
Abstract
The existing treatments for somatoform dysfunction (SfD), reaction to severe stress (RSS), and adjustment disorders (AjD) are insufficiently effective and safe. Anxiolytic drug Tenoten proved effective in clinical trials (CT). The aim of this multicenter double-blind placebo-controlled randomized CT was to investigate the safety and efficacy of Tenoten in the treatment of anxiety in adults with SfD, RSS, AjD and other neurotic disorders (oNDs). 390 adult patients with SfD, RSS and AjD or oNDs with the Hospital Anxiety and Depression scale-anxiety (HADS-A) score ≥ 11 were randomized into 4 groups (n = 127 in Tenoten group 1 (4 tablets/day); n = 131 in Tenoten group 3 (8 tablets/day), n = 132 in combined Placebo group 2 + 4). The changes from baseline in the mean Hamilton Anxiety Rating Scale (HAM-A) score in groups 1 and 3 after 12 weeks were the primary outcome. The decrease of the HAM-A score from 18.81 ± 5.81 to 7.26 ± 4.63 (in group 1) and from 18.38 ± 4.3 to 6.40 ± 4.02 (in group 3) was observed post-treatment (pgroup 1/placebo = 0.0055, pgroup 3/placebo < 0.0001). Overall, 46 adverse events (28 in the Tenoten groups and 18 in the Placebo) were reported without any difference between the study groups. Tenoten performed significantly more effective than placebo in the anxiety treatment of adults with SfD, RSS, AjD and oNDs (clinicaltrials.gov NCT03036293).
Similar content being viewed by others

Introduction
Somatic symptom disorder (SSD) or Somatoform dysfunction (SfD) is psychosomatic condition that causes one or more bodily symptoms, encompassing myalgia, anxiety, sleep deprivation, a plethora of gastrointestinal symptoms and even severe pain. The etiology of the disease is complex and the exact mechanisms are not fully unraveled. Altogether the symptoms significantly impact the quality of life of the patients who suffer from it and, often times, their mental health. The prevalence of SfD is 20–25%1,2. Being a multiorgan condition, SfD is often underdiagnosed, hence only 33–60% of patients undergo treatment3.
Post-traumatic stress disorder (PTSD) is yet another neurotic disorder (ND) caused by severe stress, trauma, violence, or violence witness, which falls under adjustment disorders (RSS). About 1.3–8.1% of people get diagnosed with PTSD at least once over lifetime4.
Comorbidity of PTSD with anxiety or depression occurs in about 60% of PTSD patients and in 20–67% of those with SD5,6. In a study, 7–48% of primary care patients with anxiety reported somatic symptoms7.
Pathological mechanisms of these NDs remain unclear and diverse. There is data that blames the SSD symptoms on the alterations in the hypothalamic–pituitary–adrenal axis, psycho-neuro-immune dysregulation and cytokine imbalance8,9. Given similar pathogeneses of SfD and anxiety and their increasing co-occurence in patients, the presentation of medically unexplained symptoms (MUS) can be considered as a marker for a risk of limited social functioning and a predictor for the development of various psychosomatic disorders10.
The efficacy of antidepressants in SfD is low and their use is complicated with a high rate of adverse events (AEs)11. There is controversial data on the efficacy of psychotherapy12,13. The same holds true for the PTSD: meta-analysis showed the trifling effect of selective serotonin reuptake inhibitors14. Hence, the search for safe and successful treatment strategies is needed.
Tenoten (NPF Materia Medica Holding, Russian Federation (RF)) is an anxiolytic drug that contains highly diluted antibodies to S100 protein (HD Abs to S100). High dilutions of substances obtained using a technological process, namely by a repeated dilution of the original substance in combination with an external physical impact, have the ability to modify the activity of the original substance15. The mechanism of action of HD is based on their ability to induce conformational changes of the original substance/target molecule16. The modifying effect of HD Abs to S100 has been demonstrated in experimental studies17,18,19,20,21,22,23,24,25,26,27.
Tenoten is manufactured under GMP conditions and has been registered as a conventional drug in the European Economic Community [marketing authorization number ЛП-N(000029)-(PГ-RU)]28. Though Tenoten is not registered in the USA, FDA experts concluded that drugs based on HD Abs should be studied and proceeded for registration using a standard regulatory approach29.
HD Abs to S100 have been comprehensively studied in pre-clinical studies where their stress-protective, anxiolytic, antidepressant, antiamnestic and neuroprotective activities have been demonstrated30. Clinical trials (CTs) have shown the efficacy and safety of HD Abs to S100 in patients with anxiety NDs as well as in patients with anxiety and concurrent neurological diseases30. Moreover, the effectiveness of HD Abs in patients with anxiety disorders was comparable to that of benzodiazepines30.
The aim of the current study was to investigate the safety and efficacy of Tenoten in the treatment of anxiety in adults suffering from SfD, RSS, AjD and other NDs.
Materials and methods
Trial design
This multicenter double-blind placebo-controlled randomized CT was conducted according to GCP at 23 sites in RF and Kazakhstan between February 2017 and March 2019. The CT was approved by the regulatory agencies of RF and Kazakhstan.
Participants
Inclusion and exclusion criteria
Outpatients (18–45 years old) with anxiety and SfD (F45.0, F45.1, F45.2, F45.4, F45.8, F45.9), RSS and AjD (F43.0, F43.1, F43.2, F43.8, F43.9) or other NDs (F48.8, F48.9) diagnosed prior to the study were enrolled in four groups stratified by the type and dosage of treatment. Anxiety scores ≥ 11 [Hospital Anxiety and Depression scale-anxiety (HADS-A)] was also included. All patients who got enrolled had to sign the informed consent form.
Exclusion criteria were: evident depression symptoms at screening (≥ 11 points according to HADS scale), organic mental disorders, mental disorders other than SfD, RSS and AjD, mental deficiency, inflammatory and traumatic brain injuries, severe somatic diseases, malignant neoplasia, drug or alcohol addiction, previous severe allergic reactions, pregnancy, breast-feeding.
Study procedures and treatment
All protocols were carried out in accordance with the relevant guidelines and regulations. After signing the informed consent form, the neurologist examined a patient, recorded demographic data and medications taken by a patient at the time of the study, administered a pregnancy test and filled out the HAM-A scale. Patients filled out the HADS and EQ-5D-3L questionnaires.
The treatment period lasted for 12 weeks.
During Visit 1 participants were randomized into 4 groups. Patients in the groups 1 and 3 were given Tenoten in the regimen of 2 pills 2 times a day or 2 pills 4 times a day, respectively. The Placebo groups 2 and 4 received placebo in dosages similar to Tenoten.
Every 4 weeks the investigator examined the patients, filled out the HAM-A scale, recorded other medications intake and assessed the safety of the protocol and patients’ compliance. The evaluation of the Clinical Global Impression-Efficacy Index (CGI-EI) and filling out of the EQ-5D-3L were performed during the last visit.
Psycholeptics, psychoanaleptics, antiepileptics, anticholinergic and dopaminergic agents, antioxidants, hormones, psychotherapy were not allowed 4 months prior to and during the study.
Outcomes
The alterations from the baseline in the mean HAM-A score in the groups 1 and 3 after 12 weeks were taken as the primary outcome.
The exploratory outcomes were: changes from the baseline in the mean HAM-A score after 4 and 8 weeks, the percentage of patients who responded to treatment (≥ 50% reduction on HAM-A) and the percentage of patients without anxiety (HAM-A < 14) after 4, 8, 12 weeks, the changes from baseline in the EQ-5D-3L scores after 12 weeks, the CGI score.
In the post-hoc analysis we assessed the effect of the particular type of diagnosis on the mean HAM-A scores, subscores for each separate question, and somatic components subscores of HAM-A scale (questions 7–13). The influence of placebo effect on the presented diagnosis was also evaluated.
Sample size determination and randomization
Sample size of the study was set in order to have desired control over the type I and II errors during investigation of the primary outcome. Overall error deviation scores were 0.05 and 0.2, respectively. There were three independent formal hypotheses: distinction of each treatment (for each Tenoten group) from placebo and equivalence of treatments. The following formal hypotheses were examined in the primary statistical analysis: distinction of the mean change (HAM-A score) from baseline between Tenoten group 1 and placebo:
where M1 is mean change of HAM-A score from baseline in the experimental group.
Similar hypothesis for comparison between Tenoten group 2 and placebo.
Hypothesis of equivalence between Tenoten group 1 and Tenoten group 2:
where δ is Equivalence margin.
Type I error level was evenly distributed and fixed across mentioned primary endpoint hypotheses during the planning stage of the study (each primary analysis was performed with critical level of α = 0.0166(6). This level also determined the estimation of the required sample size). In accordance with the study protocol, adjusted level of type I error is applicable to the primary endpoint section only. For other analyses (the exploratory endpoint section) unadjusted level of 0.05 was used.
It was assumed that the difference in the HAM-A score decrease between each Tenoten and Placebo groups would be greater than 4 points, and the difference in the HAM-A score decrease between Tenoten groups would be < 3 points. The variance of the change in the HAM-A score was a priory estimated as 44. The dropout rate during the screening was expected to be < 20%. The recruitment expectation was set at 390 patients.
Before the beginning of the study, a randomization list with randomization numbers for Tenoten or Placebo was compiled. Eligible patients were randomized into four groups via interactive system based on a random number generator. The ratio of patients between Tenoten and Placebo groups was 1:1:0.5:0.5, placebo groups 2 and 4 were combined.
The participant, the researcher and the study team of the study sponsor were not informed about the administered therapy until the study was completed. Placebo and Tenoten preparations looked the same and had similar organoleptic properties.
Statistics
Two-tailed statistical criteria were used. Changes from baseline were analyzed with ANCOVA, normality assumptions were controlled with the Kolmogorov–Smirnov test and Q-Q plot; Yeo-Johnson normalizing transformation was applied if necessary. Count data analysis was performed with Fisher test (FT) and/or conditional logistic regression. Non-gaussian data were analyzed with the Kruscall-Wallis test. Statistical inference results are presented as p-value and appropriate central tendency with confidence limits, type I error for primary outcome was controlled for exploratory data, unadjusted p-values and 95% confidence intervals are presented. The post-hoc analysis was held using multinomial or mixed ANOVA. Analyses were performed using SAS v9.4.
Ethics approval
The CT was approved by the regulatory agencies: the Ministry of Health of the RF (approval #549 August 03, 2016) and Kazakhstan (#30 February 15, 2017). The study was approved by the National Ethics Committee of the Ministry of Health of the RF (#129 July 26, 2016). All patients have signed the Informed Consent Form before they were included in the study.
Results
Study group characteristics
A total of 390 patients were enrolled in CT with 258 participants in Tenoten groups (n = 127 in group 1, n = 131 in group 3), and 132 patients in the Placebo group (group 2 + 4). Date of inclusion of the first patient—February 7th 2017, date of completion of the last patient's participation—September 22nd 2018. The study was completed in accordance with the protocol.
The patients were stratified into general groups by the type of diagnosis: F43, F45, F48. No differences between groups in baseline data were found (Table 1).
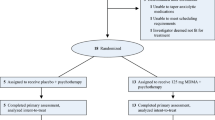
All 390 patients formed a safety population set. The monitoring revealed that 6 patients fulfilled exclusion criteria, so 384 participants with HADS-A ≥ 11 continued with the treatment and were included in the Intention-to-treat (ITT) set. Finally, 344 participants formed the PP set (Fig. 1). All results are presented for ITT and PP (showed in square brackets) sets.

Patient flow diagram. TS total set, SP safety population set.
8–12% of patients were taking permitted medications: analgesics, non-steroidal anti-inflammatory drugs and antimicrobials. FT did not reveal differences between groups.
Compliance assessment demonstrated a high level of adherence to therapy without differences between groups at 12 weeks (Kruskal–Wallis test, pgroup 1/placebo = 0.4362 [0.2506], pgroup 3/placebo = 0.1936 [0.3229], pgroup 1/group 3 = 0.0598 [0.0519]). The mean compliance index was close to 100% (Total set: pgroup 1/placebo = 0.63; pgroup 3/placebo = 0.13; pgroup 1/group 3 = 0.07).
Efficacy analysis
Primary outcome
The primary outcome were the changes from baseline in the mean HAM-A score in groups 1 and 3 after 12 weeks of treatment. The HAM-A is a clinical rating scale designed to measure the severity of a patient's anxiety disorders. It contains 14 statements with 5 response options from 0 to 4 which are correlated with the severity of anxiety. The sum of points of 13 or less means the absence of anxiety, 14–17 points—mild severity of anxiety disorder, 18–24 points—moderate severity and more than 25 points—severe anxiety disorder.
The decrease in the mean HAM-A score to 7.26 ± 4.63 [7.12 ± 4.65] in group 1 and 6.40 ± 4.02 [6.08 ± 3.78] in group 3 was observed after 12 weeks (vs 8.48 ± 5.13 [8.31 ± 4.51] in the Placebo group; ANCOVA pgroup 1/placebo = 0.0055 [0.0155], pgroup 3/placebo < 0.0001[0.0001]) (Fig. 2).

The change in the mean HAM-A score after 4, 8, 12 weeks. ITT-set. *p = 0.044 vs Placebo; **p = 0.027 vs Placebo; #p = 0.0155 vs Placebo; ##p < 0.0001 vs Placebo, t-test.
The mean changes in the scores in group 1, group 3 and Placebo group were 11.25 [11.23], 11.91 [12.36] and 9.71 [9.94], respectively.
Equivalence analysis showed no differences between Tenoten groups with different dosage regimens (ANCOVA p = 0.008 [0.008]).
Exploratory outcomes
The remission of anxiety (HAM-A < 14) was found in 46% of patients in group 1 and in 48.5% of patients in group 3 after 4 weeks. The percentage of patients in remission increased to 88.1% and 96.2% after 12 weeks in group 1 and 3, respectively. The efficacy of Tenoten in dosage of 8 tablets a day was superior to placebo (FT pgroup 3/placebo = 0.007) (Fig. 3).

The percentage of patients with remission of anxiety symptoms after 4, 8, 12 weeks. ITT-set. #p = 0.007 vs Placebo.
Dynamics of the mean HAM-A score after 4 weeks of treatment
The mean HAM-A score decreased from baseline to 13.31 ± 4.7 [13.33 ± 4.8] in group 3 (vs 13.85 ± 5.34 [13.99 ± 4.91] in Placebo group; ANCOVA t-test p = 0.044 [0.047]) (Fig. 2). The change in mean HAM-A was 4.37 [4.35] and 3.29 [3.64] points in the Tenoten 1 and Placebo groups, respectively.
The analysis found no differences between group 1 and the Placebo group (ANCOVA t-test p = 0.08 [0.14]).
Dynamics of the mean HAM-A score after 8 weeks of treatment
Tenoten administration led to positive dynamics in severity of anxiety in group 3. The decrease in mean HAM-A score to 9.88 ± 4.93 [9.82 ± 4.97] in these patients was significant (vs 10.81 ± 5.16 [9.82 ± 4.97] in the Placebo group; ANCOVA t-test pgroup 3/placebo = 0.027 [0.033]) (Fig. 2).
The percentage of patients with response (≥ 50% reduction on HAM-A scale)
There were 12.7%, 34.9% and 69.8% of responders in group 1 after 4, 8 and 12 weeks, respectively. Group 3 response rates were 13.8% at week 4, 42.3% at week 8 and 73.8% at week 12. Differences between Tenoten and Placebo groups were significant only after 12 weeks (FT pgroup 1/placebo = 0.01, pgroup 3/placebo = 0.001).
Quality of life
The European Quality of Life Instrument (EQ-5D-3L) is developed to assess the quality of life of patients and comprises the following five criteria: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. Each dimension has 3 levels: no problems (1 point), some problems (2 points), and extreme problems (3 points).
The mean EQ-5D-3L scores decreased to 5.84 ± 1.05 in the group 1, 5.71 ± 0.82 in group 3, 6.05 ± 1.07 in the Placebo group. Wilcoxon–Mann–Whitney test showed the superiority of Tenoten at a dosage of 8 tablets per day over Placebo (p = 0.031).
CGI-EI score
The Clinical Global Impressions Scale, subscale «Efficacy Index» (CGI-EI), assesses the patient’s impression of the ratio between therapeutic effects and side effects of the medication. Scores on the CGI-EI range from 0 (marked improvement and no side effects) to 4 (unchanged or worse and side effects outweigh therapeutic effects).
After 12 weeks of treatment the mean CGI-EI index was high for both Tenoten groups (4.53 ± 3.21 and 4.55 ± 2.91 for groups 1 and 3 respectively) and differed from that in the Placebo group (5.71 ± 3.28) (Wilcoxon-Mann–Whitney test pgroup 1/placebo = 0.0021, pgroup 3/placebo = 0.0056). Thus, the efficacy of Tenoten in both doses was high.
Post-hoc analysis
Type of diagnosis did not affect mean HAM-A score dynamics in groups (mixed ANOVA ptreatment × visit × diagnosis = 0.23).
Pairwise comparison showed the mean HAM-A score significantly differed between F43 and F45 patients diagnoses regardless of the group and visit (mixed ANOVA p = 0.03). The mean score during the study was higher in patients with SfD than in those with RSS and AjD (13.3 ± 0.3 vs 11.9 ± 0.5).
The dynamics of the mean HAM-A subscores for questions 1–4 didn’t depend on the diagnosis. The mean subscore changes differed between treatment groups for questions 1 and 2 (mixed ANOVAtreatment × visit p question 1 = 0.0008 and p question 2 < 0.0001).
The analysis of questions 5 and 6 showed the diagnosis affected the mean subscores (mixed ANOVAtreatment × visit × diagnosis p question 5 = 0.002 and p question 6 < 0.0001). Scores in F43 patients in group 1 differed significantly from F45 ones in group 3 for question 5 and from F45 and F48 participants in group 3 for question 6 (mixed ANOVA pF43–F45 question 5 = 0.0003 and pF43–F45 question 6 = 0.00013, pF43–F48 question 6 = 0.025).
The analysis of the relationship between diagnosis and somatic component subscores of HAM-A scale (questions 7–13) showed that the dynamics of somatic complaints was significantly different in groups regardless of ND type (ANOVA ptreatment × visit = 0.019, ptreatment × visit × diagnosis = 0.92). Nevertheless, the mean HAM-A somatic subscores in F43 patients differed significantly from other patients’ scores in pairwise comparison (pF43–F45 = 0.0002; pF43–F48 = 0.045). The mean HAM-A 7–13 questions subscore in patients with RSS and AjD during the study was 2.84 ± 0.13 and was lower than those in F45 and F48 groups (3.46 ± 0.08 and 3.23 ± 0.1, respectively). Patients with SfD performed the highest mean HAM-A subscores in questions 8 (0.94 ± 0.04; mixed ANOVA pF43–F45 = 0.0006; pF45–F48 = 0.019) and 10 (0.78 ± 0.04; mixed ANOVA pF43–F45 = 0.0004; pF45–F48 = 0.008).
Patients’ diagnosis didn’t affect the placebo effect degree in combined Placebo group and mean HAM-A score dynamics in all diagnostic groups was similar (ANOVA pdiagnosis × visit = 0.87).
Safety analysis
Investigators registered 46 AEs (10 in group 1, 18 in group 3, 18 in the Placebo group) in 37 patients (8 patients (6.3%) in group 1, 12 (9.2%) in group 3 and 17 (12.9%) in the Placebo group.
There were 6 (60.0%) and 14 (77.8%) mild AEs in group 1 and 3, respectively. There were 4 (40.0%) and 4 (22.2%) AEs of moderate severity in group 1 and 3, respectively. There were 12 (66.7%) and 6 (33.3%) AEs of mild and moderate severity registered in the Placebo group, respectively. No serious AEs were registered. The frequency of AEs did not differ between the groups (FT pgroup 1/placebo = 0.092; pgroup 3/placebo = 0.432; pgroup 1/group 3 = 0.487).
The AEs appeared irrelevant to Tenoten treatment in 70% cases in group 1 and in 100% cases in group 3. The direct relationship was unlikely in 1 case and possible in 1 case in group 1. There were no AEs unlikely or possibly related to Tenoten in group 3. One AE had a probable association with the study drug in group 1. No AEs with a definite relation to Tenoten administration were registered. The distribution of AEs by cause was significantly different between group 3 and Placebo group: the number of AEs related to placebo was higher than those related to the study drug in group 3 (FT pgroup 3/placebo = 0.009).
Discussion
In this multicenter double-blind randomized CT, we showed the efficacy of two dosage regimens of Tenoten in treatment of anxiety. According to post-hoc data, the changes in anxious mood and feeling of tension depended on the type of therapy and were not related to patients’ diagnosis.
After 12 weeks of treatment responses were registered in 69.8% and 73.8% of patients receiving Tenoten 4 and 8 pills a day, respectively, and only in 53.9% in the Placebo group (pgroup 1/placebo and pgroup 3/placebo < 0.05). More than 95% of patients getting 8 pills of Tenoten per day presented with the remission of anxiety symptoms after 12 weeks (p = 0.007 vs Placebo). The quality of life improved in group 3 after 12 weeks.
The improvement of intellectual functions due to Tenoten treatment in dosage regimen 8 tablets per day was more pronounced in patients with RSS and AjD than in F45 patients and the reduction of depressive complaints was the greatest in F43 participants.
Tenoten administration led to more significant improvement in somatic complaints in F45 and F48 patients than in F43. Decrease of sensory and respiratory symptoms was the most prominent in patients with SfD. The mean HAM-A somatic subscores were the lowest in F43 patients during the study.
We can assume that anxiety plays an important role in SfD manifestation. The influence of therapy on anxiety pathological mechanisms leads to improvement of mental state in patients with SfD and other NDs.
A fairly high placebo effect was probably due to pre-treatment conversation with an investigator regarding the cause of underlying symptoms. We can assume the conversation partially affected the results by reducing the fear of somatic disease. These assumptions stem from the findings of the role of an interview and the therapeutic relationship between physician and patient during the treatment described in other studies31,32. The high frequency of substance administration could have contributed to the degree of placebo effect, and we made an effort to lower it by combining Placebo groups 2 + 4. It is worth noticing the placebo effect was decreasing over time, while the effect of Tenoten increased by the end of the treatment.
Tenoten was well tolerated: only 20 patients in groups 1 and 3 experienced AEs. There was no difference in AEs frequency between groups. No AEs were serious and definitely related to Tenoten. Thus, the administration of Tenoten resulted in an anxiolytic effect with minimal AEs. This conclusion was in agreement with a preferable safety to efficacy ratio according to investigators’ assessments.
The key trends for the anxiolytic therapy development were reported33. First, a drug should influence several pathways of anxiety pathogenesis to avoid polypharmacy and second, it should demonstrate both high efficacy and tolerability. Possible drug interactions resulting in AEs that may occur with the antidepressants or benzodiazepines administration in patients receiving other medications was emphasized13. In accordance with these considerations, Tenoten seems to be a promising drug with anxiolytic properties. It was shown that almost 35% of all-time CTs of Tenoten were of high evidence level. No interaction between Tenoten and concomitant therapy was found34. In general, the safety profile of a drug is consistent with the results of study.
Inclusion of patients with several psychiatric diseases and the absence of dose-frequency adjustment during the therapy are the main limitations. The study was mostly carried out in neurological centers. No special psychiatric interview methods were used to establish a diagnosis. In addition, we observed a relatively high placebo effect, the possible cause of which was discussed above.
The study had some advantages contributing to bias control: multicenter double-blind randomized design, a sufficient number of participants, and 4-month washout period before the onset of the study treatment. A training session was held to master the investigators on CT procedures. Trial protocol was posted at clinicaltrials.gov prior to commencing the study and results were recorded immediately after the analysis was submitted to regulatory authorities. Altogether, these facts provide evidence of the CT data reliability without the risk of biases.
Further long-term studies with the follow-up period should be performed to apply these results to a broader population.
Data availability
The trial has been registered at clinicaltrials.gov (https://clinicaltrials.gov/ct2/show/NCT03036293) in 30/01/2017. Trial protocol can be assessed at clinicaltrials.gov NCT03036293.
Change history
14 September 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41598-022-19938-3
References
Weiland, A. et al. Training medical specialists to communicate better with patients with medically unexplained physical symptoms (MUPS): A randomized, controlled trial. PLoS ONE10(9), e0138342. https://doi.org/10.1371/journal.pone.0138342 (2015).
Haller, H., Cramer, H., Lauche, R. & Dobos, G. Somatoform disorders and medically unexplained symptoms in primary care. Dtsch Arztebl Int.112(16), 279–287. https://doi.org/10.3238/arztebl.2015.0279 (2015).
Piontek, K., Shedden-Mora, M. C., Gladigau, M., Kuby, A. & Löwe, B. Diagnosis of somatoform disorders in primary care: Diagnostic agreement, predictors, and comaprisons with depression and anxiety. BMC Psychiatry18(1), 361. https://doi.org/10.1186/s12888-018-1940-3 (2018).
Lewis, S. J. et al. The epidemiology of trauma and post-traumatic stress disorder in a representative cohort of young people in England and Wales. Lancet Psychiatry6(3), 247–256. https://doi.org/10.1016/S2215-0366(19)30031-8 (2019).
Dai, W. et al. Comorbidity of post-traumatic stress disorder and anxiety in flood survivors: prevalence and shared risk factors. Medicine96(36), e7994. https://doi.org/10.1097/MD.0000000000007994 (2017).
den Boeft, M. et al. The association between medically unexplained physical symptoms and health care use over two years and the influence of depressive and anxiety disorders and personality traits: A longitudinal study. BMC Health Serv. Res.16(1), 100. https://doi.org/10.1186/s12913-016-1332-7 (2016).
Gelenberg, A. J. Psychiatric and somatic markers of anxiety: identification and pharmacologic treatment. J. Clin. Psychiatry.2(2), 49. https://doi.org/10.4088/pcc.v02n0204 (2000).
Rief, W. & Auer, C. Cortisol and somatization. Biol. Psychol.53, 13–23. https://doi.org/10.1016/S0301-0511(00)00042-9 (2000).
Houtveen, J. H., Kavelaars, A., Heijnen, C. J. & van Doornen, L. J. Heterogeneous medically unexplained symptoms and immune function. Brain Behav. Immun.21, 1075–1082. https://doi.org/10.1016/j.bbi.2007.04.008 (2007).
De Waal, M. W. M., Arnold, I. A., Eekhof, J. A. & Van Hemert, A. M. Somatoform disorders in general practice: Prevalence, functional impairment and comorbidity with anxiety and depressive disorders. Br. J. Psychiatry184(6), 470–476. https://doi.org/10.1192/bjp.184.6.470 (2004).
Kleinstaeuber, M. et al. Pharmacological interventions for somatoform disorders in adults. Cochrane Library.7(11), 010628. https://doi.org/10.1002/14651858.CD010628.pub2 (2014).
Van Dessel, N. et al. Non-pharmacological interventions forsomatoform disorders and medically unexplained physical symptoms (MUPS) in adults. Cochrane Library.https://doi.org/10.1002/14651858.CD011142 (2014).
Wedekind, D. & Bandelow, B. Trends in the treatment of anxiety disorders. Dialog. Clin. Neurosci.31(10), 499–503 (2005).
Hoskins, M. et al. Pharmacotherapy for post-traumatic stress disorder: systematic review and meta-analysis. Br. J. Psychiatry206(2), 93–100. https://doi.org/10.1192/bjp.bp.114.148551 (2015).
Epstein, O. The spatial homeostasis hypothesis. Symmetry.10(4), 103. https://doi.org/10.3390/sym10040103 (2018).
Tarasov, S. A. et al. Insights into the mechanism of action of highly diluted biologics. J. Immunol.205(5), 1345–1354. https://doi.org/10.4049/jimmunol.2000098 (2020).
Epstein, O. I. et al. Membrane and synaptic effects of anti-S-100 are prevented by the same antibodies in low concentrations. Front Biosci.8, a79-84. https://doi.org/10.2741/1025 (2003).
Voronina, T. A., Molodavkin, G. M., Sergeeva, S. A. & Epstein, O. I. GABAergic system in the anxiolytic effect of Proproten: Experimental study. Bull. Exp. Biol. Med.135(Suppl 7), 125–127. https://doi.org/10.1023/a:1024776007215 (2003).
Kheifets, I. A. et al. Involvement of the serotoninergic system in the mechanism of action of ultralow dose antibodies to S-100 protein. Bull. Exp. Biol. Med.143(5), 598–600. https://doi.org/10.1007/s10517-007-0191-y (2007).
Ertuzun, I. A. Mekhanizmy anksioliticheskogo i antidepressatnogo dejstviya Tenotena (eksperimental'noe issledovanie). Avtoreferat Dissertacii na Soiskanie Uchenoj Stepeni Kandidata Medicinskih Nauk. (2012) [in Russian].
Larentsova, L. I. et al. Eksperimental’no-klinicheskoe izuchenie tenotena (antitela k mozgospecificheskomu belku S-100) i vozmozhnosti ego primeneniya v kachestve sredstva premedikacii na ambulatornom stomatologicheskom prieme. Rossijskaya Stomatol.1, 48–51 (2008) (in Russian).
Voronina, T. A., Kheyfets, I. A., Dugina, Y. L., Sergeeva, S. A. & Epshtein, O. I. Study of the effects of preparation containing ultralow doses of antibodies to S-100 protein in experimental hemorrhagic stroke. Bull. Exp. Biol. Med.148(3), 530–532. https://doi.org/10.1007/s10517-010-0756-z (2009).
Voronina, T. A. et al. Nootropic and antiamnestic effects of tenoten (pediatric formulation) in immature rat pups. Bull. Exp. Biol. Med.148(3), 524–526. https://doi.org/10.1007/s10517-010-0754-1 (2009).
Voronina, T. A., Sergeeva, S. A., Martyushev-Poklad, A. V., Dugina, J. L. & Epstein, O. I. Antibodies to S-100 protein in anxiety-depressive disorders in experimental and clinical conditions. Anim. Model Biol. Psychiatry137, 52 (2006).
Kheyfets, I. A., Voronina, T. A., Dugina, J. L., Molodavkin, G. M. & Sergeeva, S. A. Anxiolytic activity of tenoten and diazepam depends on conditions in Vogel conflict test. Bull. Exp. Biol. Med.151(3), 336–339. https://doi.org/10.1007/s10517-011-1324-x (2011).
Voronina, T. A. et al. Effect of ultralow doses of antibodies to S-100 protein in animals with impaired cognitive function and disturbed emotional and neurological status under conditions of experimental Alzheimer disease. Bull. Exp. Biol. Med.148(1), 533–535. https://doi.org/10.1007/s10517-010-0757-y (2009).
Voronina, T. A., Molodavkin, G. M., Sergeeva, S. A. & Epstein, O. I. Anxiolytic effect of proproten under conditions of punished and unpunished behavior. Bull. Exp. Biol. Med.1, 120–122 (2003).
Official site of the Ministry of Health of the Russian Federation, registered drugs and clinical trials information. https://grls.rosminzdrav.ru/Grls_View_v2.aspx?routingGuid=022e51ba-4296-433b-8293-4f7c1cc9b02e&t.
Andrianova, E. & Putilovskiy, M. Efficacy and safety of Ergoferon versus oseltamivir: reply to the Letter to the Editor. Int. J. Infect. Dis.89, 190–192. https://doi.org/10.1016/j.ijid.2019.09.028 (2019).
Khacheva, K. K., Khakimova, G. R., Glazunov, A. B. & Fateeva, V. V. Technologically processed highly diluted antibodies to S100 protein in the treatment of neurotic disorders: The review. Anxiety Disord.https://doi.org/10.5772/intechopen.92207 (2020).
Bandelow, B., Michaelis, S. & Wedekind, D. Treatment of anxiety disorders. Dialog. Clin. Neurosci.19(2), 93 (2017).
Brownell, A. K. W., Atkins, C., Whiteley, A., Woollard, R. F. & Kornelsen, J. Clinical practitioners’ views on the management of patients with medically unexplained physical symptoms (MUPS): a qualitative study. BMJ Open6(12), e012379. https://doi.org/10.1136/bmjopen-2016-012379 (2016).
Griebel, G. & Holmes, A. 50 years of hurdles and hope in anxiolytic drug discovery. Nat. Rev. Drug Discov.12(9), 667–687. https://doi.org/10.1038/nrd4075 (2013).
Khacheva, K. K., Glazunov, A. B. & Kamchatnov, P. R. The efficacy and safety of the most prescribed drugs with anxiolytic effect in the Russian Federation. Lechebnoye Delo.4, 15–27. https://doi.org/10.24411/2071-5315-2019-12153 (2019).
Funding
Tenoten is a preparation manufactured and marketed by OOO NPF Materia Medica Holding. OOO NPF Materia Medica Holding funded the study, including protocol development and implementation, provision of the preparations, investigator granting, data collection and processing.
Author information
Authors and Affiliations
Contributions
V.P. wrote the manuscript. All authors discussed the results of the study and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors received an investigator grant from OOO NPF Materia Medica Holding to conduct the CT of Tenoten mentioned in this article.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41598-022-19938-3"
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parfenov, V.A., Kamchatnov, P.R., Khasanova, D.R. et al. RETRACTED ARTICLE: The randomized clinical trial results of the anxiety treatment in patients with somatoform dysfunction and neurotic disorders. Sci Rep 11, 24282 (2021). https://doi.org/10.1038/s41598-021-03727-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-03727-5
This article is cited by
-
Neurological Signs of Postcovid Syndrome
Neuroscience and Behavioral Physiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
