteriochlorophyll e (Bchle) content (µg L−1) in February 2012. While this profile was not contemporary to the measurements of AOM (Fig. 1) the particulate Fe and Bacteriochlorophyll e peaks show that AOM and Archaea abundance coincided.
Accordingly, these AOM-associated archaea (AAA5) could therefore be responsible for the Fe-related AOM measured in Kabuno bay, taking into account that AOM coupled to Fe reduction is thermodynamically more favorable than coupling with other electron acceptors38. Iron-driven AOM has already been suggested as a dominant CH4 removal pathway in other ferruginous environments (i.e.38,39,40). Bacterial data reported from samples collected in 2012 evidenced that photoferrotrophs were responsible for the oxidation of 37 mmol Fe m−2 d−1 11. Comparison of this Fe oxidation rate with AOM showed that Fe3+ released via photoferrotrophy could potentially fuel an important fraction of the AOM (up to 31% in September 2013), in contrast to NO3− and SO42− reduction rates11,33, which could only fuel up to 5% of the AOM observed (Fig. 5). Furthermore, the observation of higher AOM rates when Mo was added may result from the higher availability of Fe oxides when sulfate-reducing bacteria (SRB) activity was inhibited. Indeed, SO42− reduction produces HS− that can rapidly reduce Fe oxides to form iron sulfides, which could precipitate and then be unavailable for Fe-dependent CH4 oxidizers. Fe is known to be efficiently recycled in the chemocline of ferruginous lakes38 or bioturbated marine sediments41.

Chemical evidence of Fe3+ as an important electron acceptor of AOM. Fraction (%) of the integrated AOM rates potentially sustained by Fe reduction, NO3− reduction and SO42− reduction rates measured by Llirós et al.11 and Michiels et al.33. Error bars are calculated as the standard deviation of the mean of the three sampling campaigns.
Overall, process rate measurements and metagenomics data suggest that an intense biotic regeneration of Fe3+ mediated by photoferrotrophs could provide an important fraction of the electron acceptors required to oxidize CH4 anaerobically in Kabuno Bay. Following the same approach as Jones et al.42 and assuming steady state conditions, the rate of Fe or Mn leaving the water column via sedimentation must equals the rate of Fe or Mn input in Kabuno bay’s water column via the rivers. Assuming that Mn and Fe leave the water column as a Mn2+ or Fe2+ mineral, all the oxidized Mn or Fe must ultimately be reduced, and the downward flux of particulate Fe3+ should be equivalent to the input of Fe via the rivers (3 mmol m−2 d−1) and the upward flux of Fe2+ (4–33 mmol m−2 d−1). If we consider that the totality of this downward flux of particulate Fe3+ is reduced within Kabuno bay’s chemocline, but that only 3 mmol Fe m−2 d−1 finally leave the mixolimnion, we estimated that Fe would be recycled up to 11 times before removal by sedimentation.
Consequences on representation of the Archean ocean metabolism
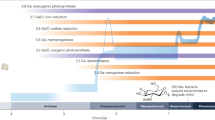
A strong interaction between photoferrotrophs and CH4 oxidizers has been hypothesized in several other modern Archean ocean analogues, such as Lake Matano38 and Lake La Cruz43. Under the ferruginous conditions of Archean oceans, photoferrotrophs would have been responsible for most of the primary production of the primitive Earth12. It is also generally assumed that a much larger fraction of the organic matter generated by primary producers would have been processed by methanogens than nowadays, given the absence of oxic and SO42− driven mineralization of organic matter in the O2 and SO42− depleted waters of the early Earth Ocean44,45.
Pavlov et al.46 estimated that up to 250 Tmol year−1 (1.9 mmol m−2 d−1, assuming an ocean area of 3.6 × 1014 m2) of CH4 would have been emitted during the Archean, hence CH4 would have been a key component of the ancient C cycle, with important consequences on the early Earth climate, as the higher CH4 concentrations (~ 100 ppmv) in the atmosphere are thought to have provided enough greenhouse warming to compensate for a 5–17% fainter Sun. The photochemical decomposition of CH4 in the atmosphere and the resulting escape of hydrogen to space may also have participated to the oxidation of the Earth surface environment47. However, geological archives do not provide constraints on the magnitude of the CH4 concentrations, and large CH4 fluxes are calculated with quantitative models that limit the existence of Fe-dependent CH4 oxidation and assume a negligible role of SO42− driven CH4 oxidizers. This paradigm has recently been challenged by Olson et al.48 and Sauterey et al.49 who showed that the combined effects of competition between methanogens and SO42− reducers and occurrence of SO42− driven CH4 oxidation would have effectively reduced the CH4 fluxes from the ocean even at modest SO42− concentrations, and regardless of O2 concentrations. Similarly, we propose that strong interactions between photoferrotrophs and Fe-dependent CH4 oxidizers might have exerted an important control on the Archean–Proterozoic CH4 cycling. For instance, modelled photoferrotrophic primary production in early Earth ferruginous ocean (3.8 mmol m−2 d−1 50) would have produced Fe oxides at a rate of 15.3 mmol m−2 d−1, assuming a 4:1 ratio between Fe oxidation and C fixation by photoferrotrophs. Although simplistic given the complexity of the C and Fe biogeochemical cycle, comparison of this rate of Fe oxides production with the sea-to-air CH4 flux (1.9 mmol m−2 d−1) proposed by Pavlov et al.46 suggests that photoferrotrophy would have been high enough to potentially support the oxidation of 100% of the CH4 flux to the early atmosphere. In Kabuno Bay, a modern analog to early Earth ocean, measured photoferrotrophy rates would have been sufficient to sustain a smaller (31 ± 18%, n = 3), but still important, part of the oxidation of the CH4 flux measured during this study (19 ± 11 mmol m−2 d−1, n = 3).
It has been hypothesized that the rise of atmospheric oxygen about 2.4 Gyr ago (Great Oxidation Event) was triggered by a decrease of atmospheric CH4 levels51. This has been attributed to an increased importance of SO42 reduction in the oceans that outcompeted the methanogenic organisms, although this explanation is not consistent with geological records52. It has also been hypothesized that the production of methane decreased in response to the decrease of the nickel inputs to oceans, as this is a key metal for methanogen enzymes53. Alternatively, it was showed that supplying a culture media with nickel does not stimulate methanogenesis54. A widespread occurrence of AOM coupled to Fe oxidation offers an alternative explanation that would have led to the decrease of dissolved CH4 in the ocean and consequently the emission of CH4 to the atmosphere, thus going in the direction of the study of Riedinger et al.39. This is consistent with the revision of the putative composition of the Archean atmosphere55 that suggests that the amount of greenhouse warming by CH4 was more limited than previously thought49,56.
While we can hypothesize that Fe(OH)3 sustaining AOM was initially provided by photoferrotrophy, the onset of oxygenic photosynthesis would also have been an ample supply of Fe(OH)3, available to sustain AOM, from the oxidation of Fe2+ with dissolved O2 released by oxygenic photoautotrophs (Fig. 6). This might also provide an explanation to the modest but significant increase in Fe-using genes from the Archean to the Proterozoic given by the phylogenomic analyses57. A decrease of dissolved CH4 concentrations due to Fe-dependent AOM could have had several other consequences on the timing and sequence of Archean and Proterozoic events. The decrease of atmospheric CH4 would have led to a decrease of H2 and CO generation by photolysis, which in turn would have led to a decrease of supply of H2 and CO to surface oceans, contributing to the demise of pelagic H2-methanogens and CO-acetogens (Fig. 6), that is usually attributed exclusively to the inhibition of these anaerobic organisms by increasing O2 level.

Potential role of iron-oxide AOM in Archean Ocean metabolism. In the Archean Ocean, anaerobic metabolism (photoferrotrophy, methanogenesis and CO-acetogenesis in dark blue) dominated before the advent of oxygenic photosynthesis (in light blue). In red, we propose anaerobic methane oxidation (AOM) using Fe oxides as electron acceptor as additional metabolic process. Before the advent of oxygenic photosynthesis, Fe3+ dependent AOM linked photoferrotrophy and H2-methanogenesis that so far have been seen as two parallel and unconnected processes. After the advent of oxygenic photosynthesis, Fe oxides-dependent AOM might have used the abundantly produced Fe(OH)3 from the oxidation of Fe2+ by O2 to further remove CH4 from the water column, facilitating the increase of O2 in the atmosphere and the great oxidation event.
Finally, the great oxygenation event was shortly afterwards followed by a low latitude glaciation that would be associated to lower atmospheric CH4 concentrations. However, the trigger of the collapse of CH4 has not been clearly identified, some authors arguing that CH4 decreased as a consequence of rising O258, while others proposed that low CH4 level preceded the transition to an O2-richer atmosphere49,51. Co-occurrence of photoferrotrophy and Fe-dependent CH4 oxidation in the Archean would support the latter hypothesis and the view of Olson et al.48 who recently proposed an alternative mechanism for the initiation of low-latitude glaciation with low baseline atmospheric CH4 levels.
References
Borges, A. V. et al. Globally significant greenhouse-gas emissions from African inland waters. Nat. Geosci. 8, 637–642. https://doi.org/10.1038/ngeo2486 (2015).
Iversen, N. & Jørgensen, B. Anaerobic methane oxidation rates at the sulfate-methane transition in marine sediments from Kattegat and Skagerrak (Denmark). Limnol. Oceanogr. 30, 944–955. https://doi.org/10.4319/lo.1985.30.5.0944 (1985).
Boetius, A. et al. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. https://doi.org/10.1038/35036572 (2000).
Jørgensen, B. B., Weber, A. & Zopfi, J. Sulfate reduction and anaerobic methane oxidation in Black Sea sediments. Deep-Sea Res. Pt. I(48), 2097–2120. https://doi.org/10.1016/S0967-0637(01)00007-3 (2001).
Knittel, K. & Boetius, A. Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. https://doi.org/10.1146/annurev.micro.61.080706.093130 (2009).
Ettwig, K. F. et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. https://doi.org/10.1038/nature08883 (2010).
Haroon, M. F. et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500, 567–570. https://doi.org/10.1038/nature12375 (2013).
Beal, E. J., House, C. H. & Orphan, V. J. Manganese-and iron-dependent marine methane oxidation. Science 325, 184–187. https://doi.org/10.1126/science.1169984 (2009).
Borges, A. V., Abril, G., Delille, B., Descy, J. P. & Darchambeau, F. Diffusive methane emissions to the atmosphere from Lake Kivu (Eastern Africa). J. Geophys. Res. Biogeosci. https://doi.org/10.1029/2011JG001673 (2011).
Ross, K. A., Gashugi, E., Gafasi, A., Wüest, A. & Schmid, M. Characterisation of the subaquatic groundwater discharge that maintains the permanent stratification within Lake Kivu; East Africa. PLoS ONE 10, e0121217. https://doi.org/10.1371/journal.pone.0121217 (2015).
Llirós, M. et al. Pelagic photoferrotrophy and iron cycling in a modern ferruginous basin. Sci. Rep. https://doi.org/10.1038/srep13803 (2015).
Camacho, A., Walter, X. A., Picazo, A. & Zopfi, J. Photoferrotrophy: remains of an ancient photosynthesis in modern environments. Front. Microbiol. https://doi.org/10.3389/fmicb.2017.00323 (2017).
Planavsky, N. J. et al. Widespread iron-rich conditions in the mid-Proterozoic ocean. Nature 477, 448–451. https://doi.org/10.1038/nature10327 (2011).
Morana, C. et al. Chemoautotrophy and anoxygenic photosynthesis within the water column of a large meromictic tropical lake (Lake Kivu, East Africa). Limnol. Oceanogr. 61, 1424–1437. https://doi.org/10.1002/lno.10304 (2016).
Roland, F. A. E. Biogeochemical Processing of Greenhouse Gases (Methane and Nitrous Oxide) in Meromictic Lakes. Ph.D thesis, University of Liège, (2017).
Weiss, R. F. Determinations of carbon dioxide and methane by dual catalyst flame ionization chromatography and nitrous oxide by electron capture chromatography. J. Chromatogr. Sci. 19, 611–616 (1981).
APHA. Standard Methods for the Examination of Water and Wastewater. Vol. 2 (American Public Health Association, Washington DC, 1998).
Miranda, K. M., Espey, M. G. & Wink, D. A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide-Biol. Ch. 5, 62–71. https://doi.org/10.1006/niox.2000.0319 (2001).
Westwood, D. in Methods for the Examination of Waters and Associated Materials (ed HMSO) (Stationery Office Books, London, United Kingdom, 1981).
Cline, J. D. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnol. Oceanogr. 14, 454–458. https://doi.org/10.4319/lo.1969.14.3.0454 (1969).
Roland, F. A. E. et al. Anaerobic methane oxidation and aerobic methane production in an east African great lake (Lake Kivu). J. Great Lakes Res. 44, 1183–1193. https://doi.org/10.1016/j.jglr.2018.04.003 (2018).
Pasche, N. et al. Physical and biogeochemical limits to internal nutrient loading of meromictic lake kivu. Limnol. Oceanogr. 54, 1863–1873. https://doi.org/10.4319/lo.2009.54.6.1863 (2009).
Raghoebarsing, A. A. et al. A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440, 918–921. https://doi.org/10.1038/nature04617 (2006).
Sarmento, H., Isumbisho, M. & Descy, J.-P. Phytoplankton ecology of Lake Kivu (eastern Africa). J. Plankton Res. 28, 815–829. https://doi.org/10.1093/plankt/fbl017 (2006).
İnceoğlu, Ö. et al. Distribution of bacteria and archaea in meromictic tropical Lake Kivu (Africa). Aquat. Microb. Ecol. 74, 215–233. https://doi.org/10.3354/ame01737 (2015).
Llirós, M., Casamayor, E. O. & Borrego, C. High archaeal richness in the water column of a freshwater sulfurous karstic lake along an interannual study. FEMS Microbiol. Ecol. 66, 331–342. https://doi.org/10.1111/j.1574-6941.2008.00583.x (2008).
Shah, V. et al. Bacterial and Archaea community present in the Pine Barrens Forest of Long Island, NY: unusually high percentage of ammonia oxidizing bacteria. PLoS ONE 6, e26263. https://doi.org/10.1371/journal.pone.0026263 (2011).
Pruesse, E., Peplies, J. & Glockner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics (Oxford, England) 28, 1823–1829. https://doi.org/10.1093/bioinformatics/bts252 (2012).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. https://doi.org/10.1093/nar/gkh293 (2004).
Milucka, J. et al. Methane oxidation coupled to oxygenic photosynthesis in anoxic waters. ISME J. 9, 1991–2002. https://doi.org/10.1038/ismej.2015.12 (2015).
Canfield, D. E., Erik, K. & Bo, T. in Advances in Marine Biology Vol. 48 (eds D.E. Canfield, E. Kristensen, & B. Thamdrup) 313–381 (Academic Press, New York, 2005).
Overmann, J., Cypionka, H. & Pfennig, N. An extremely low-light adapted phototrophic sulfur bacterium from the Black Sea. L&O 37, 150–155. https://doi.org/10.4319/lo.1992.37.1.0150 (1992).
Michiels, C. C. et al. Iron-dependent nitrogen cycling in a ferruginous lake and the nutrient status of Proterozoic oceans. Nat. Geosci. 10, 217–221. https://doi.org/10.1038/ngeo2886 (2017).
Crowe, S. A. et al. Draft Genome Sequence of the Pelagic Photoferrotroph Chlorobium phaeoferrooxidans. Genome Announc. https://doi.org/10.1128/genomeA.01584-16 (2017).
Weber, H. S., Habicht, K. S. & Thamdrup, B. Anaerobic methanotrophic Archaea of the ANME-2d cluster are active in a low-sulfate, iron-rich freshwater sediment. Front. Microbiol. https://doi.org/10.3389/fmicb.2017.00619 (2017).
Ettwig, K. F. et al. Archaea catalyze iron-dependent anaerobic oxidation of methane. PNAS 113, 12792–12796. https://doi.org/10.1073/pnas.1609534113 (2016).
Cai, C. et al. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J. https://doi.org/10.1038/s41396-018-0109-x (2018).
Crowe, S. et al. The methane cycle in ferruginous Lake Matano. Geobiology 9, 61–78. https://doi.org/10.1111/j.1472-4669.2010.00257.x (2011).
Riedinger, N. et al. An inorganic geochemical argument for coupled anaerobic oxidation of methane and iron reduction in marine sediments. Geobiology 12, 172–181. https://doi.org/10.1111/gbi.12077 (2014).
Sivan, O. et al. Geochemical evidence for iron-mediated anaerobic oxidation of methane. Limnol. Oceanogr. 56, 1536–1544. https://doi.org/10.4319/lo.2011.56.4.1536 (2011).
Canfield, D. E., Thamdrup, B. & Hansen, J. W. The anaerobic degradation of organic matter in Danish coastal sediments: iron reduction, manganese reduction, and sulfate reduction. Geochim. Cosmochim. Acta 57, 3867–3883 (1993).
Jones, C. et al. Biogeochemistry of manganese in ferruginous Lake Matano, Indonesia. Biogeosciences 8, 2977–2991. https://doi.org/10.5194/bg-8-2977-2011 (2011).
Oswald, K. et al. Methanotrophy under versatile conditions in the water column of the ferruginous Meromictic Lake La Cruz (Spain). Front. Microbiol. https://doi.org/10.3389/fmicb.2016.01762 (2016).
Lovley, D. R. & Klug, M. J. Sulfate reducers can outcompete methanogens at freshwater sulfate concentrations. Appl. Environ. Microbiol. 45, 187–192 (1983).
Roberson, A. L., Roadt, J., Halevy, I. & Kasting, J. F. Greenhouse warming by nitrous oxide and methane in the Proterozoic Eon. Geobiology 9, 313–320. https://doi.org/10.1111/j.1472-4669.2011.00286.x (2011).
Pavlov, A. A., Hurtgen, M. T., Kasting, J. F. & Arthur, M. A. Methane-rich proterozoic atmosphere?. Geology 31, 87–90. https://doi.org/10.1130/0091-7613(2003)031%3c0087:MRPA%3e2.0.CO;2 (2003).
Catling, D. C., Claire, M. W. & Zahnle, K. J. Anaerobic methanotrophy and the rise of atmospheric oxygen. Philos. Trans. A Math. Phys. Eng. Sci. 365, 1867–1888. https://doi.org/10.1098/rsta.2007.2047 (2007).
Olson, S. L., Reinhard, C. T. & Lyons, T. W. Limited role for methane in the mid-Proterozoic greenhouse. PNAS 113, 11447–11452. https://doi.org/10.1073/pnas.1608549113 (2016).
Sauterey, B., Charnay, B., Affholder, A., Mazevet, S. & Ferrière, R. Co-evolution of primitive methane-cycling ecosystems and early Earth’s atmosphere and climate. Nat. Commun. 11, 2705. https://doi.org/10.1038/s41467-020-16374-7 (2020).
Canfield, D. E., Rosing, M. T. & Bjerrum, C. Early anaerobic metabolisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 361, 1819–1834. https://doi.org/10.1098/rstb.2006.1906 (2006).
Zahnle, K., Claire, M. & Catling, D. The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology 4, 271–283. https://doi.org/10.1111/j.1472-4669.2006.00085.x (2006).
Papineau, D., Mojzsis, S. J. & Schmitt, A. K. Multiple sulfur isotopes from Paleoproterozoic Huronian interglacial sediments and the rise of atmospheric oxygen. Earth Planet. Sci. Lett. 255, 188–212. https://doi.org/10.1016/j.epsl.2006.12.015 (2007).
Konhauser, K. O. et al. Oceanic nickel depletion and a methanogen famine before the Great Oxidation Event. Nature. https://doi.org/10.1038/nature07858 (2009).
Bray, M. S. et al. Shifting microbial communities sustain multiyear iron reduction and methanogenesis in ferruginous sediment incubations. Geobiology 15, 678–689. https://doi.org/10.1111/gbi.12239 (2017).
Haqq-Misra, J. D., Domagal-Goldman, S. D., Kasting, P. J. & Kasting, J. F. A revised, hazy methane greenhouse for the Archean Earth. Astrobiology 8, 1127–1137. https://doi.org/10.1089/ast.2007.0197 (2008).
Pavlov, A. A., Kasting, J. F., Brown, L. L., Rages, K. A. & Freedman, R. Greenhouse warming by CH4 in the atmosphere of early Earth. J. Geophys. Res. 105, 11981–11990 (2000).
David, L. A. & Alm, E. J. Rapid evolutionary innovation during an Archaean genetic expansion. Nature. https://doi.org/10.1038/nature09649 (2010).
Kopp, R. E., Kirschvink, J. L., Hilburn, I. A. & Nash, C. Z. The Paleoproterozoic snowball Earth: a climate disaster triggered by the evolution of oxygenic photosynthesis. PNAS 102, 11131–11136. https://doi.org/10.1073/pnas.0504878102 (2005).
Acknowledgements
We thank the team of the Observatoire volcanologique de Goma (OVG) for their involvement in sampling, Bo Thamdrup (University of Southern Denmark) for the access to his laboratory, Bruno Leporcq (University of Namur), Renzo Biondo (University of Liège), Dina Holmgaard Skov and Heidi Grøn Jensen (University of Southern Denmark) for help in measurements, Jack Middelburg and Steven Bouillon for comments on the draft and the three anonymous reviewers and associated editor for their valuable contribution. We also thank Thibault Lambert (University of Liège) for the elaboration of the map (Figure S1). This study was funded by the Belgian Federal Science Policy Office (BELSPO, Belgium) under the EAGLES (East African Great lake Ecosystem Sensitivity to Changes, SD/AR/02A) project, by the Fonds National de la Recherche Scientifique (FNRS) under the MICKI (Microbial diversity and processes in Lake Kivu, 1715859) project, and contributes to the European Research Council (ERC) starting grant project AFRIVAL (African river basins: Catchment-scale carbon fluxes and transformations, 240002). GC apparatus was acquired with funds from the FNRS (Contract No. 2.4.598.07). AVB is a senior research associate at the FNRS. FAER had a PhD grant from FNRS (« Fonds pour la formation à la Recherche dans l’Industrie et dans l’Agriculture »—FRIA) and is now post-doctoral researcher at the FNRS (Project No. X.3007.17 funded by the Walloon Institute of Sustainable Development). This study shows results mainly obtained during FAER's Ph.D., and the “Material and methods” section has been mostly described in the resulting unpublished thesis.
Author information
Authors and Affiliations
Contributions
F.A.E.R., A.V.B and F.D designed the study, and F.A.E.R and F.D. carried out the field data collection. J.-P.D. carried out the HPLC measurements, M.Ll. carried out the archaeal diversity analyses. F.A.E.R. and C.M. drafted the manuscript, which was substantially commented upon and amended by A.V.B. All co-authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Roland, F.A.E., Borges, A.V., Darchambeau, F. et al. The possible occurrence of iron-dependent anaerobic methane oxidation in an Archean Ocean analogue. Sci Rep 11, 1597 (2021). https://doi.org/10.1038/s41598-021-81210-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81210-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
