
Abstract
Antibiotics are failing fast, and the development pipeline remains alarmingly dry. New drug research and development is being urged by world health officials, with new antibacterials against multidrug-resistant Gram-negative pathogens as the highest priority. Antivirulence drugs, which inhibit bacterial pathogenicity factors, are a class of promising antibacterials, however, their development is stifled by lack of standardised preclinical testing akin to what guides antibiotic development. The lack of established target-specific microbiological assays amenable to high-throughput, often means that cell-based testing of virulence inhibitors is absent from the discovery (hit-to-lead) phase, only to be employed at later-stages of lead optimization. Here, we address this by establishing a pipeline of bacterial cell-based assays developed for the identification and early preclinical evaluation of DsbA inhibitors, previously identified by biophysical and biochemical assays. Inhibitors of DsbA block oxidative protein folding required for virulence factor folding in pathogens. Here we use existing Escherichia coli DsbA inhibitors and uropathogenic E. coli (UPEC) as a model pathogen, to demonstrate that the combination of a cell-based sulfotransferase assay and a motility assay (both DsbA reporter assays), modified for a higher throughput format, can provide a robust and target-specific platform for the identification and evaluation of DsbA inhibitors.
Similar content being viewed by others
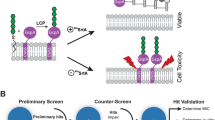


Introduction
In 2014 the World Health Organisation (WHO) released a statement declaring antimicrobial resistance (AMR) as a public health priority that demands decisive global action1. Although WHO’s statement has increased AMR awareness, at the time of writing over half a decade has passed, and little progress has been made in developing effective solutions2,3; meanwhile, AMR rates continue to rise. The current AMR crisis demands the urgent development of effective strategies to tackle bacterial infections. One actively researched strategy is the development of antivirulence therapeutics, which have recently been gaining momentum as effective antibacterials that can circumvent the mechanisms of antibiotic resistance and eliminate or reduce resistance selection4. Antivirulence drugs target bacterial virulence factors and are designed to disarm pathogens, unlike conventional antibiotics which either kill or inhibit bacterial growth5,6. Targeting virulence factors can attenuate a pathogen’s ability to cause infection and render bacteria susceptible to the host's defence systems7. Consequently, virulence factors present a plethora of attractive targets for the development of new therapeutics.
Although several antivirulence drugs are currently under various stages of development, (e.g. toxin, adhesin, enzyme, secretion and quorum sensing inhibitors6,8,9) the potential of any antivirulence drug candidate for further clinical development relies on having established robust assays for evaluating their efficacy in vitro and in vivo10. While the development of antibiotics over the past several decades has benefited from standardised and comprehensive preclinical and clinical evaluation methods, the field of antivirulence drugs has had minimal guidelines for consistent testing, with only a few general guidelines reported for some types of inhibitors, e.g. for quorum sensing10,11. In addition, antivirulence inhibitor screening campaigns often utilise biophysical and/or biochemical assays (when the target is known), which do not allow early evaluation of inhibitor effects on bacterial cells12, or on cell-based virulence assays (target agnostic), which might be prone to bias by reporting non-specific inhibitor effects11. Here we develop a pipeline of robust cell-based assays for the in vivo evaluation of inhibitors against the DsbA antivirulence target.
In Gram-negative pathogens, the biogenesis and function of many virulence factors are intrinsically linked to the redox enzyme pair of DsbA and DsbB13,14,15,16. DsbA is a periplasmic oxidoreductase which catalytically introduces disulfide bonds into secreted and outer membrane proteins17, while its inner membrane partner DsbB reoxidises DsbA18,19. Intramolecular disulfide bonds are often essential for the native folding and subsequent function of multiple secreted or surface proteins, including fimbriae, flagellar motor, secretion systems, and secreted toxins13,16. Given that many of these proteins are bona fide virulence factors or form integral components of machinery for virulence factor assembly, this makes DsbA and DsbB ideal targets for the development of antivirulence drugs13,16,20. Recently, several classes of small molecule inhibitors of DsbA, as well as inhibitors of its cognate DsbB, have been reported, primarily through screening campaigns involving biophysical and/or biochemical assays12,21,22,23,24,25,26. Any in vivo assessment of promising hits was typically conducted as part of subsequent testing, often at a stage where significant efforts into the chemical elaboration of initial hits had already taken place. Incorporation of cell-based testing at an earlier stage of inhibitor screening, as conducted for DsbB and its homologue VKOR24, could be used to complement early hit selection by biophysical/biochemical approaches and likely save time and money, by informing which hits should be prioritised and what properties should be optimised (e.g. solubility, cell permeability, toxicity etc.).
For monitoring DsbA function in vivo, the bacterial motility assay on soft agar has been most commonly used27,28,29 and more recently this method was applied to DsbA inhibitor testing in vivo12,30. In many pathogens, such as uropathogenic Escherichia coli (UPEC), and Salmonella enterica serovar Typhimurium (S. Typhimurium), motility requires the production of functional flagella, with DsbA playing a central role in the biogenesis of these surface appendages27,31,32,33,34. The standard bacterial motility assay format (performed in Petri dishes) is however relatively low-throughput and requires large inhibitor quantities and manual data collection30, thus, limiting its utility for high-throughput inhibitor screening and testing. A second method recently utilised for DsbA inhibitor testing monitors the enzymatic activity of ASST30, an arylsulfate sulfotransferase encoded by several pathogens (e. g. UPEC, S. Typhimurium, Klebsiella28,29,35,36), which is proposed to play a role in the intracellular detoxification of phenolic substances37,38,39. ASST is a native substrate for DsbA and its homologue DsbL40 as it requires the formation of a disulfide bond for its correct function41. Consequently, ASST’s sulfotransferase activity can be used to measure DsbA activity in vivo, and can be monitored either in solution40 or using an agar-based assay28. Although very informative, previously used ASST assays have not been amenable to high-throughput inhibitor screening and testing. Here, we present a comprehensive pipeline of cell-based assays that provide an accurate and high throughput platform for the identification of DsbA inhibitors and their subsequent development from hits to leads, and from lead optimisation to early preclinical candidate validation.
Results
Establishing a high-throughput assay for in vivo monitoring of ASST enzyme activity in pathogenic bacteria
Enzymatic assays are well suited to high-throughput inhibitor screening campaigns. Thus, we sought to develop a cell-based assay for monitoring the activity of the ASST enzyme, which is a known DsbA substrate in UPEC. We first determined if ASST’s sulfotransferase activity could be assayed in solution using live UPEC cells cultured in standard laboratory conditions. We specifically wanted an assay that is easy to perform using standard laboratory reagents and equipment (i.e. streamlined protocol, minimal number of steps, use of standard growth media and conditions) so that it could be easily adopted for high-throughput screening (HTS) in various settings. As the ASST gene in UPEC (astA) is not expressed under standard growth conditions29 and in order to make our assay transferrable to non ASST-encoding bacteria, we chose to produce ASST from a plasmid vector previously shown to depend on DsbA for activity in UPEC and S. Typhimurium28,29,42. The ASST overexpressing strain CFT073/pASST was cultured overnight in LB, and culture aliquots were mixed in a 96-well plate with the aryl sulfate phenolic donor, potassium 4-methylumbelliferyl sulfate (MUS) and the phenol acceptor, phenol. ASST catalysed the cleavage of the sulfate group from the non-fluorescent substrate MUS to the highly fluorescent product 4-methylumbelliferone (MU) (Fig. 1)43. MUS and similar substrates have been extensively used to study the activity of the ASST enzyme from various bacterial species28,36,39,40,43,44. A steady increase in fluorescence was observed over time for strain CFT073/pASST, but not for CFT073 carrying the empty vector control (Fig. 2A). Using these strains, we confirmed (a) the production of functional ASST which catalysed the conversion of MUS to fluorescent MU in the UPEC periplasm (where ASST and DsbA localise) and (b) determined the assay’s dynamic range. Because the chromosomal astA gene copy in CFT073/pSU2718 is not expressed under the assay conditions29, we further explored the lower limit of the assay’s dynamic range by utilising two previously characterised CFT073 mutants lacking either DsbA (CFT073ΔdsbA) or both DsbA and DsbL (CFT073ΔdsbAΔdsbLI). Overexpression of ASST in these mutants (CFT073ΔdsbA/pASST and CFT073ΔdsbAΔdsbLI/pASST) resulted in equally low sulfotransferase activity, displaying 23% normalised fluorescence intensity in the ASST assay (Fig. 2A). While DsbA, or one of its homologues, is required to rapidly introduce a functional disulfide bond into the ASST enzyme43, the disulfide bond could also form, albeit at a much slower rate, via background oxidation. This could lead to low levels of functional ASST in the cell, which would account for the activity reported for CFT073ΔdsbA/pASST (Fig. 2A). This 23% increase over baseline strain CFT073/pSU2718 (that does not express ASST) represents the background level of sulfotransferase activity in our assay, which likely results from environmental oxidation of ASST during overnight growth in rich media. Thus, the maximum reduction in fluorescence that can be achieved in our assay is 77%. Conducting the assay in phosphate buffer instead of media gave the same basal levels of fluorescence, suggesting that environmental oxidation of ASST could be neglected during the assay timescale (data not shown). The suitability of our assay for HTS was further evaluated by calculating the Z′-factor using the positive and negative genetic controls, CFT073/pASST and CFT073/pSU2718, respectively. Our assay has a Z′-factor of 0.89 ± 0.02 indicating excellent assay quality related to screening45. Taken together, these results demonstrate an effective dynamic range for the assay and confirm that it can be used to monitor DsbA-mediated ASST activity in live bacteria under standard laboratory growth conditions.

ASST catalysed conversion of MUS to MU.

Cell-based ASST activity in UPEC CFT073 in varying concentrations of DsbA inhibitor F1. Sulfotransferase activity of (A) CFT073/pASST, CFT073/pSU2718 (vector control), CFT073ΔdsbA/pASST, and CFT073ΔdsbAΔdsbLI/pASST cultured in 0.4% DMSO in the absence of DsbA inhibitors. (B) Sulfotransferase activity of CFT073/pASST cultured in the presence of F1 (1–0.125 mM) or 0.4% DMSO (vehicle control) and (C) corresponding F1 dose–response curve calculated at 40-min time point. F1 treated bacterial cultures were mixed with MUS and phenol and immediately monitored spectrofluorometrically (excitation 360 nm and emission 450 nm) for 60 min at room temperature (21 °C). Data are shown as normalised fluorescence intensity units (calculated by normalising the treatment data against the DMSO control (set as 100%) (A and B: slopes reported in (B) were calculated from the 15 to 30 min region), and normalised fluorescence intensity at 40 min (C) (calculated from the 40-min time point of B), with the mean ± SD of 3 biological replicates plotted at each time point.
The cell-based ASST assay offers a first high-throughput step in our DsbA inhibitor screening and development pipeline
We hypothesised that inhibition of DsbA in CFT073/pASST would result in misfolding of the ASST enzyme and loss of sulfotransferase activity. To examine this hypothesis, we repeated the ASST assay with CFT073/pASST cells treated with various concentrations of the previously described DsbA inhibitor F1 (1–0.125 mM)42. Sulfotransferase activity was decreased at all tested F1 concentrations (Fig. 2B), with the lowest fluorescence measured from cells cultured at 1 mM F1 (55% reduction compared to vehicle (DMSO) control). The vehicle solvent (DMSO) was found to have no effect on bacterial growth or ASST activity at the administered concentration of 0.4% (data not shown). Reduction of ASST sulfotransferase activity by F1 was dose-dependent with a half maximal inhibitory concentration (IC50) value in the 0.14–0.17 mM range (Fig. 2C). IC50 values in the mM range are indicative of modest affinity inhibitor hits that represent good candidates for synthetic optimisation. To further explore the sensitivity of the assay we also examined two other previously documented DsbA inhibitors, F2 and F4, both of which are structural isomers of F1 (Fig. 3A)30. Like F1, both F2 and F4 deceased sulfotransferase activity in a dose dependent manner (Fig. 3B,C). However, both inhibitors (F2 and F4) were less potent than F1 at similar concentrations, a result which is consistent with our previous in vivo findings for these compounds30. Taken together, these results demonstrate that DsbA inhibition results in loss of ASST sulfotransferase activity, confirming that our cell-based ASST sulfotransferase assay can be used for indirectly assessing DsbA inhibition in a high-throughput format. Furthermore, the assay is sensitive enough to distinguish small differences in inhibitor efficacy, suggesting that weaker inhibitors of DsbA can also be identified using this assay.

Cell-based ASST activity in UPEC CFT073 in varying concentrations of DsbA inhibitors F2 and F4. (A) Chemical structures of DsbA inhibitors F1, F2, and F4. Sulfotransferase activity of CFT073/pASST cultured in the presence of (B) F2 (0.5–0.125 mM) (C) F4 (0.5–0.125 mM) or 0.4% DMSO (vehicle control). Treated bacterial cultures were mixed with MUS and phenol and immediately monitored spectrofluorometrically (excitation 360 nm and emission 450 nm) for 60 min at room temperature (21 °C). Data are shown as normalised fluorescence intensity units calculated by normalising the treatment data against the DMSO control (set as 100%), with the mean ± SD of 3 biological replicates plotted at each time point.
The cell-based ASST assay allows target-specific testing of DsbA inhibitor activity
With assay protocol and conditions established, we next sought to confirm the specificity of our ASST assay for the DsbA target. To investigate this, we utilised the previously described strain CFT073ΔdsbAΔdsbLI/pASST. This strain lacks both DsbA homologues and showed decreased fluorescence compared to the wild-type strain (CFT073/pASST). In trans complementation with DsbA fully restored the mutant’s fluorescence back to wild-type levels (Fig. 4A), confirming that in our assay DsbA is required for the production of functional ASST enzyme. In addition, both the control strain CFT073/pASST (WT) and the complemented mutant CFT073ΔdsbAΔdsbLI/pASST/pEcDsbA (KO/pDsbA) were equally attenuated for ASST function when treated with 0.5 mM F1 inhibitor (Fig. 4A). In contrast, the mutant CFT073ΔdsbAΔdsbLI/pASST (KO) was unresponsive to F1 treatment, and its fluorescence profile remained unaltered upon treatment with 0.5 mM F1 or with DMSO (Fig. 4A). These results indicate that our assay can identify inhibitors that target DsbA.

F1 inhibitor effects on DsbA cellular function and UPEC growth. (A) Cell-based sulfotransferase activity of CFT073/pASST (WT) grown in the presence of 0.4% DMSO or 0.5 mM F1; CFT073ΔdsbAΔdsbLI/pASST (KO) grown in the presence of 0.4% DMSO or 0.5 mM F1; and CFT073ΔdsbAΔdsbLI/pASST/pEcDsbA (KO/pDsbA) grown in the presence of 0.4% DMSO or 0.5 mM F1. Treated bacterial cultures were mixed with MUS and phenol and immediately monitored spectrofluorometrically for 60 min at room temperature (21 °C). (B) Growth curves of CFT073/pASST cultured in LB medium containing F1 (1–0.125 mM) or 0.4% DMSO (vehicle control) and monitored spectrophotometrically (Optical Density (O.D.) at 600 nm) for at least 8 h at 37 °C. Data are normalised fluorescence intensity units (calculated by normalising the treatment data against the DMSO control (set as 100%) (A) or absorbance at 600 nm (B), with mean ± SD of 3 biological replicates plotted at each time point.
Adding a growth analysis step prior to assessing sulfotransferase activity in the cell-based ASST assay simultaneously screens for inhibitor effects on DsbA cellular function and bacterial growth
DsbA is not required for UPEC CFT073 growth in rich media and standard laboratory culture conditions29. As such, inhibitors specific to DsbA would be predicted to have no effect on CFT073 growth under these conditions. On the other hand, large libraries of low affinity compounds, such as those typically used in early inhibitor screens, could contain several compounds with bacterial growth toxicity. To incorporate growth testing as part of our high-throughput cell-based ASST assay, CFT073/pASST growth was continuously monitored (step 1) during culture for the preparation of live-cell samples for sulfotransferase activity testing (step 2). Testing F1 in the growth analysis step of the sulfotransferase assay, revealed that under these conditions CFT073/pASST growth was slightly reduced in the presence of 1 mM F1, with no growth defects observed at lower F1 concentrations (0.5–0.125 mM) (Fig. 4B). We have previously examined the effects of F1 on the growth of wild type UPEC CFT073 and found no significant inhibition of growth at concentrations up to 1 mM. However, the ASST assay utilises a CFT073 strain which is overexpressing ASST (CFT073/pASST) that is subjected to growth conditions that differ from those used in our original study (microaerobic plate format in this study versus aerobic in tubes in our previous study)30. In addition to uncovering this small growth defect at high F1 concentrations, incorporating the growth step in our assay allowed us to account for any potential reduction in viable cells present in culture samples tested for sulfotransferase activity. Having an accurate O.D. 600 nm reading at the time of culture collection, ensured that all samples tested in the ASST assay could be easily adjusted to contain the same number of live cells, which was confirmed by plating for viable CFU (3–4 × 108 CFU/mL). These results demonstrate that adding a growth analysis step to the cell-based sulfotransferase enzyme assay allows growth related inhibitor effects to be identified and adjusted prior to downstream inhibitor testing.
Establishing a plate-reader cell-based assay for monitoring bacterial motility over time
Screening for DsbA inhibitors using cell-based assays, requires reporter phenotypes that are mediated by DsbA substrates proteins, such as ASST. Using such an approach for inhibitor screening, unavoidably could lead to hits that either target DsbA (our intended target) or the reporter protein itself (e.g. ASST). Including genetic controls can control for this, as demonstrated above for inhibitor F1 and our DsbA knock-out mutants, however, in a high-throughput screen of thousands of compounds this would require a parallel screen to be conducted with the control strain. While feasible this would not present a cost-effective design. Instead, most drug screening approaches combine orthogonal techniques for hit validation. Here we have modified a second cell-based DsbA reporter assay that relies on a different reporter substrate (the flagella motor protein FlgI) and can be used as a second stage screen of the hits identified by the high-throughput ASST assay. This not only ensures target specificity for identified hits but also presents a standalone medium-throughput assay for evaluating compounds further elaborated and developed as leads. As bacterial motility assays are the most common method of assessing DsbA inhibitor efficacy, we first sought to develop a motility assay which would circumvent the limitations of the standard assay format and provide a platform which could be employed directly after a HTS campaign. We adapted a microtiter plate-based assay which had previously been reported for screening antimicrobial compounds using bacterial motility46. We first confirmed that bacterial swimming motility could be accurately monitored spectrophotometrically. As bacteria radially migrated through the soft agar, a zone of motility corresponding to an increase in absorbance at 600 nm was observed, and a motility curve could be generated over time (Fig. 5A,B). Using this method, the start, end, and zone of motility of several test species (E. coli (JCB816), Fig. 5A; Pseudomonas aeruginosa (PAO1) and S. Typhimurium (SL1344), Fig. 5C), were accurately measured under a set of specific culture conditions. In these conditions, an E. coli DsbA null mutant (JCB817) remained immotile throughout the assay, demonstrating the lower end of the assay’s dynamic range (Fig. 5A).

Absorbance-based monitoring of bacterial motility. (A) Motility curves of E. coli JCB816 and E. coli ΔdsbA JCB817 monitored spectrophotometrically during incubation on soft LB agar at 37 °C over 13 h. (B) Digital images tracking the swimming motility of E. coli JCB816 on soft LB agar in a 24-well plate. E. coli was inoculated at the left edge of each well, and by 12 h incubation at 37 °C the zone of motility (boundary marked in red) had reached the opposite edge of the well. (C) Motility curves of P. aeruginosa (PAO1) and S. Typhimurium (SL1344) monitored spectrophotometrically during incubation on soft LB agar at 37 °C over 15 h. Data are shown as absorbance at 600 nm (A and C), with mean ± SD of 3 biological replicates plotted at each time point.
The absorbance-based bacterial motility assay offers a second step for hit validation and further DsbA inhibitor development in our pipeline
To demonstrate the value of a plate reader-based motility assay in assessing DsbA inhibitor hits, we generated motility curves for UPEC strain CFT073 in the presence and absence of DsbA inhibitor F1 (Fig. 6A), which we have previously shown to inhibit DsbA in CFT073 using the traditional petri-dish motility assay30. The motility of CFT073 in soft agar containing F1 at a concentration gradient (1–0.1 mM) was reduced compared to the vehicle control in a dose-dependent manner (Fig. 6A). Maximum motility inhibition was observed at 1 mM F1 were CFT073 remained immotile similar to the double dsbA dsbL null mutant (97% compared to DMSO control at 10 h post-inoculation) (Fig. 6A). Analysis of the longitudinal motility data revealed that both the start time and the rate of motility were directly related to F1 inhibitor concentration, with higher concentrations resulting in longer motility start times and slower motility rates (Table 1), effects that were not previously evident with the conventional motility assay methodology27. Furthermore, the high reproducibility of our assay allowed for even small changes in motility rate to be robustly detected (P < 0.0001, one-way ANOVA test) between different F1 treatment groups (Table 1). Using the calculated rate of motility for each F1 concentration, an F1 dose–response curve could be generated (Fig. 6B) with an IC50 for F1 calculated at the 0.35–0.47 mM range. To further confirm the assay’s suitability for validating and evaluating DsbA inhibitors, two additional inhibitors were tested. Like F1, DsbA inhibitors F2 and F4 were found to inhibit the motility of CFT073 compared to the DMSO control (Fig. 6C). Taken together, our absorbance-based motility assay proved to be of value in generating accurate and highly reproducible motility curve data that could be used to identify and characterise early hits from DsbA inhibitor screening campaigns, such as inhibitors F1, F2, and F412. Moreover, this motility assay format (24-well plate) required almost 29-fold less inhibitor than the standard petri-dish assay (0.14 mg/well versus 4 mg/petri-dish) and used an automated data collection pipeline that markedly reduced the assay’s hands-on time.

Absorbance-based UPEC motility in the presence of DsbA inhibitors. (A) Motility curves and (B) motility dose–response curve of UPEC CFT073 on LB agar (0.25%) containing DsbA inhibitor F1 (1–0.1 mM) or (C) DsbA inhibitors F2 (0.5 mM), F4 (0.5 mM) and 0.4% DMSO (vehicle control), monitored spectrophotometrically during incubation at 37 °C over 15 h. The immotile isogenic dsbA null strain (KO) is shown as a control (A). Data are shown as absorbance 600 nm (A and C), and normalised gradient (calculated from the treatment slopes of (A), listed in Table 1) (B), with the mean ± SD of 3 biological replicates plotted at each time point.
Discussion
Antimicrobial drug development typically starts with screening large fragment or compound libraries to identify initial hits and the subsequent chemical elaboration of different hit series. Such screening campaigns represent a big investment, in terms of time and resources, both for the industry and for the academic lab. Success relies heavily on the use of well-established, accurate reporter assays that can identify hits with some degree of target-specificity and are amenable to high-throughput testing of several thousands of compounds at once. For antibiotics, such testing is now considered routine and follows global standards and guidelines47,48. For non-traditional antibacterials, however, which are currently being actively explored as viable solutions to the pressing problem of AMR, consistency in drug testing and reporting is far from achieved. For antivirulence drugs in particular, a major challenge lies in standardising preclinical testing for a largely diverse set of targets that potentially mediate multiple different phenotypes in bacterial pathogens. Measuring virulence target inhibition reliably and at large-scale is often difficult when using microbiological assays, so when the target is known, inhibitor screening and early evaluation typically relies on biochemical/biophysical approaches. This is the case for DsbA inhibitors that have been reported to date, with hits from several chemical classes having been identified as part of fragment-based screening campaigns primarily using saturation transfer difference NMR spectroscopy12,21,22,23. Later-stage microbiological evaluation has validated some but not all hits, and in some cases, even chemically elaborated analogues have failed to show activity in cell-based assays12,30. In this study, we have optimised two cell-based assays previously used to monitor DsbA function in vivo for an accurate, streamlined, and high-throughput testing pipeline of DsbA inhibitors. When combined, these assays could support DsbA inhibitor development from hit identification to lead optimisation and preclinical candidate validation.
For developing a high throughput assay, we chose to use a read-out that is a native virulence substrate of DsbA in UPEC. ASST is a large periplasmic enzyme encoded by UPEC and other intestinal bacteria30 that was reported to be upregulated in the urine of UPEC-infected mice49,50, but was not required for colonisation of the murine bladder30. The gene encoding ASST (astA) is found in a tri-cistronic operon with the dsbL and dsbI genes, which encode an accessory redox protein pair in UPEC with specificity for ASST40,43,51, although the DsbA and DsbB redox pair was also shown to functionally fold ASST28,30. The ASST activity assay was previously performed in liquid medium using bacterial cell lysates40 or on solid medium using whole live cells28. To evaluate DsbA inhibitors, we have previously utilised the solid medium cell-based method to successfully quantify DsbA inhibition of ASST activity30. Despite this being an accurate cell-based assay, its petri-dish format has several limiting factors, which are preventing its use in inhibitor screens: (i) a relatively low-throughput capacity, (ii) the requirement of high inhibitor quantities, and (iii) manual data collection by endpoint imaging28,30. The modified ASST sulfotransferase enzyme assay presented here operates on the same principle, yet its application is quite different. The assay is conducted in liquid media using live UPEC cells treated with minimal quantities of DsbA inhibitor, and the activity of ASST is assessed in an automated fashion by monitoring the MUS-phenol sulfotransferase reaction spectrofluorometrically in real-time (rather than as an endpoint30). In addition, conducting the enzyme assay in liquid medium significantly increased scalability while drastically reduced reaction volumes and the amount of substrate and inhibitor needed. In fact, by performing the assay in 96-well plates the amount of substrate and inhibitor used was reduced by 100-fold compared to previous assay methodology30. While we showcased scalability by conducting the assay in a 96-well format, the assay can be additionally downscaled to suit a 384-well plate, which would further reduce the amount of substrate and inhibitor required (1000-fold reduction over the previous method30).
Our HTS ASST enzyme assay not only functions as a method for identifying DsbA inhibitors, but it could potentially be utilised for the identification of inhibitors for other targets, as it monitors both bacterial growth and sulfotransferase activity. Sulfotransferase enzymes are widespread in pathogenic bacteria (e.g. Klebsiella sp., S. Typhimurium, Mycobacterium tuberculosis)35,38,52 and eukaryotic cells, where they play a pivotal role in the detoxification of xenobiotics, drugs, and other endogenous compounds53. Most importantly, sulfotransferases have been linked to several disease states (e.g. cancer and infection) which has prompted studies into their specific functions54,55,56 and search for selective inhibitors57,58. However, very few HTS methods are available for identifying sulfotransferase inhibitors59. Our HTS ASST assay could be optimised for use with different sulfotransferase enzymes thus facilitating the development of selective sulfotransferase inhibitors. However, for the purposes of a DsbA targeted inhibitor screen, we aimed to ensure that our assay could differentiate between inhibitors targeting DsbA vs. growth and/or ASST. To identify and exclude potential growth inhibitors we added a bacterial growth analysis step (prior to measuring ASST activity and during bacterial treatment with inhibitors), which required no additional inhibitor, and is also easily scalable to fit the 384-well format, a benefit for future fragment-based drug design approaches. Identifying inhibitors that block ASST’s sulfotransferase activity without inhibiting DsbA required the use of genetic controls. While our ASST assay can be easily modified to run in various strain backgrounds in parallel (as demonstrated by the use of various knock-out mutants), doing so in a compound screening scenario is not ideal as it would significantly increase compound quantity requirements. As such, controlling for hit target specificity by this approach was not considered cost-effective. Instead we opted for an orthogonal technique to confirm hits from our primary screen, an approach commonly used in drug discovery campaigns. The development of a second cell-based assay that could be run subsequently to the ASST assay and was based on a different DsbA reporter phenotype (bacterial motility) presented with several added advantages.
Flagella-mediated bacterial motility is a well-established reporter phenotype for DsbA activity and thus the standard petri-dish soft agar motility assay has been successfully used to evaluate DsbA inhibitors in vivo12,30. However, the current format of the bacterial motility assay in petri-dishes has the same limitations of relatively low-throughput capacity, high inhibitor quantity requirement and manually intensive data acquisition of other petri-dish based assays. Our modified plate-reader motility assay utilises the same soft agar methodology, however, instead of relying on incremental images and manual measurements of motility zones, it uses a fully-automated system (plate-reader absorbance measurements) and requires no human intervention throughout the assay period making it less labour-intensive and less prone to bias or human error in motility assessments. In addition, automating the assay ensures conditions (e.g. temperature) are better controlled and can remain constant from start to finish. An important improvement was in that downscaling the assay from a petri-dish to a multi-well plate format, drastically reduced the quantity of inhibitor required (30-fold reduction in 24-well plate and 60-fold reduction in 48-well plate, compared to previous method30). While others have demonstrated that an agar-based motility assay can be performed in 96-well or even a 384-well plate46,60, these assays require specific and/or specialised equipment not typically present in the average research lab or do not allow for small differences in motility inhibition to be accurately observed and assessed. For these reasons, our absorbance-based motility assay is better suited to inhibitor confirmation and evaluation post-hit discovery.
In conclusion, our study describes the establishment of a microbiological assay pipeline that can support DsbA inhibitor development all the way from screening to early preclinical candidate validation. In addition, our assays are also well suited for screening and evaluating other types of inhibitors (e.g. for flagella components, motility regulators, quorum sensing, sulfotransferase activity), and may find multiply uses in other inhibitor screening projects. Importantly, we hope our study will serve as a paradigm for the development of similarly accurate, easy to perform, and high throughput cell-based assays that can advance the discovery and preclinical development of other antivirulence drugs that could offer future solutions to curbing the AMR crisis.
Methods
Bacterial strains, plasmids, and culture conditions
All bacterial strains utilised in this study (Table 2) were routinely cultured at 37 °C in liquid or on solid lysogeny broth (LB-Lennox) medium supplemented, when required, with chloramphenicol (34 µg/mL) or ampicillin (100 µg/mL), or both. CFT073 mutants were constructed previously using λ-red-mediated homologous recombination as described elsewhere29,61. Plasmids pASST28, pEcDsbA29, and pSU2718 were routinely transformed into strains using electroporation.
Chemicals and stock solutions
Chloramphenicol, ampicillin, phenol, and MUS were purchased from Sigma-Aldrich (Australia), F1 and F2 were purchased from Thermo Fisher Scientific (Australia), and F4 was purchased from Synthesis Med Chem (Australia). MUS (10 mM) and phenol (50 mM) solutions were prepared in sodium chloride (0.9%), and F1 (250 mM) solution was prepared in dimethyl sulfoxide (DMSO). All stock solutions were stored in the absence of light at − 20 °C. Working solutions were prepared in LB-Lennox and were used on the same day.
Absorbance-based bacterial motility assay
Bacterial strains were grown by static 24-h culture in LB-Lennox media at 37 °C. Cultures were normalized to an O.D. 600 nm of 2 (~ 2 × 109 CFU/mL) using a spectrophotometer. The multi-well soft agar plates were prepared by adding a volume of 700 µL (24-well) or 350 µL (48-well) of soft LB-Lennox agar (0.25% [wt/vol]), containing either DMSO (0.4%, vehicle control) or inhibitors F1, F2 or F4 at various concentrations (1–0.1 mM), to each well of the plate. The soft agar was allowed to solidify for at least 2 h at room temperature (21 °C), before being inoculated in the left-hand corner of each well with 1 µL of normalized culture (~ 2 × 109 CFU/mL). Inoculated plates were incubated at room temperature for 20 min to allow the inoculum to dry. The zone of motility was measured by incubating plates at 37 °C in a CLARIOstar plate reader (BMG, Australia) programmed to measure absorbance (O.D. 600 nm) at each hour over 15 h (Fig. 7). Absorbance measurements were made using the inbuilt spiral averaging function with orbital averaging producing similar results (data not shown). Instrument data were normalized (with DMSO vehicle control data set at 100%) and plotted using GraphPad Prism 8. Mean motility values were calculated from 3 biological replicates of each strain tested under each specific condition. The start and end of motility were estimated from motility curves and were defined as the beginning and endpoints, respectively, of the exponential phase (zone of motility). The slope of each zone of motility was calculated in Excel and group means were compared for statistical differences by one-way ANOVA (p < 0.05) in GraphPad Prism 8. F1 dose–response curves were generated using the motility slopes, and the corresponding IC50 value was calculated by applying a non-linear regression (curve fit) Y = 100/(1 + 10^((LogIC50-X) × HillSlope))).

Overview of absorbance-based bacterial motility assay. (A) Bacterial strains were cultured statically in LB-Lennox media for 24 h. (B) Overnight cultures were normalized to an O.D. 600 nm of 2. (C) 24-well motility plates were prepared by pipetting 700 µL of warm (55 °C) soft LB agar (0.25%) in each well supplemented with F1 inhibitor or vehicle control (DMSO). The soft agar was allowed to solidify at room temperatures for at least 2 h. (D) 1 µL of bacterial culture fixed at O.D. 600 nm = 2 was inoculated onto the surface of triplicate soft agar wells by depositing the inoculum at the left edge of the well (with care not to penetrate the agar). The inoculum was allowed to dry onto the agar for 20 min at room temperature before the plate was covered with a plastic lid. (E) Bacterial swimming motility was monitored spectrophotometrically for 15-h in a plate reader (BMG Australia) at 37 °C (using orbital, spiral, or matrix averaging to ensure optimal well coverage). (F) Data acquisition and analysis.
Cell-based ASST sulfotransferase enzyme assay
Bacterial strains were cultured in LB media, supplemented with antibiotics as appropriate, at 37 °C overnight with shaking at 200 rpm. Overnight cultures were used as inocula in bacterial growth assays (step 1) conducted in a 96-well plate by preparing two-fold serial dilutions of F1, F2 or F4 inhibitor compound or DMSO (vehicle control) at twice the desired final concentration in LB-Lennox medium (100 µL final volume). Each well was then inoculated with 100 µL of 1 × 107 CFU/mL inoculum, to give a total well volume of 200 µL and a final cell concentration of 5 × 106 CFU/mL. The growth analysis plate was covered with a breathable sealing membrane (Breathe-Easy sealing membrane, Sigma, Australia), and incubated at 37 °C for 15 h with shaking (300 rpm) in a CLARIOstar plate reader (BMG, Australia) programmed to obtained O.D. 600 nm measurements every 15 min over the 15-h period. At the end of the culture period, each well was normalized to an O.D. 600 nm of 0.4 (~ 3.5 × 108 CFU/mL) in a fresh 96-well plate (step 2), to ensure that each well contained an equal number of cells. Wells were then supplemented with potassium 4-methylumbelliferyl sulfate (MUS, Sigma, Castle Hill, Australia) (0.5 mM final concentration), and phenol (Sigma, Castle Hill, Australia) (1 mM final concentration) and sulfotransferase activity was monitored immediately in a CLARIOstar plate reader (BMG, Australia) by measuring fluorescence emitted at 450–480 nm (excitation wavelength at 360–380 nm) and measurements acquired every 5 min over a 60–90 min time period (Fig. 8). MUS and other similar substrates are typically used in the mM range when examining ASST function where whole cells or whole cell lysates are used28,39,40,44. In our analysis, we found 0.5 mM of MUS and 1 mM of phenol gave the most consistent results (data not shown). Instrument data were normalized (with DMSO vehicle control set at 100%) and analysed using GraphPad Prism 8. The F1 dose–response curve was generated using fluorescence data from the 40-min time point, and the corresponding IC50 value was calculated by applying a non-linear regression (curve fit) Y = 100/(1 + 10^((LogIC50-X) × HillSlope))).

Overview of cell-based ASST sulfotransferase assay. (A) Bacterial strains were cultured in LB-Lennox media overnight at 37 °C with aeration (200 rpm). (B) Growth analysis plates were prepared by subculturing the O/N cultures from (A) into a 96-well plate containing the test inhibitors (akin to preparing an MIC challenge plate). (C) Growth plates were incubated at 37 °C, 300 rpm, in a microplate reader programmed to take O.D. 600 nm readings every 15 min for 15 h. (D) Growth plate cultures were transferred in a fresh 96-well plate with each culture well normalised at an O.D. 600 nm of 0.4. (E) A master mix containing 4 µL of phenol (50 mM), 10 µL of MUS (10 mM), and 126 µL of LB-Lennox per reaction well was prepared. (F) The reaction master mix (140 µL) was added to normalized cultures (60 µL) and mixed. (G) Fluorescence at 450–480 nm was immediately monitored in a plate reader (BMG, Australia) with measurements obtained every 5 min for up to 90 min. (H): Data acquisition and analysis.
Method and formula for calculating Z′-factor62
The Z´-factor was calculated using the formula below and positive and negative genetic controls, CFT073/pASST and CFT073/pSU2718, respectively. The mean Z´-factor was calculated across the 10–60 min time range (Fig. 2A), and positive and negative control values were obtained from two separate experiments each consisting of 4 biological and 3 technical replicates.
References
World Health Organization. Antimicrobial resistance: Global report on surveillance. (2014). https://apps.who.int/iris/handle/10665/112642.
World Health Organization. Antibacterial agents in clinical development: An analysis of the antibacterial clinical development pipeline. (2019). https://apps.who.int/iris/bitstream/handle/10665/330420/9789240000193-eng.pdf.
World Health Organization. Antibacterial agents in preclinical development: An open access database. (2019). https://apps.who.int/iris/bitstream/handle/10665/330290/WHO-EMP-IAU-2019.12-eng.pdf.
Allen, R. C., Popat, R., Diggle, S. P. & Brown, S. P. Targeting virulence: Can we make evolution-proof drugs?. Nat. Rev. Microbiol. 12, 300–308. https://doi.org/10.1038/nrmicro3232 (2014).
Escaich, S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol. 12, 400–408. https://doi.org/10.1016/j.cbpa.2008.06.022 (2008).
Dickey, S. W., Cheung, G. Y. C. & Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug. Discov. 16, 457–471. https://doi.org/10.1038/nrd.2017.23 (2017).
Heras, B., Scanlon, M. J. & Martin, J. L. Targeting virulence not viability in the search for future antibacterials. Br. J. Clin. Pharmacol. 79, 208–215. https://doi.org/10.1111/bcp.12356 (2015).
Fleitas Martínez, O., Cardoso, M. H., Ribeiro, S. M. & Franco, O. L. Recent advances in anti-virulence therapeutic strategies with a focus on dismantling bacterial membrane microdomains, toxin neutralization, quorum-sensing interference and biofilm inhibition. Front. Cell. Infect. Microbiol. 9, 74–74. https://doi.org/10.3389/fcimb.2019.00074 (2019).
Lyons, B. J. E. & Strynadka, N. C. J. On the road to structure-based development of anti-virulence therapeutics targeting the type III secretion system injectisome. Med. Chem. Commun. 10, 1273–1289. https://doi.org/10.1039/C9MD00146H (2019).
Totsika, M. Benefits and challenges of antivirulence antimicrobials at the dawn of the post-antibiotic era. Curr. Med. Chem. 6, 30–37. https://doi.org/10.2174/2210303106666160506120057 (2016).
Defoirdt, T., Brackman, G. & Coenye, T. Quorum sensing inhibitors: How strong is the evidence?. Trends Microbiol. 21, 619–624. https://doi.org/10.1016/j.tim.2013.09.006 (2013).
Adams, L. A. et al. Application of fragment-based screening to the design of inhibitors of Escherichia coli DsbA. Angew. Chem. Int. Ed. Engl. 54, 2179–2184. https://doi.org/10.1002/anie.201410341 (2015).
Heras, B. et al. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225. https://doi.org/10.1038/nrmicro2087 (2009).
Lasica, A. M. & Jagusztyn-Krynicka, E. K. The role of Dsb proteins of Gram-negative bacteria in the process of pathogenesis. FEMS Microbiol. Rev. 31, 626–636. https://doi.org/10.1111/j.1574-6976.2007.00081.x (2007).
Yu, J. & Kroll, J. S. DsbA: A protein-folding catalyst contributing to bacterial virulence. Microbes Infect. 1, 1221–1228. https://doi.org/10.1016/S1286-4579(99)00239-7 (1999).
Landeta, C., Boyd, D. & Beckwith, J. Disulfide bond formation in prokaryotes. Nat. Microbiol. 3, 270–280. https://doi.org/10.1038/s41564-017-0106-2 (2018).
Bardwell, J. C. Building bridges: Disulphide bond formation in the cell. Mol. Microbiol. 14, 199–205. https://doi.org/10.1111/j.1365-2958.1994.tb01281.x (1994).
Bardwell, J. C. et al. A pathway for disulfide bond formation in vivo. Proc. Natl. Acad. Sci. USA 90, 1038. https://doi.org/10.1073/pnas.90.3.1038 (1993).
Missiakas, D., Georgopoulos, C. & Raina, S. Identification and characterization of the Escherichia coli gene dsbB, whose product is involved in the formation of disulfide bonds in vivo. Proc. Natl. Acad. Sci. USA 90, 7084. https://doi.org/10.1073/pnas.90.15.7084 (1993).
Smith, R. P., Paxman, J. J., Scanlon, M. J. & Heras, B. Targeting bacterial Dsb proteins for the development of anti-virulence agents. Molecules https://doi.org/10.3390/molecules21070811 (2016).
Halili, M. A. et al. Small molecule inhibitors of disulfide bond formation by the bacterial DsbA–DsbB dual enzyme system. ACS Chem. Biol. 10, 957–964. https://doi.org/10.1021/cb500988r (2015).
Duncan, L. F. et al. The fragment-based development of a benzofuran hit as a new class of Escherichia coli DsbA inhibitors. Molecules https://doi.org/10.3390/molecules24203756 (2019).
Mohanty, B. et al. Fragment library screening identifies hits that bind to the non-catalytic surface of Pseudomonas aeruginosa DsbA1. PLoS ONE 12, e0173436. https://doi.org/10.1371/journal.pone.0173436 (2017).
Landeta, C. et al. Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 11, 292–298. https://doi.org/10.1038/nchembio.1752 (2015).
Früh, V. et al. Application of fragment-based drug discovery to membrane proteins: Identification of ligands of the integral membrane enzyme DsbB. Chem. Biol. 17, 881–891. https://doi.org/10.1016/j.chembiol.2010.06.011 (2010).
Landeta, C. et al. Inhibition of Pseudomonas aeruginosa and Mycobacterium tuberculosis disulfide bond forming enzymes. Mol. Microbiol. 111, 918–937. https://doi.org/10.1111/mmi.14185 (2019).
Dailey, F. E. & Berg, H. C. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. P. Natl. A. Sci. USA 90, 1043–1047. https://doi.org/10.1073/pnas.90.3.1043 (1993).
Heras, B. et al. Structural and functional characterization of three DsbA paralogues from Salmonella enterica serovar typhimurium. J. Biol. Chem. 285, 18423–18432. https://doi.org/10.1074/jbc.M110.101360 (2010).
Totsika, M., Heras, B., Wurpel, D. J. & Schembri, M. A. Characterization of two homologous disulfide bond systems involved in virulence factor biogenesis in uropathogenic Escherichia coli CFT073. J. Bacteriol. 191, 3901. https://doi.org/10.1128/JB.00143-09 (2009).
Totsika, M. et al. Inhibition of diverse DsbA enzymes in multi-DsbA encoding pathogens. Antioxid. Redox Signal. 29, 653–666. https://doi.org/10.1089/ars.2017.7104 (2018).
Agudo, D., Mendoza, M. T., Castanares, C., Nombela, C. & Rotger, R. A proteomic approach to study Salmonella typhi periplasmic proteins altered by a lack of the DsbA thiol: Disulfide isomerase. Proteomics 4, 355–363. https://doi.org/10.1002/pmic.200300554 (2004).
Arts, I. S. et al. Dissecting the machinery that introduces disulfide bonds in Pseudomonas aeruginosa. mBio 4, e00912-00913. https://doi.org/10.1128/mBio.00912-13 (2013).
Coulthurst, S. J. et al. DsbA plays a critical and multifaceted role in the production of secreted virulence factors by the phytopathogen Erwinia carotovora subsp. atroseptica. J. Biol. Chem. 283, 23739–23753. https://doi.org/10.1074/jbc.M801829200 (2008).
Hayashi, S., Abe, M., Kimoto, M., Furukawa, S. & Nakazawa, T. The dsbA-dsbB disulfide bond formation system of Burkholderia cepacia is involved in the production of protease and alkaline phosphatase, motility, metal resistance, and multi-drug resistance. Microbiol. Immunol. 44, 41–50. https://doi.org/10.1111/j.1348-0421.2000.tb01244.x (2000).
Kim, D.-H., Kim, H.-S. & Kobashi, K. Purification and Characterization of novel sulfotransferase obtained from Klebsiella K-36, an intestinal bacterium of Rat1. J. Biochem. 112, 456–460. https://doi.org/10.1093/oxfordjournals.jbchem.a123921 (1992).
Malojcić, G. et al. A structural and biochemical basis for PAPS-independent sulfuryl transfer by aryl sulfotransferase from uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 105, 19217–19222. https://doi.org/10.1073/pnas.0806997105 (2008).
Kang, J. W. et al. Cloning, sequence analysis, and characterization of the astA gene encoding an arylsulfate sulfotransferase from Citrobacter freundii. Arch. Pharm. Res. 24, 316–322. https://doi.org/10.1007/bf02975099 (2001).
Kang, J. W., Kwon, A. R., Kim, D. H. & Choi, E. C. Cloning and sequencing of the astA gene encoding arylsulfate sulfotransferase from Salmonella typhimurium. Biol. Pharm. Bull. 24, 570–574. https://doi.org/10.1248/bpb.24.570 (2001).
Kwon, A. R., Oh, T. G., Kim, D. H. & Choi, E. C. Molecular cloning of the arylsulfate sulfotransferase gene and characterization of its product from Enterobacter amnigenus AR-37. Protein Expr. Purif. 17, 366–372. https://doi.org/10.1006/prep.1999.1129 (1999).
Grimshaw, J. P. et al. DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J. Mol. Biol. 380, 667–680. https://doi.org/10.1016/j.jmb.2008.05.031 (2008).
Kwon, A. R. & Choi, E. C. Role of disulfide bond of arylsulfate sulfotransferase in the catalytic activity. Arch. Pharm. Res. 28, 561–565. https://doi.org/10.1007/bf02977759 (2005).
Totsika, M. et al. Inhibition of diverse DsbA enzymes in multi-DsbA encoding pathogens. Antioxid. Redox. Sign. 29, 653–666. https://doi.org/10.1089/ars.2017.7104 (2018).
Malojcic, G., Owen, R. L. & Glockshuber, R. Structural and mechanistic insights into the PAPS-independent sulfotransfer catalyzed by bacterial aryl sulfotransferase and the role of the DsbL/Dsbl system in its folding. Biochemistry 53, 1870–1877. https://doi.org/10.1021/bi401725j (2014).
Yun, H. J., Kwon, A. R. & Choi, E. C. Bacterial arylsulfate sulfotransferase as a reporter system. Microbiol. Immunol. 45, 673–678. https://doi.org/10.1111/j.1348-0421.2001.tb01301.x (2001).
Zhang, J. H., Chung, T. D. & Oldenburg, K. R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73. https://doi.org/10.1177/108705719900400206 (1999).
Malapaka, V. R., Barrese, A. A., Tripp, B. C. & Tripp, B. C. High-throughput screening for antimicrobial compounds using a 96-well format bacterial motility absorbance assay. J. Biomol. Screen. 12, 849–854. https://doi.org/10.1177/1087057107304478 (2007).
CLSI. in CLSI Document M07-A10 Vol. Approved Standard—10th Edition (Wayne, PA, 2015).
Reller, L. B., Weinstein, M., Jorgensen, J. H. & Ferraro, M. J. Antimicrobial susceptibility testing: A review of general principles and contemporary practices. Clin. Infect. Dis. 49, 1749–1755. https://doi.org/10.1086/647952 (2009).
Lloyd, A. L., Rasko, D. A. & Mobley, H. L. Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J. Bacteriol. 189, 3532–3546. https://doi.org/10.1128/jb.01744-06 (2007).
Snyder, J. A. et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect. Immun. 72, 6373–6381. https://doi.org/10.1128/IAI.72.11.6373-6381.2004 (2004).
Malojčić, G. & Glockshuber, R. The PAPS-independent aryl sulfotransferase and the alternative disulfide bond formation system in pathogenic bacteria. Antioxid. Redox. Sign. 13, 1247–1259. https://doi.org/10.1089/ars.2010.3119 (2010).
Mougous, J. D., Green, R. E., Williams, S. J., Brenner, S. E. & Bertozzi, C. R. Sulfotransferases and sulfatases in mycobacteria. Chem. Biol. 9, 767–776. https://doi.org/10.1016/S1074-5521(02)00175-8 (2002).
Gamage, N. et al. human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 90, 5–22. https://doi.org/10.1093/toxsci/kfj061 (2006).
Qian, Y., Deng, C. & Song, W.-C. Expression of estrogen sulfotransferase in MCF-7 cells by cDNA transfection suppresses the estrogen response: Potential role of the enzyme in regulating estrogen-dependent growth of breast epithelial cells. J. Pharmacol. Exp. Ther. 286, 555 (1998).
Kaltenbach, D. D. et al. Sulfotransferase and heparanase: Remodeling engines in promoting virus infection and disease development. Front. Pharmacol. 9, 1315–1315. https://doi.org/10.3389/fphar.2018.01315 (2018).
Frame, L. T., Gatlin, T. L., Kadlubar, F. F. & Lang, N. P. Metabolic differences and their impact on human disease sulfotransferase and colorectal cancer. Environ. Toxicol. Pharmacol. 4, 277–281. https://doi.org/10.1016/s1382-6689(97)10023-0 (1997).
Armstrong, J. I. et al. A library approach to the generation of bisubstrate analogue sulfotransferase inhibitors. Org. Lett. 3, 2657–2660. https://doi.org/10.1021/ol0162217 (2001).
Gesteira, T. F. et al. Inhibitory peptides of the sulfotransferase domain of the heparan sulfate enzyme, N-deacetylase-N-sulfotransferase-1. J. Biol. Chem. 286, 5338–5346. https://doi.org/10.1074/jbc.M110.100719 (2011).
Paul, P., Suwan, J., Liu, J., Dordick, J. S. & Linhardt, R. J. Recent advances in sulfotransferase enzyme activity assays. Anal. Bioanal. Chem. 403, 1491–1500. https://doi.org/10.1007/s00216-012-5944-4 (2012).
Rasmussen, L. et al. A high-throughput screening assay for inhibitors of bacterial motility identifies a novel inhibitor of the Na+-driven flagellar motor and virulence gene expression in Vibrio cholerae. Antimicrob. Agents Chemother. 55, 4134–4143. https://doi.org/10.1128/AAC.00482-11 (2011).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640. https://doi.org/10.1073/pnas.120163297 (2000).
Sui, Y. & Wu, Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J. Biomol. Screen. 12, 229–234. https://doi.org/10.1177/1087057106296498 (2007).
Bardwell, J. C., McGovern, K. & Beckwith, J. Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589. https://doi.org/10.1016/0092-8674(91)90532-4 (1991).
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. https://doi.org/10.1038/35023079 (2000).
Holloway, B. W. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbiol. 13, 572–581. https://doi.org/10.1099/00221287-13-3-572 (1955).
Wray, C. & Sojka, W. J. Experimental Salmonella typhimurium infection in calves. Res. Vet. Sci. 25, 139–143 (1978).
Hoiseth, S. K. & Stocker, B. A. D. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239. https://doi.org/10.1038/291238a0 (1981).
Mobley, H. L. et al. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect. Immun. 58, 1281–1289 (1990).
Acknowledgements
This work was supported by a National Health and Medical Research Council Project Grant (GNT1144046), a Clive and Vera Ramaciotti Health Investment Grant (2017HIG0119) and an ARC Discovery Early Career Researcher Award (DE130101169). MT was supported by a Queensland University of Technology Vice-Chancellor’s Research Fellowship.
Author information
Authors and Affiliations
Contributions
M.T. designed the project, obtained the funding, contributed to experimental design and data analysis, and wrote and edited the manuscript. A.D.V. designed and carried out the experiments, analysed the data, prepared figures and wrote the original manuscript. R.D., Y.H., and T.K.A. assisted with experimental design, carrying out experiments, analysing data and proofreading the manuscript. B.H. provided expert advice, contributed to obtaining the funding and study design and proofread the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verderosa, A.D., Dhouib, R., Hong, Y. et al. A high-throughput cell-based assay pipeline for the preclinical development of bacterial DsbA inhibitors as antivirulence therapeutics. Sci Rep 11, 1569 (2021). https://doi.org/10.1038/s41598-021-81007-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-81007-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
