
Abstract
The STRC gene, located on chromosome 15q15.3, is one of the genetic causes of autosomal recessive mild-to-moderate sensorineural hearing loss. One of the unique characteristics of STRC-associated hearing loss is the high prevalence of long deletions or copy number variations observed on chromosome 15q15.3. Further, the deletion of chromosome 15q15.3 from STRC to CATSPER2 is also known to be a genetic cause of deafness infertility syndrome (DIS), which is associated with not only hearing loss but also male infertility, as CATSPER2 plays crucial roles in sperm motility. Thus, information regarding the deletion range for each patient is important to the provision of appropriate genetic counselling for hearing loss and male infertility. In the present study, we performed next-generation sequencing (NGS) analysis for 9956 Japanese hearing loss patients and analyzed copy number variations in the STRC gene based on NGS read depth data. In addition, we performed Multiplex Ligation-dependent Probe Amplification analysis to determine the deletion range including the PPIP5K1, CKMT1B, STRC and CATSPER2 genomic region to estimate the prevalence of the STRC-CATSPER deletion, which is causative for DIS among the STRC-associated hearing loss patients. As a result, we identified 276 cases with STRC-associated hearing loss. The prevalence of STRC-associated hearing loss in Japanese hearing loss patients was 2.77% (276/9956). In addition, 77.1% of cases with STRC homozygous deletions carried a two copy loss of the entire CKMT1B-STRC-CATSPER2 gene region. This information will be useful for the provision of more appropriate genetic counselling regarding hearing loss and male infertility for the patients with a STRC deletion.
Similar content being viewed by others

Introduction
Congenital hearing loss is one of the most common sensory disorders, occurring in 1 in 700–1000 newborns, with approximately 60% of cases attributable to genetic causes1. The STRC gene, which is located on chromosome 15q15.3 in what is known as the DFNB16 locus, is one of the genetic causes of autosomal recessive mild-to-moderate sensorineural hearing loss2,3.
One of the unique characteristics of STRC-associated hearing loss is the high prevalence of long deletions or copy number variations observed on chromosome 15q15.3. Chromosome 15q15.3 contains segmental duplications including five functional genes and three pseudo-genes, PPIP5K1 (MIM: 610,979), CKMT1B (MIM: 123,290), STRC (MIM: 606,440), CATSPER2 (MIM: 607,249), PPIP5K1P1, CKMT1A (MIM: 613,415), STRCP1, and CATSPER2P1. Pseudo-STRC (i.e., STRCP1) is located downstream of the STRC gene and shows 98% homology to the functional STRC gene. This high degree of homology makes variant interpretation in the analysis of next-generation sequencing results challenging. STRC-associated hearing loss is a relatively common genetic cause of non-syndromic hearing loss, with an incidence of 5.4–16.1% in hearing loss populations with mixed ethnicity4,5, 6–11.2% in American hearing loss patients6,7, 6% in Italian hearing loss patients8, 1.7–2.4% in Japanese hearing loss patients9,10, 2.6% in Turkish autosomal recessive hearing loss patients11, 10% in Czech mild-to-moderate hearing loss patients12, 10.8% in Korean mild-to-moderate hearing loss patients13, and 6% in GJB2-negative Germany hearing loss patients14.
Further, the deletion of chromosome 15q15.3 from STRC to CATSPER2, is also known to be a genetic cause of deafness infertility syndrome (DIS: OMIM 611,102), which is associated with not only hearing loss but also male infertility, as CATSPER2 plays crucial roles in sperm motility15,16. Thus, data regarding the deletion range for each patient is important for appropriate genetic counseling in terms of hearing loss and male infertility. Several patterns regarding the deletion range of chromosome 15q15.3 have been reported (HGMD professional), with some patients harboring a deletion lacking only the STRC gene with the CATSPER2 gene intact. Thus, the prevalence of the STRC-CATSPER deletion among STRC-associated hearing loss patients is thought to be valuable information.
In the present study, we performed next-generation sequencing (NGS) analysis for a large number of Japanese hearing loss patients (n = 9956) and analyzed copy number variations (CNVs) in the STRC gene using NGS read depth data. In addition, we performed Multiplex Ligation-dependent Probe Amplification (MLPA) analysis to determine the deletion range including the PPIP5K1, CKMT1B, STRC and CATSPER2 genomic regions to estimate the prevalence of the STRC-CATSPER deletion, which is causative for DIS among STRC-associated hearing loss patients.
Methods
Subjects
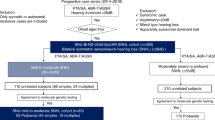
We performed target re-sequencing analysis for 9956 unrelated Japanese sensorineural hearing loss patients (2069 autosomal dominant or mitochondrial inheritance cases, 5831 autosomal recessive inheritance or sporadic cases, 1944 unknown family history cases and 112 unilateral hearing loss cases) enrolled in this study from 90 otorhinolaryngology departments nationwide. Peripheral blood samples were obtained and DNA was extracted using a DNeasy blood and tissue kit (QIAGEN, Hilden, Germany). Evaluation of HL was performed by pure-tone audiometry for patients aged 4 years or older. The pure-tone average (PTA) was calculated from the audiometric thresholds at four frequencies (500, 1000, 2000 and 4000 Hz). The severity of HL was classified into 4 categories: mild (PTA 20-40 dB), moderate (41-70 dB), severe (71-90 dB) and profound (> 91 dB). The average age of participants was 26.5 (range 0 to 105), with 3812 males and 4819 females and 1325 without gender information.
Informed written consent was obtained from all subjects (or guardians in the case of minors) prior to participation. This study was approved by the Shinshu University Ethics Committee (Approval no.: 576) and the ethics committees of all other participating institutions. All procedures were performed in accordance with the Guidelines for Genetic Tests and Diagnoses in Medical Practice of the Japanese Association of Medical Sciences and the Declaration of Helsinki.
Next-generation sequencing
Next-generation sequencing was performed for the 63 genes reported to cause non-syndromic hearing loss as described in a previous report17. In brief, amplicon libraries were prepared by using the Ion AmpliSeq Custom Panel, with the Ion AmpliSeq Library Kit 2.0 and the Ion Xpress Barcode Adapter 1–96 Kit (Life Technologies) according to the manufacturer’s instructions. After amplicon library preparation, equal amounts of the libraries for forty-five patients per one sequence run were pooled. After library preparation, next-generation sequencing analysis was performed using the Ion Proton system with an Ion P1 chip or the Ion S5 system with an Ion 540 chip according to the manufacturer’s instructions. We performed about 230 runs for the sequencing of 9956 patients. The averaged number of reads per sample was 1,684,289 reads (range 275,889–24,777,837) and the average depth of coverage was 667 (range 106–10,126).
The sequence data were aligned to the human reference genome sequence (build GRCh37/hg19) using the Torrent Mapping Alignment Program (TMAP). Subsequently, DNA variants were piled up with the Torrent Variant Caller plug-in software included in the Torrent Suit (Life Technologies).
The effects of the variants were analyzed by using ANNOVAR software18. The missense, nonsense, insertion/deletion, and splicing variants were selected from among the identified variants. Variants were further filtered to retain only those with a minor allele frequency under 1% in several control databases including the 1000 genome database (http://www.1000genomes.org/), the 6500 exome variants (http://evs.gs.washington.edu/EVS/), The Genome Aggregation Database (https://gnomad.broadinstitute.org), the human genetic variation database (dataset for 1208 Japanese exome variants) (http://www.genome.med.kyoti-u.ac.jp/SnpDB/index.html), the 8300 Japanese genome variation database (https://jmorp.megabank.tohoku.ac.jp/202102/) and the 333 in-house Japanese normal hearing controls. All filtering procedures were performed using our previously described original database software19. The pathogenicity of the identified variants was evaluated in accordance with the American College of Medical Genetics (ACMG) standards and guidelines20 with the ClinGen hearing loss clinical domain working group expert specification21. Copy number variant (CNV) analysis was performed according to our previous report22.
MLPA analysis
MLPA analysis was performed to confirm the CNVs identified from the read depth data obtained by next-generation sequencing analysis. MLPA was performed using SALSA MLPA Probe mix P461-A1 and the SALSA MLPA EK1 reagent kit FAM according to the manufacturer’s instructions (MRC-Holland, Amsterdam, Netherlands). MLPA amplicon fragment lengths were analyzed by ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, USA) with the GeneScan 500 LIZ Size Standard (Applied Biosystems, Foster City, USA). The fragment data were analyzed using Coffalyser.net software (MRC-Holland). We performed two duplicate tests for each sample.
Results
Prevalence and clinical features of STRC-associated hearing loss
In this study, we performed large-cohort next-generation sequencing analysis and CNV analysis for 9956 Japanese hearing loss patients. As a result, we identified 1258 (12.6%) patients carrying CNVs in 63 previously reported deafness genes. The most prevalent CNVs identified in this study were those in the STRC gene, which were observed in 1093 cases (231 cases carrying a 2 copy number loss, 612 cases carrying a 1 copy number loss, 243 cases carrying a 1 copy number gain and 7 cases carrying a 2 copy number gain). Among the 612 cases with a 1 copy number loss of the STRC gene, 41 cases carried single nucleotide variants (SNVs) or small insertion/deletion variants in the trans allele configuration. In addition to the 41 cases with SNVs or small ins/dels in the trans allele with the single copy loss of STRC, 1 case had compound heterozygous SNVs and 3 cases had homozygous SNVs. The SNVs or small insertion/deletion variants identified in this study are summarized in Table 1. Thus, a total of 276 cases were identified with STRC-associated hearing loss in this study, with the prevalence in Japanese hearing loss patients being 2.77% (276/9956) among all patients, 3.29% (192/5831) among autosomal recessive or sporadic hearing loss cases and 0.87% (18/2069) among autosomal dominant hearing loss cases. The STRC-associated hearing loss identified from autosomal dominant families is caused by pseudodominance as we previously reported9. When we restricted the patients to those with mild-to-moderate hearing loss, the prevalence of STRC-associated hearing loss was increased to 4.27% (132/3088). In addition, we identified only 2 cases with STRC-associated hearing loss among severe-to-profound hearing loss patients (0.07%; 2/2948). These two cases carried both a homozygous STRC deletion and a biallelic mutation of the GJB2 gene. The severe-to-profound hearing loss observed in these two cases reflects the clinical phenotype of GJB2-associated hearing loss. This observed overlapping of both of the GJB2 and STRC biallelic mutation is thought to result from the high carrier frequency of both of the GJB2 mutations and the STRC deletion.
In this study, we collected the hearing thresholds for 7 frequencies (125 Hz, 250H, 500 Hz, 1000 Hz, 2000 Hz, 4000 Hz and 8000 Hz) by pure-tone audiometry for 159 patients with STRC-associated hearing loss. Many patients in this study were too young to undergo pure-tone audiometry. We also excluded the patients without hearing thresholds information and two severe-to-profound hearing loss patients carrying both of GJB2 and STRC biallelic mutations. Figure 1 shows the averaged hearing thresholds for every 10 years of age. These results clearly indicate that the characteristic phenotype of STRC-associated hearing loss is congenital-onset, non-progressive, mild-to-moderate hearing loss.

Averaged audiograms of STRC-associated hearing loss patients. The hearing threshold data were obtained from 159 probands and show the averaged hearing thresholds with standard deviation for every 10 years of age.
Types and frequencies of STRC-CATSPER2 deletions identified in STRC-associated hearing loss patients
To confirm the STRC gene deletions identified using the NGS data, we performed MLPA analysis for 140 randomly selected patients with a 2 copy number loss and 18 patients with a 1 copy number loss of the STRC gene and SNVs using a commercially available MLPA probe set (SALSA MLPA Probemix P461-A1 DIS). As a result of the MLPA analysis, we identified 11 types of deletion pattern for cases with a 2 copy number loss (Fig. 2 and Supplemental Table 1). The most prevalent deletion pattern was a 2 copy number loss of the whole CKMT1B-STRC-CATSPER2 gene region, which was identified in 77.1% of cases (108/140), followed by a 2 copy number loss of the CKMT1B-STRC gene region with a 1 copy number loss of the CATSPER2 gene in 12.9% of cases (18/140). In addition, several complex deletion or duplication patterns were observed in some cases. Among 108 cases with a 2 copy number loss of the whole CKMT1B-STRC-CATSPER2 gene region, 56 cases were female and 52 cases were male. Thus, the prevalence of DIS in patients with hearing loss associated with a 2 copy number loss of the STRC gene was 37.1% (52/140).

Types of homozygous STRC gene deletion identified by MLPA analyses. In this study, we identified 11 types of CKMT1B-STRC-CATSPER2 gene deletion by NGS and MLPA analyses. NGS-based CNV analysis (left) was performed in accordance with our previous report22. Blue indicates the estimated copy number for each amplicon and red indicates the smoothing value for five relative amplicons. The vertical axis shows the copy number for each amplicon. MLPA (right) was performed using a commercially available MLPA probe (SALSA MLPA Probe mix P461-A1; MRC-Holland, Amsterdam, Netherlands). Box plots and error bars indicated the estimated copy number range and standard deviation for each MLPA probe, respectively. The vertical axis shows the copy number for each probe and the horizontal axis indicates the probes used in the MLPA analysis. *The MLPA probe set used in this study was only designed for exons 19, 23, 24 and 25 of the STRC gene and did not cover exons 1 to 18. Some patients carried CNVs in these exons and we could detect CNVs by NGS data analysis, but not by MLPA (such as Pattern 5 or 6), in these cases.
Discussion
In this study, we identified homozygous deletions of the STRC gene (a 2 copy number loss) in 231 of 9956 Japanese hearing loss patients (2.32%; 231/9956), and 41 cases with a 1 copy number loss of the STRC gene which also carried SNVs in the trans allele configuration. We also identified 4 cases with homozygous or compound heterozygous SNVs; thus, the prevalence of STRC-associated hearing loss in Japanese hearing loss patients was 2.77% (276/9956). In our previous study, we identified 17 of 1025 (1.7%) Japanese hearing loss patients carrying homozygous STRC deletions9. Based on micro-droplet digital PCR analysis, Ito et al. reported that 2 of 84 (2.4%) Japanese mild-to-moderate hearing loss patients carried a homozygous STRC deletion10. Similarly, the prevalence of STRC-associated hearing loss has been reported to range from 5.4 to 16.1% in hearing loss patients of mixed ethnicity, and in American and Italian populations4,5,6,7,8, 2.6% in Turkish autosomal recessive hearing loss patients11, and 10% and 10.8%, respectively, in Czech and Korean mild-to-moderate hearing loss patients12,13. From these reports, STRC-associated hearing loss is concluded to be a relatively common cause of hearing loss in every ethnic population. We previously reported the carrier frequency of the STRC deletion in Japanese controls with normal hearing to be 2.6% (4/152)9. Ito et al. reported the carrier frequency of the STRC deletion in Japanese controls with normal hearing to be 0.9% of (1/107)10, whereas Hoppman et al. estimated the carrier frequency of the STRC deletion in controls with normal hearing to be 1%23.
In the present study, we showed that most STRC-associated hearing loss patients exhibited mild-to-moderate hearing loss that did not progress up to the age of 60 (Fig. 1). This type of hearing loss is consistent with that described in previous studies9,10,13,14,23,24,25.
We also identified 11 types of deletion pattern for the homozygous STRC gene deletion (Fig. 2), with the most prevalent deletion pattern being the homozygous deletion of the whole CKMT1B-STRC-CATSPER2 gene region, which was identified in 77.1% of cases (108/140). Thus, most of the homozygous STRC deletions associated hearing loss cases carried a 2 copy number loss of the whole CKMT1B-STRC-CATSPER2 gene region. Among 108 cases with a 2 copy loss of the whole CKMT1B-STRC-CATSPER2 gene region, 52 cases were male and the prevalence of DIS in patients with hearing loss associated with a 2 copy number loss of the STRC gene was estimated to be 37.1% (52/140). Unfortunately, the main symptom of our study cohort was hearing loss and the data were collected from otolaryngology departments; thus, only limited information was obtained regarding male infertility. The phenotype-genotype correlation between our MLPA results and male infertility, therefore, remains unclear. However, a non-negligible portion of patients with homozygous STRC gene deletions carried the intact CATSPER2 gene. Thus, the determination of the deletion range for STRC-associated hearing loss patients should be performed to allow more appropriate genetic counseling in terms of hearing loss and male infertility. It is noteworthy that the commercially available MLPA probes for DIS are designed for only exons 19 to 25 of the STRC gene and did not identify a partial deletion of exons 1 to 18 of the STRC gene. Indeed, we could not identify STRC gene deletions for 4 cases with partial deletions of the STRC gene (Fig. 2, deletion pattern number 5 and 6). Thus, NGS-based analysis is more powerful and appropriate for the identification of STRC deletions.
In conclusion, we herein report that the prevalence of STRC-associated hearing loss was 2.77% in a large cohort of Japanese hearing loss patients. In addition, 77.6% of cases (108/140) with a homozygous STRC deletion carried a two copy number loss of the whole CKMT1B-STRC-CATSPER2 gene region. This information will be useful in allowing more appropriate genetic counseling regarding hearing loss and male infertility for patients with STRC deletions.
References
Morton, C. C. & Nance, W. E. Newborn hearing screening–a silent revolution. N. Engl. J. Med. 354, 2151–2164 (2006).
Campbell, D. A. et al. A new locus for non-syndromal, autosomal recessive, sensorineural hearing loss (DFNB16) maps to human chromosome 15q21-q22. J. Med. Genet. 34, 1015–1017 (1997).
Villamar, M., del Castillo, I., Valle, N., Romero, L. & Moreno, F. Deafness locus DFNB16 is located on chromosome 15q13-q21 within a 5-cM interval flanked by markers D15S994 and D15S132. Am. J. Hum. Genet. 64, 1238–1241 (1999).
Shearer, A. E. et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6, 37 (2014).
Sloan-Heggen, C. M. et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450 (2016).
Mandelker, D. et al. Comprehensive diagnostic testing for stereocilin: An approach for analyzing medically important genes with high homology. J. Mol. Diagn. 16, 639–647 (2014).
Amr, S. S. et al. Allele-specific droplet digital PCR combined with a next-generation sequencing-based algorithm for diagnostic copy number analysis in genes with high homology: Proof of concept using stereocilin. Clin. Chem. 64, 705–714 (2018).
Morgan, A. et al. Lights and shadows in the genetics of syndromic and non-syndromic hearing loss in the Italian population. Genes (Basel) 11, 1237 (2020).
Yokota, Y. et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 9, 4408 (2019).
Ito, T. et al. Rapid screening of copy number variations in STRC by droplet digital PCR in patients with mild-to-moderate hearing loss. Hum. Genome Var. 6, 41 (2019).
Bademci, G. et al. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet. Test. Mol. Biomarkers. 18, 658–661 (2014).
Plevova, P. et al. STRC deletion is a frequent cause of slight to moderate congenital hearing impairment in the Czech Republic. Otol. Neurotol. 38, e393–e400 (2017).
Kim, B. J. et al. Significant Mendelian genetic contribution to pediatric mild-to-moderate hearing loss and its comprehensive diagnostic approach. Genet. Med. 22, 1119–1128 (2020).
Vona, B. et al. DFNB16 is a frequent cause of congenital hearing impairment: Implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 87, 49–55 (2015).
Avidan, N. et al. CATSPER2, a human autosomal nonsyndromic male infertility gene. Eur. J. Hum. Genet. 11, 497–502 (2003).
Zhang, Y. et al. Sensorineural deafness and male infertility: A contiguous gene deletion syndrome. J. Med. Genet. 44, 233–240 (2007).
Nishio, S. Y., Hayashi, Y., Watanabe, M. & Usami, S. Clinical application of a custom AmpliSeq library and ion torrent PGM sequencing to comprehensive mutation screening for deafness genes. Genet. Test. Mol. Biomarkers. 19, 209–217 (2015).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids Res. 38, e164 (2010).
Nishio, S. Y. & Usami, S. I. The clinical next-generation sequencing database: A tool for the unified management of clinical information and genetic variants to accelerate variant pathogenicity classification. Hum. Mutat. 38, 252–259 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613 (2018).
Nishio, S. Y., Moteki, H. & Usami, S. I. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeq™ custom panel. Mol. Genet. Genomic Med. 6, 678–686 (2018).
Hoppman, N. et al. Genetic testing for hearing loss in the United States should include deletion/duplication analysis for the deafness/infertility locus at 15q15.3. Mol. Cytogenet. 6, 19 (2013).
Čada, Z. et al. Moderate sensorineural hearing loss is typical for DFNB16 caused by various types of mutations affecting the STRC gene. Eur. Arch. Otorhinolaryngol. 276, 3353–3358 (2019).
Markova, T. G. et al. Clinical features of hearing loss caused by STRC gene deletions/mutations in Russian population. Int. J. Pediatr. Otorhinolaryngol. 138, 110247 (2020).
Acknowledgements
We would like to thank the following 102 collaborative hospitals for participating in the Deafness Gene Study Consortium; Asahikawa Medical University, Shinoro ENT Clinic, Hokkaido University, Sapporo Medical University, Hakodate Central General Hospital, Hirosaki University, Iwate Medical University, Tohoku Rosai Hospital, Tohoku University, Akita University, Omagari Kousei Medical Center, Yamagata University, Fukushima Medical University, Fukushima Medical University Aizu Medical Center, Hoshi General Hospital, Hitachinaka General Hospital, University of Tsukuba, Dokkyo Medical University, Jichi Medical University, Gunma University, TAKASAKI Ear Nose & Throat Clinic, Kawagoe Otology Institute, Jichi Medical University Saitama Medical Center, National Rehabilitation Center for Persons with Disabilities, National Defense Medical College, Showa General Hospital, Nippon Medical School, Juntendo University, Tokyo Medical and Dental University, Kamio Memorial Hospital, Keio University, Tokyo Medical University, Tokyo Metropolitan Children's Medical Center, Akasaka Toranomon Clinic, Toranomon Hospital, The Jikei University School of Medicine, International University of Health and Welfare, Abe ENT Clinic, Kitasato University, Yokohama City University Medical Center, Kanagawa Children's Medical Center, Tokai University, Yokohama City University, Niigata University, Kanazawa University, University of Fukui, University of Yamanashi, Miyagawa ENT Clinic, Shinshu University, Gifu University, Nagara Medical Center, Kizawa Memorial Hospital, Kyoai Clinic, Japanese Red Cross Shizuoka Hospital, Hamamatsu University School of Medicine, Nagoya University, Chubu Rosai Hospital, Aichi Children's Health and Medical Center, National Mie Hospital, Mie University, Shiga Medical Center for Children, Shiga University School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto University, Kyoto Teishin Hospital, Osaka Medical College, Osaka University, Kansai Medical University, Kitano Hospital, National Hospital Organization Osaka National Hospital, Japanese Red Cross Osaka Hospital, Osaka Women's and Children's Hospital, Izumi Clinic, Hyogo Prefectural Amagasaki General Medical Center, Hyogo Medical University, Kobe University, Kobe City Medical Center General Hospital, Nara Medical University, Wakayama Medical University, Tottori University, Okayama University, Hiroshima City Hiroshima Citizens Hospital, Hiroshima University, Hiroshima Prefectural Hospital, Yamaguchi University, Tokushima University, Ehime University, Kochi University, Fukuoka Children's hospital, Kyushu University, Fukuoka University, Kurume University, Saga University, Kanda ENT Clinic, Nagasaki University, Oita University, University of Miyazaki, Kagoshima City Hospital, Kagoshima University, University of the Ryukyus. We also thank Fumiko Tomioka and Sachiko Matsuda for their technical assistance with this research.
Funding
This research was funded by a Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Japan (S.U. 20FC1048), and Grants-in-Aid from the Japan Agency for Medical Research and Development (AMED) (S.U.: 17kk0205010h0002, 18ek0109363h0001).
Author information
Authors and Affiliations
Contributions
Study conception and design: S.N. and S.U.; Bioinformatics analysis: S.N.; Data analysis and interpretation: S.N.; Writing of the manuscript: S.N. and S.U.; Study supervision: S.U. All authors read and approved of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishio, Sy., Usami, Si. Frequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients. Sci Rep 12, 634 (2022). https://doi.org/10.1038/s41598-021-04688-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-04688-5
This article is cited by
-
Dispersed DNA variants underlie hearing loss in South Florida’s minority population
Human Genomics (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
