
Abstract
The electronic properties, including bandgap and conductivity, are critical for nearly all applications of multifunctional perovskite oxide ferroelectrics. Here we analysed possibility to induce semiconductor behaviour in these materials, which are basically insulators, by replacement of several percent of oxygen atoms with nitrogen, hydrogen, or vacancies. We explored this approach for one of the best studied members of the large family of ABO3 perovskite ferroelectrics — strontium titanate (SrTiO3). The atomic and electronic structure of defects were theoretically investigated using the large-scale first-principles calculations for both bulk crystal and thin films. The results of calculations were experimentally verified by studies of the optical properties at photon energies from 25 meV to 8.8 eV for in-situ prepared thin films. It was demonstrated that substitutions and vacancies prefer locations at surfaces or phase boundaries over those inside crystallites. At the same time, local states in the bandgap can be produced by vacancies located both inside the crystals and at the surface, but by nitrogen substitution only inside crystals. Wide-bandgap insulator phases were evidenced for all defects. Compared to pure SrTiO3 films, bandgap widening due to defects was theoretically predicted and experimentally detected.
Similar content being viewed by others
Introduction
Strontium titanate (SrTiO3, or STO) is one of the best studied materials belonging to a broad class of perovskite-structure oxide ferroelectrics1. Spontaneous polarization, large dielectric permittivity, strong piezoelectric, pyroelectric, electrocaloric, photovoltaic, and electro-optic effects enable numerous applications of ferroelectrics in diverse fields of electronics and photonics1,2,3,4,5,6,7,8. Requirements to the electronic properties are application-specific and vary significantly. For instance, perfect insulator properties are demanded for majority of the mainstream ferroelectric applications (e.g., in capacitors, electro-mechanical, and electro-optical devices), whereas bandgap narrowing and increased semiconductor- or metal-like conductivity are desirable for innovative photovoltaic, thermoelectric, and resistive-switching devices. Remarkably, such immense variations of the electronic properties are feasible using appropriate doping within the class of perovskite oxide ferroelectrics. However, although there are successful practical strategies, explicit mechanisms of the properties’ changes are far from understood. In particular, one of the methods relies on the possibility to remove oxygen atoms and/or substitute oxygen with hydrogen or nitrogen9,10,11,12,13,14,15,16. Compared to a wide-bandgap insulator state in pure crystalline STO, semiconductor-type behaviour was reported for SrTiO3-xHx (x = 0.05–0.45) and SrTiO3-xNx (x = 0.20)10,15,16. Substitution-induced shallow donor states were hypothesized to be responsible for the bandgap narrowing and large band conductivity therein. Yet, this hypothesis has not been proven.
It is worth mentioning that the removal and/or replacement of oxygen is often implemented by high-temperature processing that can last for several hours up to several days10,11. Under such conditions, one cannot exclude the formation of local compositional and/or structural inhomogeneities and, correspondingly, different types of phase boundaries, or interfaces. Importantly, the presence of interfaces is known to alter the properties of regular perovskite oxide ferroelectrics17,18. This presence additionally suggests the coexistence of two distinct types of substitutional locations: inside single crystal and at interfaces. The location may have an influence on the electronic structure and properties of STO. Furthermore, high-temperature processing may generate structural defects, such as dislocations19, whose role may be essential.
In this paper, we combined theoretical and experimental methods to elucidate the electronic properties, that originate from substitutions for oxygen either inside crystal or at interfaces in STO. Substitution of several percent of oxygen atoms with nitrogen, hydrogen, or vacancies was investigated.
We theoretically analysed crystalline STO with substitutions/vacancies inside the crystal. Because of uncertainties related to possible multiple states of hydrogen in STO20,21,22, we focused on substitutional nitrogen. Additionally, we examined the possibility for substitution/vacancy to be stabilized at a surface location. We employed large-scale first-principles calculations, which ensured high accuracy of the predicted electronic structure and bandgaps23,24,25,26,27,28,29,30,31,32.
We verified theoretical calculations by experimental studies of the optical properties of STO films, which were doped in-situ with nitrogen or hydrogen, or contained oxygen vacancies32,33,34,35,36. Contrary to often employed long-lasting high-temperature processing, the films were prepared without such processing. To ensure substitution locations inside a crystal, we grew single-crystal epitaxial films, whereas to introduce locations at boundaries, we deposited polycrystalline films.
Our observations suggest that boundary or surface locations for substitution/vacancy are more energetically favourable than locations inside the crystal. Deep in-gap states were theoretically predicted for substitutions inside crystals and experimentally detected in epitaxial films, whereas such states were absent for boundary locations as verified in polycrystalline films. Broad in-gap donor bands were found for oxygen vacancies. For all cases, only wide-bandgap insulator phases and bandgap widening were experimentally evidenced. The observations of wide-bandgap phases agree with theoretical predictions, and, furthermore, the calculations reveal some widening of bandgap for systems with in-gap defect states.
Results and discussion
First, we summarize the results of simulations and discuss the properties of the neutral oxygen vacancies and nitrogen substitution atoms in non-magnetic STO. Both of these defects are characterized by unpaired spins. In calculations of such defects, all electrons are divided into two groups: those with spins up (alpha-electrons) and those with spins down (beta-electrons). The total spin of a defect is defined by the difference in the numbers of alpha- and beta-electrons. The computational details are presented in “Methods” section. Compared to the stoichiometric number of oxygen atoms, the concentration of the defects was 4.17% in the bulk crystal, 12.5% in the layer with defect in the thin-film slab, or 3.125% with respect to the whole slab.
Oxygen vacancies
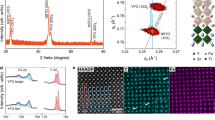
The oxygen vacancy is a common point defect in perovskite oxides. The majority of ab initio calculations conventionally focused on vacancies in the bulk STO37,38,39,40,41,42,43, with a few studies considering surface defects41,42,43,44. However, most of these calculations were performed within plane wave basis set using GGA + U approximation, which depends on the parameter choice. Here we investigated neutral vacancies VO placed not only inside ultrathin films, but also on the surface, using the identical, parameter free, approach with full structures optimization. As a model, we used free standing symmetric 2D slabs built of 5 atomic layers and having similar TiO2–TiO2 terminations on both sides. The two unpaired electron spins arise after the removal of an oxygen atom. The spins are distributed among Ti atoms, which are not necessarily the nearest ones to the vacancy, as we noted before for BaTiO331. An additional occupied local energy level or a band emerge in the band gap due to the formation of defects. The band appears because of interaction of periodically repeated defects.
First, we consider a neutral oxygen vacancy VO in the central layer of the slab. The calculated density of states (DOS) for the total spin state Sz = 1 (Fig. 1a) reveals a modified bandgap, which is the direct transition for beta-electrons. The bandgap energy is Eg = 3.1 eV, larger than Eg = 2.5 eV in the 5-layer pristine slab with TiO2 terminations. Compared to an isolated energy level in the limit of a single defect, we observe a relatively broad donor defect band in the periodic model. The minimum energy difference between the bottom of the conduction band and the top of the in-gap vacancy band, or the defect ionization energy, is found to be Eion = 0.2 eV. The corresponding transition is indirect for alpha-electrons.

The electronic DOS for slabs with oxygen vacancy, total spin Sz = 1. (a) VO in the central layer of the slab (space group SG 1); (b) VO on the surface of the slab (SG 6). α_Total, β_Total—total DOS for alpha- and beta- (spin up/down) electrons; the zero value of the energy corresponds to the Fermi level.
The formation energy for the vacancy is defined by
where E(VO) and E(perfect) are the total energies of the system with and without vacancy, respectively, and E(O) is the half energy of a gas-phase O2 molecule (that is, O atom) in the triplet state. The calculated formation energy is Evac = 6.7 eV. It should be noted that the energies Eion and Evac do not vary for systems with either Sz = 0 or Sz = 1.
Next, we inspect a neutral vacancy on the TiO2 surface of the slab. The calculations (Fig. 1b) give a bandgap energy Eg = 2.9 eV (the direct transition for beta-electrons) and an ionization energy Eion = 0.1 eV (indirect transition for alpha-electrons), with a rather delocalized electron density around the vacancy. The defect formation energy is Evac = 5.9 eV. Again, we note that Eion and Evac do not depend on the spin state of the system.
The calculations uncovered an interesting spin localization. For a vacancy in the centre of the slab (Fig. 1a), spins are localized on the Ti ions in the layer containing the vacancy. However, for a vacancy on the surface (Fig. 1b), spins are localized on the Ti ions in both the surface layer and central layer.
Overall, compared to the pristine slab, neutral vacancies slightly widen the bandgap and produce in-gap donor energy levels, at least by 0.1–0.2 eV below the conduction band. Importantly, the formation energy is by 0.8 eV smaller for the vacancy on the surface with respect to that inside the slab. This significant difference points to a preferential location of the vacancies at phase boundaries, interfaces, or surfaces in real systems.
Substitutional nitrogen atoms
We modelled lattices with substitutional nitrogen atoms in three distinct locations: in the bulk STO (Supplementary Fig. S1), in the centre of the free standing slab (Fig. 2a), and lastly, on the surface of the slab (Fig. 2b).

The electronic DOS for slabs with substitutional N atom. (a) N in the central layer of the slab (SG 25); (b) N on the surface of the slab (SG 6). α_Total, β_Total—total DOS for alpha- and beta-electrons, α_N, β_N—DOS projected onto N atom (alpha- and beta-electrons); the zero value of the energy corresponds to the Fermi level.
For nitrogen in the bulk STO, the calculated direct bandgap for alpha-electrons is Eg = 3.0 eV, which is somewhat smaller than the gap Eg = 3.36 eV in the pristine cubic STO. Calculations reveal an unoccupied in-gap defect energy level, which is mainly produced by the nitrogen atom. The minimum energy difference between the lowest defect levels and the top of the valence band, or optical absorption energy here, is Eabs = 1.5 eV (indirect transitions of beta-electrons).
The defect formation energy Edop is defined by
where E(Ndop) is the total energy of the crystal with nitrogen dopant and E(N) is the half energy of a gas-phase N2 molecule. The simulation of defective STO by SC 2 × 2 × 2 gives the energy Edop = 5.7 eV.
Like nitrogen in the bulk STO, the nitrogen substitution in the centre of the slab also produces an unoccupied defect energy level in the band gap (Fig. 2a). The calculated bandgap energy is Eg = 2.8 eV for direct transition of alpha-electrons (cf. with 2.5 eV for pristine slab). The found absorption energy is Eabs = 1.6 eV for indirect transition of beta-electrons and the formation energy is Edop = 5.5 eV.
Contrary to nitrogen in the bulk STO or inside the slab, substitutional atom on the surface of the slab does not create any defect levels in the band gap, but modifies only the edges of the valence and conduction bands (Fig. 2b). The modified bandgap is Eg ~ 2.4 eV, which is close to that in the pristine slab. The formation energy for nitrogen substitution on the surface of the slab is Edop = 5.2 eV, which is by 0.3 eV smaller than substitution energy inside the slab. Thus, also substitutional nitrogen could prefer surface locations analogously to vacancies, although with a lesser energy gain.
It is worth emphasizing that the in-gap defect levels can be produced by the substitutional nitrogen located either in the slab or bulk, but not on the surface. This behaviour contrasts the presence of the in-gap levels for any location of the oxygen vacancy: either inside the slab or on its surface. Both the oxygen vacancies and substitutional atoms prefer surface locations over those inside the slabs.
For all defects, the wide bandgap insulator phases are predicted. Interestingly, compared to pure 5-layer STO slab with TiO2 terminations, in systems, where the in-gap states arise, the calculated band gaps are wider, by 0.6 eV (VO in the central layer), 0.4 eV (VO on the surface) and 0.3 eV (nitrogen in the central layer) respectively. At the same time, simulations reveal that the electronic transitions, related to the in-gap states, are indirect. According to the calculations, neither oxygen substitutions, nor vacancies can lead to often reported large band conductivity and significant optical absorption in the visible spectral range. To verify the theoretical predictions, we experimentally investigated the optical properties of STO films with oxygen substitutions or vacancies.
Optical properties
Epitaxial and polycrystalline STO films were grown by pulsed laser deposition, during which anion dopants and/or oxygen vacancies were introduced in-situ by varying the ambient gas (“Methods”)33. The content δ of oxygen vacancies/substitutions was to ~ 0.3 in SrTiO3-δXδ (Supplementary section S1). The prepared films are marked here by S- and S*- for stoichiometric films, O- and O*- for oxygen deficient films, and N- and N*- (and H- and H*) for nitrogen-(hydrogen)-doped epitaxial and polycrystalline (*) ones, respectively. The optical properties were inspected by spectroscopic ellipsometry using very small intensity of light, that ensures probing of authentic, and not photoinduced properties.
All films exhibited profound absorption peaks centred at 5 eV, which is consistent with the fundamental absorption in regular STO (Fig. 3a–f).

Absorption coefficient α as a function of photon energy. Spectral range is (a–f) from 0.7 to 7.5 eV, (g–l) from 2.5 to 4.0 eV, and (m,n) from 0.7 to 4.0 eV. Dashed lines show Urbach absorption in (g–n). STO films are marked as follows: S epitaxial stoichiometric, O epitaxial with oxygen vacancies, N epitaxial with nitrogen substitution, H epitaxial with hydrogen substitution, O* polycrystalline with oxygen vacancies, N* polycrystalline with nitrogen substitution, H* polycrystalline with hydrogen substitution.
In all films, a steep raise of the absorption coefficient α at photon energy E ~ 4 eV is caused by a direct band-to-band transition. We note that compared to the indirect bandgap in pure unstressed cubic STO, the theoretically predicted bandgaps are direct in the presence of oxygen vacancies/substitutions (see above) or epitaxial lattice strain45. The energy Ed of the direct bandgap was found using Tauc-type plots [(αE)2 ∝ (E – Ed)] (Supplementary Fig. S2). The energy E0 of the lowest-energy critical point, corresponding to the strongest band-to-band transition, was extracted using second derivatives of the dielectric function (Supplementary Fig. S3). When compared to the lowest energy direct transition in bulk cubic STO, the energies Ed and E0 appeared to be clearly larger in the films (Table 1). The blueshift up to ~ 0.4 eV was detected in the polycrystalline doped N*- and H* films. The blueshift of ~ 0.2 eV was also found in stoichiometric epitaxial film (S), whereas it was absent in stoichiometric polycrystalline film (S*) (Supplementary Fig. S4).
As theoretically demonstrated, the lattice strain can lift or lower the conduction band45, whereas oxygen substitutions/vacancies can widen the bandgap (as shown in this work). Both mechanisms coexist and are difficult to separate in the epitaxial O-, N-, and H-films. However, the behaviour of the polycrystalline S*-, O*-, N*-, and H*-films points to the bandgap widening due to oxygen substitutions/vacancies. Importantly, our observations imply that commonly believed bandgap narrowing is very unlikely for several percent of oxygen substitutions /vacancies in STO.
Next, we closer inspected the absorption coefficient α at photon energies E < 4 eV. Semilog (α–E) plots (Fig. 3g–l) revealed the presence of an Urbach-type absorption46 [α ∝ exp(E)] in all films. Previously, we suggested that Fröhlich-type electron–phonon interactions may lead to steep Urbach tails in pure STO films47. This mechanism may also explain the behaviour here.
The Urbach-type absorption is superimposed with an additional contribution in the epitaxial N- and H-films (Fig. 3m,n). The additional peak at ~ 2 eV in the N-film and the peak at ~ 1.5 eV in the H-film evidence the presence of anion-specific deep in-gap states in these films. According to the calculations, these observations indicate substitutional locations inside the crystal for the epitaxial films. Importantly, the N- and H-specific absorption peaks were not detected in the polycrystalline N*- and H*-films. Because in-gap states were not expected for boundary substitutional locations, the difference between the epitaxial and polycrystalline films proves that anionic dopants are mainly located at phase boundaries, and not inside crystallites, in the polycrystalline films.
In the visible spectral range (photon energies ~ 1.8–3.1 eV), the absorption coefficient is α < 104 cm−1 in the epitaxial O-, N-, and H-films and α < 103 cm−1 in the rest of the films. This small magnitude of α indicates that visual observations of changes in colour and/or transmittance, that are often reported for long-lasting high-temperature processing of STO at reduced oxygen pressure or in the presence of hydrogen or nitrogen9,10,11,12,13,14,15,16, can be hardly caused by oxygen substitutions/vacancies. We stress that here we avoided such high-temperature processing, during which structural and/or compositional inhomogeneity, as well as networks of dislocations or other structural defects can easily form in STO19.
For the photon energies E < 1 eV, spectral features related to the presence of in-gap states or free carriers were not detected in any of the films (Fig. 4a). The dielectric and loss functions in the infrared range (Fig. 4b,c) were consistent with regular STO behaviour and indicated lattice vibrations at approximately 59, 68, and 98 meV (475, 550, 790 cm−1)48. The strongest lines at ~ 550 cm−1 (Fig. 4c) correspond to transverse optical phonons present both in the cubic and tetragonal phases of STO. The lines at ~ 475 and 790 cm−1 (Fig. 4b) are related to longitudinal optical phonons.

Optical properties in the infrared spectral range. (a) Absorption coefficient, (b) dielectric loss function, and (c) imaginary part of the dielectric function as a function of (a) photon energy or (b,c) wavenumber in different STO films. Data for all films overlap in (a,b). Data for reference crystal are shown in (c) for comparison.
Thus, our theoretical calculations and experimental observations are in excellent agreement with each other and demonstrate that the substitution of several percent of oxygen atoms with nitrogen, hydrogen, and/or vacancies leads to the wide-bandgap insulator phases for all substitution types and locations in STO. The detected anion-specific absorption in the epitaxial N- and H-films evidences the presence of in-gap states and is consistent with the theoretical picture of substitutional locations inside the crystals.
The obtained results imply that the bandgap narrowing and semiconductor-like band conductivity cannot be caused by oxygen substitutions/vacancies in STO. Contrary to the previous expectations of bandgap narrowing, our theoretical calculations and experimental observations point to the bandgap widening. Furthermore, the minimum calculated energy for carriers’ excitation from the in-gap states to the conduction band is at least 0.1 eV for oxygen vacancies, whereas other defects produce deeper states. Although the magnitude of 0.1 eV is small compared to the bandgap in STO, and the term “shallow” is often used to describe such in-gap states in STO, this magnitude is generally rather large. For instance, a gap of only 20–50 meV is associated with high-resistivity insulator phase in perovskite nickelates. Our observations suggest a minor if any carriers’ excitation at room-temperature in STO films.
Finally, we note that semiconductor-like behaviour is usually reported for STO subjected to a high-temperature processing for several hours to days. Our work evidences the lack of semiconductor-like properties in the absence of such processing. Concurrently, in the last years, it has been found that structural or compositional defects, other than oxygen vacancies/substitutions, can drive the electronic properties of STO towards semiconductor state49,50,51,52,53,54. We believe that during high-temperature processes, such defects can unintentionally form and lead to the increased optical absorption and electrical conductivity.
Conclusions
Substitutions of several percent of oxygen atoms with nitrogen, hydrogen, or vacancies were studied in perovskite oxide SrTiO3 films. The large-scale theoretical first-principles calculations of the atomic and electronic structure of the crystals and ultrathin films were combined with experimental investigations of the optical properties of in-situ prepared epitaxial and polycrystalline films. It was theoretically predicted and experimentally verified, that the location of the substitutions and vacancies is more energetically favourable at phase boundaries, interfaces, or surfaces than inside a crystallite. Deep in-gap states were detected for substitutional atoms only inside crystallites, whereas in-gap donor bands were suggested for all locations of oxygen vacancies. The theoretical calculations and experimental observations evidenced wide-bandgap insulator phases for all cases. Furthermore, a tendency to bandgap widening was observed.
Methods
First-principles calculations
Ab initio first-principles computer simulations were performed to investigate neutral oxygen vacancies and substitutional nitrogen atoms in both bulk STO crystals and ultrathin freestanding STO (001) films. The structural and electronic properties of these systems, as well as the formation energy of the defects were examined within the linear combination of atomic orbitals approximation. We employed the B1WC advanced hybrid exchange–correlation functional of the density-functional-theory (DFT), as implemented in the CRYSTAL17 computer code23. Basis sets with Hay and Wadt small core pseudopotential were used for Sr and Ti atoms24, and the all-electron basis sets were applied for the description of oxygen atoms25 and nitrogen atoms26. The periodic supercell (SC) approach was employed to simulate point defects. More computational details can be found in our previous studies of perovskite (Ba,Sr)TiO3 and (Ba,Ca)TiO3 solid solutions27,28,29 and BaTiO3/SrTiO3 heterostructures28,30.
For analysis of substitutional nitrogen atom in the bulk STO crystal, we used SC with extended lattice constants 2 × 2 × 2 (40 atoms). In turn, to simulate thin STO films, we employed free-standing slabs in the form of 2D supercells. The surface parameter extension was 2 × 2 along x and y directions of the slab, which contained 5-layers (001) along the z axis and possessed TiO2 terminations on its both sides (52 atoms). The defects were then placed either in the central layer of the slab or on its surface.
Various possible spin states were considered by means of unrestricted open shell DFT calculations23. When an oxygen atom is removed from STO and a neutral vacancy VO is formed, the spins of the two unpaired electrons are distributed among the Ti atoms near the vacancy. This system may have two different spin states: either Sz = 0 (the singlet state; two antiparallel spins) or Sz = 1 (the triplet state; two spins are parallel)31,32. Concurrently, the removal of an O2− anion produces a charged oxygen vacancy VO2+, around which all chemical bonds are closed32. Nitrogen atom, which substitutes oxygen in STO, has one unpaired electron (here, N atom mimics missing O2− ion) and, hence, the spin is Sz = 1/2.
An important issue is related to the symmetry of the crystal or film containing point defects. Here, the defects—substitutional nitrogen atoms and oxygen vacancies—were created in the SC constructed from a cubic STO (space group SG 221) with the Wickoff positions of atoms as follows: Sr:1b(1/2,1/2,1/2), Ti:1a(0,0,0), O:3d(1/2,0,0). The symmetry of the system is lowered upon the formation of a defect. Because the result of a full structural optimization depends on the symmetry of the system, the choice of symmetry affects the total energy, spin distribution, and the formation energy of defects, along with other important parameters. In turn, the symmetry is affected by both the dimension of the SC and the exact position of the defect. For example, the presence of defect may lower the cubic SG 221 symmetry of SC 2 × 2 × 2 down to tetragonal SG 99 or orthorhombic SG 25. Additionally, different atom spins can further lower the symmetry. Here, for all systems, we performed calculations with the lowest symmetry for each SC or, even, without any symmetry (SG 1). In this approach, artificial constraints imposed by symmetry are minimal or completely absent and the results only depend on atomic interaction potentials. Unfortunately, the computational cost of such calculations strongly increases.
Experimental
Thin STO films (thicknesses of 80–100 nm) were grown by pulsed laser deposition, during which anion dopants and/or oxygen vacancies were introduced in-situ by varying the ambient gas33. Epitaxially polished (001) (La0.3Sr0.7)(Al0.65Ta0.35)O3 substrates (LSAT) and silicon substrates covered with native surface oxide (Si/SiO2) were used to prepare epitaxial and polycrystalline films, respectively. The films were deposited in pairs (one on LSAT and another one on Si/SiO2) within a single process, thus ensuring similar chemical composition for both films in each pair. The different microstructures of these films were determined by the underlying substrates.
The spectroscopic ellipsometry measurements were performed using J. A. Woollam ellipsometers at room temperature. The measurements at photon energies from 0.75 to 8.8 eV were carried out on a VUV ellipsometer in dry nitrogen atmosphere and those at the energies from 25 meV to 1.0 eV were performed on an IR ellipsometer. The data were processed using a commercial WVASE32 software package. The dielectric function and optical constants (index of refraction, extinction coefficient, and absorption coefficient) were determined as a function of photon energy. Specific methodological aspects were presented in our previous studies35,36.
Data availability
The datasets generated and/or analyzed during the current study are available from the authors on reasonable request.
References
Lines, M. E., Glass, A. M. Principles and Applications of Ferroelectrics and Related Materials (Clarendon Press 2004).
Uchino, K. Ferroelectric Devices (Marcel Dekker, 2000).
Scott, J. F. Ferroelectric Memories (Springer-Verlag, 2000).
Huang, H., Scott, J. F. (eds.) Ferroelectric Materials for Energy Applications (Wiley-VCH Verlag, 2018).
Nanoelectronics and Information Technology: Advanced Electronic Materials and Novel Devices (ed. Waser, R.) (Wiley, 2012).
Scott, J. F. Application of modern ferroelectrics. Science 315, 954 (2007).
Mikolajick, T. et al. Next generation ferroelectric materials for semiconductor process integration and their applications. J. Appl. Phys. 129, 100901 (2021).
Zhang, S., Malič, B., Li, J.-F. & Rödel, J. Lead-free ferroelectric materials: Prospective applications. J. Mater. Res. 36, 985 (2021).
Meng, Y. et al. Review: recent progress in low-temperature proton-conducting. J. Mater. Sci. 54, 9291 (2019).
Kobayashi, Y. et al. An oxyhydride of BaTiO3 exhibiting hydride exchange and electronic conductivity. Nat. Mater. 11, 507 (2012).
Yajima, T. et al. A labile hydride strategy for the synthesis of heavily nitridized BaTiO3. Nat. Chem. 7, 1017 (2015).
Kageyama, H. et al. Expanding frontiers in materials chemistry and physics with multiple anions. Nat. Commun. 9, 772 (2018).
Eklöf-Österberg, C. et al. The role of oxygen vacancies on the vibrational motions of hydride ions in the oxyhydride of barium titanate. J. Mater. Chem. A 8, 6360 (2020).
Liu, X. et al. Highly correlated hydride ion tracer diffusion in SrTiO3-xHx oxyhydrides. J. Am. Chem. Soc. 141, 4653 (2019).
Yajima, T. et al. Epitaxial thin films of ATiO(3–x)H(x) (A = Ba, Sr, Ca) with metallic conductivity. J. Am. Chem. Soc. 134, 8782 (2012).
Bouilly, G. et al. Electrical properties of epitaxial thin films of oxyhydrides ATiO3–xHx (A = Ba and Sr). Chem. Mater. 27, 6354 (2015).
Arlt, G., Hennings, D. & de With, G. Dielectric properties of fine-grained barium titanate ceramics. J. Appl. Phys. 58, 1619 (1985).
Ghosh, D. et al. Domain wall displacement is the origin of superior permittivity and piezoelectricity in BaTiO3 at intermediate grain sizes. Adv. Func. Mater. 24, 885 (2014).
Burian, M. et al. Buried moiré supercells through SrTiO3 nanolayer relaxation. Phys. Rev. Res. 3, 013225 (2021).
Iwazaki, Y., Gohda, Y. & Tsuneyuki, S. Diversity of hydrogen configuration and its roles in SrTiO3−δ. APL Mater. 2, 012103 (2014).
Takeyasu, K., Fukada, K., Ogura, S., Matsumoto, M. & Fukutani, K. Two charged states of hydrogen on the SrTiO3 (001) surface. J. Chem. Phys. 140, 084703 (2014).
Varley, J. B., Janotti, A. & Van de Walle, C. G. Hydrogenated vacancies and hidden hydrogen in SrTiO3. Phys. Rev. B 89, 075202 (2014).
Dovesi, R. et al. CRYSTAL17 User’s Manual (University of Torino, 2017).
Piskunov, S., Heifets, E., Eglitis, R. I. & Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comput. Mater. Sci. 29, 165–178 (2004).
Bredow, T., Jug, K. & Evarestov, R. A. Electronic and magnetic structure of ScMnO3. Phys. Status Solidi B 243, R10–R12 (2006).
Gatti, C., Saunders, V. R. & Roetti, C. Crystal field effects on the topological properties of the electron density in molecular crystals: The case of urea. J. Chem. Phys. 101, 10686 (1994).
Rusevich, L. L., Zvejnieks, G., Erba, A., Dovesi, R. & Kotomin, E. A. Electromechanical properties of Ba(1–x)SrxTiO3 perovskite solid solutions from first-principles calculations. J. Phys. Chem. A 121, 9409–9414 (2017).
Rusevich, L. L. et al. Theoretical and experimental study of (Ba, Sr)TiO3 perovskite solid solutions and BaTiO3/SrTiO3 heterostructures. J. Phys. Chem. C 123, 2031–2036 (2019).
Rusevich, L. L., Zvejnieks, G. & Kotomin, E. A. Ab initio simulation of (Ba, Sr)TiO3 and (Ba, Ca)TiO3 perovskite solid solutions. Solid State Ionics 337, 76–81 (2019).
Zvejnieks, G., Rusevich, L. L., Gryaznov, D. & Kotomin, E. A. Interface-induced enhancement of piezoelectricity in the (SrTiO3)m/(BaTiO3)M−m superlattice for energy harvesting applications. Phys. Chem. Chem. Phys. 21, 23541–23551 (2019).
Rusevich, L. L., Kotomin, E. A., Zvejnieks, G. & Popov, A. I. Ab initio calculations of structural, electronic and vibrational properties of BaTiO3 and SrTiO3 perovskite crystals with oxygen vacancies. Low Temp. Phys. 46, 1185–1195 (2020).
Tyunina, M. et al. Epitaxial growth of perovskite oxide films facilitated by oxygen vacancies. J. Mater. Chem. C 9, 1693–1700 (2021).
Tyunina, M. et al. In situ anion-doped epitaxial strontium titanate films. Phys. Chem. Chem. Phys. 22, 24796 (2020).
Tyunina, M. et al. Oxygen vacancy dipoles in strained epitaxial BaTiO3 films. Phys. Rev. Res. 2, 023056 (2020).
Tyunina, M. et al. Multiple optical impacts of anion doping in epitaxial barium titanate films. APL Mater. 8, 071107 (2020).
Dejneka, A. et al. Optical effects induced by epitaxial tension in lead titanate. Appl. Phys. Lett. 112, 031111 (2018).
Evarestov, R. A., Kotomin, E. A. & Zhukovskii, Yu. F. DFT study of a single F center in cubic SrTiO3 perovskite. Int. J. Quantum Chem. 106, 2173–2183 (2006).
Evarestov, R. et al. Jahn-Teller effect in the phonon properties of defective SrTiO3. Phys. Rev. B 85, 174303 (2012).
Gryaznov, D. et al. A comparative ab initio thermodynamic study of oxygen vacancies in ZnO and SrTiO3: Emphasis on phonon contribution. J. Phys. Chem. C 117, 13776–13784 (2013).
Janotti, A., Varley, J. B., Choi, M. & Van de Walle, C. G. Vacancies and small polarons in SrTiO3. Phys. Rev. B 90, 085202 (2014).
Carrasco, J. et al. First-principles calculations of the atomic and electronic structure of F centers in the bulk and on the (001) surface of SrTiO3. Phys. Rev. B 73, 064106 (2006).
Alexandrov, V. E., Kotomin, E. A., Maier, J. & Evarestov, R. A. First-principles study of bulk and surface oxygen vacancies in SrTiO3 crystal. Eur. Phys. J. B 72, 53–57 (2009).
Al-Zubi, A., Bihlmayer, G. & Blügel, S. Electronic structure of oxygen-deficient SrTiO3 and Sr2TiO4. Crystals 9, 580 (2019).
Jeschke, H. O., Juan Shen, J. & Valentí, R. Localized versus itinerant states created by multiple oxygen vacancies in SrTiO3. New J. Phys. 17, 023034 (2015).
Berger, R. F., Fennie, C. J. & Neaton, J. B. Band gap and edge engineering via ferroic distortion and anisotropic strain: The case of SrTiO3. Phys. Rev. Lett. 107, 146804 (2011).
Kurik, N. V. Urbach rule. Phys. Stat. Sol. (a) 8, 9 (1971).
Tyunina, M., Nepomniashchaia, N., Vetokhina, V. & Dejneka, A. Optics of epitaxial strained strontium titanate films. Appl. Phys. Lett. 117, 082901 (2020).
Zollner, S., Demkov, A. A., Liu, R., Fejes, P. L. & Gregory, R. B. Optical properties of bulk and thin-film SrTiO3 on Si and Pt. J. Vac. Sci. Technol. B 18, 2242 (2000).
Weber, W. J. et al. Synergy of elastic and inelastic energy loss on ion track formation in SrTiO3. Sci. Rep. 5, 7726 (2015).
Liu, B., Cooper, V. R., Zhang, Y. & Weber, W. J. Segregation and trapping of oxygen vacancies near the SrTiO3 Σ3 (112)[110] tilt grain boundary. Acta Mater. 90, 394–399 (2015).
Lee, W. et al. Resistance switching behavior of atomic layer deposited SrTiO3 film through possible formation of Sr2Ti6O13 or Sr1Ti11O20 phases. Sci. Rep. 6, 20550 (2016).
Crespillo, M. L., Graham, J. T., Agulló-López, F., Zhang, Y. & Weber, W. J. Role of oxygen vacancies on light emission mechanisms in SrTiO3 induced by high-energy particles. J. Phys. D: Appl. Phys. 50, 155303 (2017).
Liu, B. et al. Composition dependent intrinsic defect structures in SrTiO3. Phys. Chem. Chem. Phys. 16, 15590 (2014).
Baki, A. et al. Influence of Sr deficiency on structural and electrical properties of SrTiO3 thin films grown by metal-organic vapor phase epitaxy. Sci. Rep. 11, 7497 (2021).
Acknowledgements
The authors would like to thank R. Dittmann for useful discussions, T. Kocourek, O. Pacherova, S. Cichon, V. Vetokhina, and P. Babor for their contributions to sample preparation and characterization. The authors (M.T., A.D.) acknowledge support from the Czech Science Foundation (Grant No. 19-09671S), the European Structural and Investment Funds and the Ministry of Education, Youth and Sports of the Czech Republic through Programme “Research, Development and Education” (Project No. SOLID21 CZ.02.1.01/0.0/0.0/16-019/0000760). This study was partly supported by FLAG-ERA JTC project To2Dox (L.R. and E.K.). Calculations have been performed on the LASC Cluster in the Institute of Solid State Physics (ISSP), University of Latvia, and at the HLRS supercomputer centre, Stuttgart (Project DEFTD). The ISSP has received funding as a Centre of Excellence through the EU Horizon 2020 Programme H2020-WIDESPREAD-01-2016-2017-Teaming-Phase2 (CAMART2, Grant No. 739508).
Funding
This article was funded by FLAG-ERA JTC project To2Dox, Czech Science Foundation (Grant no. 19-09671S), Ministry of Education, Youth and Sports of the Czech Republic, programme “Research, Development and Education” (Grant no. SOLID21 CZ.02.1.01/0.0/0.0/16-019/0000760).
Author information
Authors and Affiliations
Contributions
L.R. and E.K. performed calculations; M.T. and A.D. supervised experimental work; N.N. carried out optical measurements; M.T. analyzed results; L.R. and M.T. wrote the manuscript; L.R., M.T., E.K., and A.D. acquired funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rusevich, L.L., Tyunina, M., Kotomin, E.A. et al. The electronic properties of SrTiO3-δ with oxygen vacancies or substitutions. Sci Rep 11, 23341 (2021). https://doi.org/10.1038/s41598-021-02751-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02751-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
