
Abstract
Total spikelet number per spike (TSN) is a major component of spike architecture in wheat (Triticum aestivum L.). A major and consistent quantitative trait locus (QTL) was discovered for TSN in a doubled haploid spring wheat population grown in the field over 4 years. The QTL on chromosome 7B explained up to 20.5% of phenotypic variance. In its physical interval (7B: 6.37–21.67 Mb), the gene FLOWERING LOCUS T (FT-B1) emerged as candidate for the observed effect. In one of the parental lines, FT-B1 carried a non-synonymous substitution on position 19 of the coding sequence. This mutation modifying an aspartic acid (D) into a histidine (H) occurred in a highly conserved position. The mutation was observed with a frequency of ca. 68% in a set of 135 hexaploid wheat varieties and landraces, while it was not found in other plant species. FT-B1 only showed a minor effect on heading and flowering time (FT) which were dominated by a major QTL on chromosome 5A caused by segregation of the vernalization gene VRN-A1. Individuals carrying the FT-B1 allele with amino acid histidine had, on average, a higher number of spikelets (15.1) than individuals with the aspartic acid allele (14.3) independent of their VRN-A1 allele. We show that the effect of TSN is not mainly related to flowering time; however, the duration of pre-anthesis phases may play a major role.
Similar content being viewed by others


Introduction
In light of a growing world population, the increase of wheat yield and the genetic mechanisms behind are of immediate interest. It was shown that grain yield in German winter wheat cultivars was highly correlated with grain number per spike1 and, therefore, the genes and mechanisms determining inflorescence and spike architecture in cereals have been in the focus of research (reviewed in Gauley and Boden2; Koppolu and Schnurbusch3). The unbranched spike of wheat contains one spikelet per node and is terminated with a single terminal spikelet at the apical end of the inflorescence. Each spikelet contains 8–12 florets and produces usually 3–5 grains per spikelet1. Important determinants of grain number are the floret fertility4 as well as spike fruiting efficiency (grains per unit spike dry weight at anthesis)5,6. Among the complex network of spike-related traits, total spikelet number per spike (TSN) was studied in several reports. In genetic approaches, TSN appears usually as a quantitative trait and several quantitative trait loci (QTL) were described7,8,9. A few genes influencing TSN are known; among them are (1) the Q gene involved in wheat domestication10,11, (2) a putative ortholog to rice MOC112, and (3) a wheat ortholog (TaAPO-A1 or WAPO-A1) to rice ABERRANT PANICLE ORGANIZATION (APO1)13,14,15.
Besides the modulation of spikelet meristem identity genes, the genetic regulation of spikelet initiation through timing and florigenic signals plays a major role in the determination of TSN. Homologs of Arabidopsis FLOWERING LOCUS T (FT)16,17,18, such as HEADING DATE 3a (Hd3a) and RICE FLOWERING LOCUS T 1 (RFT1) in rice promote transition to flowering and tillering19,20,21. In wheat, the FT-B1 gene on chromosome 7B was originally reported as VRN-B3 that affects the vernalization requirement in wheat and—besides the three VRN-1 genes—that determines the spring or winter growth type of hexaploid wheat22,23. In wheat FT-B1 acts downstream of PHOTOPERIOD-1 (PPD-1) gene to influence spikelet number, with loss of FT-B1 resulting in increased spikelet number in a thermally responsive manner2,24,25. An ortholog of the maize domestication gene TEOSINTE BRANCHED 1 (TB1) was reported to interact with FT-B1 in a dosage-dependent manner to promote the formation of paired spikelets in wheat by activating a cascade of floral meristem identity genes26.
FT-B1 is a member of an evolutionarily well-conserved gene family [phosphatidylethanolamine-binding protein (PEBP)] that is present in all taxa including bacteria, animals, and plants27. Among angiosperms, three subfamilies are described FT-like, MFT-like (MOTHER OF FT AND TFL), and TFL1-like genes (TERMINAL FLOWER1)17. Phylogenetic analyses identified MFT—the only member of the PEBP family in mosses—as the ancestral gene, and its duplication in FT/TFL1-like genes and their divergence coincided with the evolution of vascular plants28,29. Although sharing a high homology, the genes have been described to be partially redundant as flowering promoters (FT and MFT) or to act in an antagonist manner (TFL1)17,30. Compared to the two other gene families, FT-like genes, evolving via small- and large-scale duplications, are the most numerous among monocots with 13 paralogs identified in rice, five in barley, and nine in wheat based on expressed sequences tags (ESTs) data31,32. Due to their impact on the phenology and flowering architecture of crops, FT-like genes have been a target of selection during domestication and subsequent range expansion to adapt to new environments33,34.
In this study, we performed linkage mapping to investigate the genetic basis of total spikelet number per spike and its associated traits viz., spike length, heading date, and flowering time in a doubled haploid spring wheat population. Our analyses identified a large-effect locus that harbored FT-B1—a candidate gene for TSN. The origin and frequency of a non-synonymous mutation associated with an increased spikelet number was investigated by analyzing a large diversity of land plants and a panel of hexaploid wheat varieties. Moreover, we also investigated the interaction of FT-B1 with the vernalization gene VRN-A1 that segregated in the studied population.
Results
Description of phenotypic data
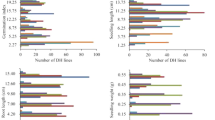
The doubled haploid (DH) population-2 described in Muqaddasi et al.35 was grown on the field and characterized for 4 years (2016–2019) for total spikelet number (TSN) per spike, spike length (SL), heading date (HD), and flowering time (FT). The best linear unbiased estimations (BLUEs) showed a wide variation within and across years for all traits, with TSN and SL being normally distributed (Shapiro–Wilk test: P = 0.82 for TSN, P = 0.06 for SL, P = 0.001 for HD, and P = 0.003 for FT). Although the parental lines did not differ much for their flowering traits (HD and FT), the population presented a bimodal distribution (Fig. 1) resulting from the segregation of the vernalization genes VRN-A1 and VRN-B135. The parental line TRI-10703 carried the dominant allele Vrn-A1a and the recessive allele vrn-B1, while TRI-5310 harbored the recessive allele vrn-A1 and the dominant allele Vrn-B1. For the spike morphology traits (TSN and SL), the parental genotype TRI-10703 (parent-A) systematically performed better than parental genotype TRI-5310 (parent-B; Fig. 1, Supplementary Figure S1). The ANOVA indicated significant (P < 0.001) genotypic and environmental differences for all traits. High repeatability values (H2 = 0.89–0.96) were obtained for biological replicates of TSN and SL. Broad sense heritability values calculated over 4 years ranged between 0.71 for SL and 0.88 for FT and HD (Table 1). The Pearson correlation coefficient calculated based on BLUEs indicated a positive and significant (P < 0.001) correlation of TSN with SL, HD and FT, and between HD and FT, while the correlation between SL and the flowering traits was null (Fig. 2).

Phenotypic distribution of the best linear unbiased estimations (BLUEs) of the investigated traits. Arrows indicate the phenotypes of the parental lines with parent A corresponding to TRI-10703 and parent B to TRI-5310.

Pearson’s correlation coefficient of the best linear unbiased estimations (BLUEs) of total spikelet number (TSN), spike length (SL), heading date (HD), and flowering time (FT). Significance of the correlations is indicated with ***P < 0.001.
Mapping of quantitative trait loci (QTL)
Composite interval mapping identified a total of 56 individual QTL at 28 loci for all years, all traits and their resulting BLUEs (Table 2, Supplementary Table S1). However, only a single major QTL per trait was found consistent, i.e., present in all the years and their corresponding BLUEs, on chromosome 5A for FT and HD and on chromosome 7B for TSN (Fig. 3, Table 2). No consistent QTL were identified for SL. The FT (QFt.ipk-5A) and HD (QHd.ipk-5A) QTL collocated at 145.01–171.23 cM and explained 29.7–66.4% and 23–63.6% of phenotypic variance, respectively. These QTL most probably correspond to the vernalization gene VRN-A1 (TraesCS5A02G391700). For QTsn.ipk-7B—located at 0–33.46 cM—the phenotypic variance amounted to 13.1–20.5%. Interestingly, minor QTL for the three other traits (viz., QHd.ipk-7B, QFt.ipk-7B.1, QFt.ipk-7B.2, and QSl.ipk-7B.1) were identified within or overlapping the QTsn.ipk-7B interval.

Quantitative trait loci (QTL) map of total spikelet number (TSN), spike length (SL), heading date (HD), and flowering time (FT) in a DH-population of spring wheat grown over 4 years and their corresponding best linear unbiased estimations (BLUEs). The horizontal dashed lines indicate the corresponding logarithm of odds (LOD) threshold estimated at α = 0.05 with 1000 permutations to assess the QTL significance.
Identification of candidate gene FT-B1 in the physical region
The mapping onto the IWGSC RefSeq v1.1 of the most significant markers defining the QTsn.ipk-7B interval identified for the BLUEs resulted in a physical interval spanning from 6.37 to 21.67 Mb. The region harbored 121 high-confidence genes (Supplementary Table S2). Potential candidates included two genes annotated as CLAVATA3/ESR (CLE)-related protein, six annotated as MADS box transcription factors, and the FLOWERING LOCUS T (VRN-B3 or FT-B1, TraesCS7B02G013100) located at 9.7 Mb (9,702,354–9,704,354 bp). Among the 121 high-confidence genes, 26 harbored a total of 56 non-synonymous mutations between both parental lines. All non-synonymous mutations were analyzed with PROVEAN to evaluate their potential effect on the proteins. This analysis identified eight potentially deleterious mutations distributed among six genes including FT-B1 (Supplementary Table S3). This gene—an ortholog of Arabidopsis FT and rice RFT1—has three predicted exons encoding a phosphatidylethanolamine-binding protein (PEBP). FT-B1 had a single non-synonymous substitution close to the start codon. At position 19 in the coding sequence, the parental genotype TRI-5310 (parent-B) and the reference Chinese Spring have a G, while TRI-10703 (parent-A) has a C. This mutation modifying an aspartic acid (D) into a histidine (H), although located outside of predicted domains, occurred in a highly conserved position. The 1.5 kb upstream region of FT-B1 was also searched for transcription factors using the bread wheat epigenomic map (http://bioinfo.sibs.ac.cn/cs_epigenome)36. The region harbored potential binding sites for RAP211, ERF2 and RAMOSA1, however, none were polymorphic between both parents.
Development of a KASP marker significantly associated with TSN
To confirm FT-B1 as a candidate gene for TSN, a KASP marker was developed for the described mutation in FT-B1 and genotyping was performed across the whole DH-population. The marker was included in the original matrix for a second round of composite interval mapping for TSN. The KASP marker was the most significant marker for QTsn.ipk-7B in the BLUEs and all years but 2017 and explained 10.6–22.9% of phenotypic variance (Supplementary Figure S2, Supplementary Table S4).
Interaction between VRN-A1 and FT-B1 on phenotypic expression of traits
The genotyping for VRN-A1 was done according to Zhang et al.23 and alleles were coded according to the rest of the data with TRI-10703 as parent A and TRI-5310 as parent B. A two-way factorial ANOVA for the effects of VRN-A1 and FT-B1 on all traits revealed a highly significant effect of FT-B1 but not VRN-A1 on TSN, while both genes were found significant for the three other traits (Table 3, Supplementary Table S5). The interaction of both genes was not significant. Indeed, individuals carrying the FT-B1 A allele with amino acid histidine had on average a higher number of spikelets (15.1) than individuals with the B allele (14.3) independent of their VRN-A1 allele (Fig. 4). Individuals carrying the dominant Vrn-A1a allele (A) flowered on average 3 days earlier than individuals with the recessive vrn-A1 allele (B), while the difference in flowering time caused by the respective FT-B1 allele was only 1 day. However, this difference was only significant for individuals carrying the dominant Vrn-A1a allele (Wilcoxon test, P = 0.002). For SL, individuals with the recessive vrn-A1 allele (B), the spike was on average 0.48 cm (Wilcoxon test, P = 0.005) shorter than for the Vrn-A1a allele (A).

Allele-wise phenotypic distribution of the KASP marker designed for FT-B1 of the best linear unbiased estimations (BLUEs) for the investigated traits in dominant (A) or recessive (B) VRN-A1 background. Significance of the mean differences of the marker alleles obtained from a Wilcoxon rank-sum test.
Analysis of frequency of the observed mutation in FT-B1
In the wheat reference genome, a total of 67 genes, including genes annotated as MFT-like and TFL1-like, were classified as FT-B1 paralogs in the EnsemblPlants database with 27 identified in the B genome, whereas, 20 in each of the A- and D-genomes. Among those 70 PEBP genes, including FT-B1 and its homoeologues on chromosomes 7A and 7D (TraesCS7A02G115400 and TraesCS7D02G111600), the majority possessed an aspartic acid (D) but none had a histidine (Fig. 5b). The homoeologues in the parental lines were identical to each other and to the reference (Fig. 6). In a set of 377 homologs retrieved by PROVEAN from the NCBI database across a large diversity of flowering plants as well as a few Gymnosperms, 86.4% had a D, 7.1% possessed an asparagine (N), and 5.5% had a glutamic acid (E). Three sequences had another amino acid but none had an H (Fig. 5c). This mutation obtained a PROVEAN score of − 4.267 and is therefore considered deleterious. However, in a set of 135 wheat varieties and landraces genotyped with the high-density array Axiom37, ca. 68% carried the mutation (marker AX-94810990; Fig. 5a) while none of the bread wheat progenitors (eight T. turgidum and one Aegilops speltoides accession) included in this genotyping effort had it.

Intraspecific, intragenomic and interspecific diversity for the FT-B1 mutation. All values are percentages. (a) Allele frequency across 135 wheat varieties sequenced with the Axiom marker AX-94810990 of the nucleotide substitution, with C translating to histidine (H) and G translating to aspartic acid (D). (b) Allele frequency for the corresponding amino acid position across 70 PEBP genes annotated in the wheat reference genome. (c) Allele frequency for the corresponding amino acid position across 377 homologs retrieved from the PROVEAN analysis across a large diversity of plants.

Sequence alignment of the 20 first amino acids of a subset of FT-B1 homologs including the homoeologues on chromosome 7A and 7D. The wheat reference sequence (Chinese Spring), the sequences of the two parental lines, and FT-B1 homologs in diverse species viz. Hordeum vulgare, Brachypodium distachyon, Oryza sativa, Zea mays, and Arabidopsis thaliana. The blue arrow indicates the position of the aspartic acid (D) to histidine (H) mutation present in the parent A and associated with elevated total spikelet number. The amino acids are color coded according to their side chain polarity with nonpolar (gold), polar (green), basic polar (blue), and acidic polar (red).
Discussion
FLOWERING LOCUS T is a likely candidate for a major TSN-QTL
A consistent major QTL for TSN was discovered over 4 years in the field-grown doubled haploid population TRI-10703 × TRI-5310 on chromosome 7BS explaining up to 20.5% of phenotypic variance. The FLOWERING LOCUS T (FT-B1) gene located in the most significant QTL interval (7B: 6.3–21.7 Mb) was considered as most likely candidate for the observed effect on TSN, because (1) FT-B1 carried a non-synonymous mutation causing an amino acid exchange in the parental lines, (2) a QTL analysis including the KASP marker based on the mutation confirmed its significant association with TSN, and (3) FT-B1 was previously associated with effects on spikelet number in the literature. Moreover, the analysis of all non-synonymous mutations within the QTL interval confirmed FT-B1 as a likely candidate. Overexpression of FT-B1 did not have an effect on spikelet number but accelerated flowering time in warm conditions while deletion of FT-B1 nearly doubled the flowering time and increased spike length and spikelet number24,25. According to Boden et al.38 the expression of FT-B1 is regulated by the pseudo-response regulator gene Ppd-1 and a reduced expression of FT-B1 was associated with paired spikelet formation and inflorescence architecture.
FT-B1 is usually associated with flowering time or heading date. Higher FT-B1 transcripts levels controlled by the insertion of a retrotransposon in the promoter region (“Hope” allele) accelerated flowering time and transgenic winter wheat transformed with the Hope promoter behaved like spring types. The retrotransposon characteristic of the highly expressed FT-B1 allele from variety “Hope”22,39 was not present in our parental lines. Changes in the expression of the wild type FT-B1 were shown to coincide with major transition of the inflorescence meristem2.
Different effects of FT-B1 and VRN-A1 on flowering time and total spikelet number
Our population is segregating for the vernalization genes VRN-A1 and VRN-B1, as well as for the photoperiodism gene Ppd-D1. The heading and flowering time were in all years governed by a major QTL on chromosome 5A which represented the effect of VRN-A1, while on chromosome 7B representing FT-B1 only minor QTL were observed for HD and FT (Table 2, Supplementary Table S1). VRN-A1 had highly significant effects on HD, FT and SL but not on TSN. On the other hand, FT-B1 was highly significant for TSN and showed small effects on the other three traits (Fig. 4, Table 3). In the correlations, increased TSN was correlated with increased SL as well as with delayed HD and FT. However, FT-B1 affected spikelet formation without a major effect on the flowering time. It may, therefore, be concluded that the FT-B1 effects on pre-flowering phases play a major role. The effects of sub-phases in plant growth of wheat on yield and yield components were highlighted by Guo et al.40,41, where spikelet initiation and spikelet number was associated with double ridge to terminal spikelet stage. Dixon et al.24 reported that flowering time, in contrast to spikelet number, was not responsive to changes in ambient temperature in a ft-b1 NIL, suggesting that the promotion of inflorescence development between the double ridge and terminal spikelet stages is regulated by FT-independent thermally responsive factors. In contrast to Arabidopsis, where FT induces flowering at elevated growth temperature42, the accelerated development pace of domesticated wheat under high temperature is largely independent of FT24,43. Li et al.44 investigated the interactions between the three wheat MADS-box genes VRN1, FUL2 and FUL3 and showed delayed formation of the terminal spikelet and increased number of spikelets per spike in VRN1-null and ful2-null mutants. Flowering delays in VRN1-null mutants were associated with reduced FT1 transcript levels in leaves. Also changes in expression level of FLOWERING LOCUS T2 (FT2), the closest paralog of the FT1 gene in temperate grasses, were associated with changes in spikelet number, flowering time and fertility45,46. Another QTL-study reported three pleiotropic QTL regions associated with spikelet number and heading date on chromosomes 2A, 7A and 7D, with FT-A1 considered as candidate gene for QTspn/Hd.cau-7A9.
A non-synonymous mutation in exon1 of FT-B1 is widespread in the species T. aestivum
Sequencing of the parental lines of the DH-population allowed us to screen for mutations and indels affecting the genes annotated in the physical interval corresponding to the major QTL. Evaluating all non-synonymous mutations affecting the 121 high-confidence genes present in the QTL interval, only eight were considered as potentially deleterious. Among those, we identified a non-synonymous substitution in exon1 of FT-B1 in our parental lines changing the aspartic acid (D) into a histidine (H). Analyzing a collection of more than 300 sequences of FT-B1 homologs, including homoeologues and paralogs, covering a large diversity of land plants revealed that the mutation was novel in hexaploid wheat. However, it appeared that the mutation, present with the array code AX-94810990 on the high-density genotyping arrays Axiom 820 K and 35 K, is probably widespread (68%) in wheat varieties and landraces but absent in the few tetraploid and Ae. speltoides accessions screened to validate the 820 K array37. The mutation from D to H confers a phenotype similar but weaker than the complete loss of FT-B1 (and 400 other genes) described in Dixon et al.24 and Finnegan et al.25. We conclude that this could be a mutation potentially only slightly impairing the protein which remains suitable in breeding. Taken together, our results suggest that this variant is a novelty private to hexaploid wheat where it was favored potentially for its positive impact on the grain yield. It may also have been selected for winter hardiness as the histidine mutation was found to be associated with an increased frost tolerance47. Interestingly, analysis of transgenic rice showed that Hd3a—a rice FT homolog—accumulated in axillary meristems and promoted the formation of branches21. Analyzing FT-like genes in maize and rice, Zheng et al.34 have shown that those genes had been under positive selection during domestication allowing the plants to adapt to different environments during the range expansion of by modulating their flowering time.
Conclusion
Linkage mapping revealed the effects of FT-B1 on total spikelet number in the genetic background of a doubled haploid spring wheat population, while the heading and flowering time was dominated by the effects of a segregating VRN-A1 gene. A non-synonymous mutation in FT-B1 causing an amino acid exchange from aspartic acid to histidine was observed in the parental lines of the mapping population. The observed mutation is widespread in hexaploid wheat, but could not be detected in any other orthologs and paralogs within the higher plants. It may be speculated that effects of FT-B1 on subphase duration of wheat affect the final spikelet number rather than the flowering time. This is a first report about a structural variation in FT-B1 influencing TSN, which is also inheritable, while other reports were mainly based on expression analysis or the complete loss described in Dixon et al.24 and Finnegan et al.25 consisting of the first 23 Mb of chromosome 7B encompassing more than 400 genes.
Materials and methods
Plant material and field trials
The doubled haploid (DH) spring wheat population-2 consisting of 159 lines was previously described in Muqaddasi et al.35. The parental lines, conserved at the IPK Genebank, were TRI-10703 (landrace from Greece) and TRI-5310 (variety Eureke from France). The accessions were originally selected for their contrasting anther extrusion phenotypes but exhibited a large difference for total spikelet number per spike (TSN). The DH population and its parental lines were sown at the end of March and grown in 2 m2 plots on the field in IPK Gatersleben for four consecutive cropping seasons (2016–2019) and characterized each year for TSN, spike length (SL), heading date (HD), and flowering time (FT).
Phenotyping and data analysis
The spike traits (TSN and SL) were recorded from ten spikes per plot as the total number of spikelets and spike length in centimeters (cm) from basal spikelet to the top of a spike but excluding the awns. The flowering traits (HD and FT) were recorded for each year as the number of days between the first of January and when approximately half of the spikes in a plot emerged and flowered, respectively.
Phenotypic data were analyzed following Muqaddasi et al.35. Specifically, best linear unbiased estimations (BLUEs) were calculated per year for the spike traits and across years for all traits by assuming fixed effects for the intercept and the genotype and all others as random. All phenotyping data analyses, including ANOVAs, Pearson’s correlation coefficient analyses, repeatability among replicates, broad-sense-heritability across years, and statistical tests, were performed using the software R48.
Genotyping and QTL mapping
The 159 lines were genotyped with a custom-designed 15 k Infinium array49 by the company TraitGenetics GmbH in Gatersleben, Germany, that resulted in 3,457 polymorphic markers. The 15 k Infinium array was created as a cost-effective option based on the 90 k Infinium array described by Wang et al.50. Data filtering and linkage map construction were previously described in Muqaddasi et al.35.
The quantitative trait loci (QTL) underlying the four traits were analyzed by composite interval mapping (CIM)51 in Windows QTL Cartographer 2.552. For each trait, single year and across year BLUEs were used to calculate both forward and backward regression. The step size was set at 2 cM. To identify significant QTL at α = 0.05, thresholds for the logarithm of odds (LOD) values were estimated with 1000 permutation53. Confidence interval for the significant QTL were defined as a continuous genomic region comprised within a 1.5-LOD from the marker with the highest LOD per chromosome passing the threshold54. QTL repeatedly identified in the same interval each year and in their BLUEs were defined as major and consistent.
Analysis of the physical region
All genomic analyses were performed in Geneious Prime 2020.0.5 (https://www.geneious.com). First the sequences of the markers corresponding to the consistent QTL’s genetic intervals were obtained from Wang et al.50 and BLASTed on their assigned chromosome of the wheat genome reference sequence55 to define genomic intervals. The physical interval defined for QTsn.ipk-7B was retrieved from the assemblies of the parental lines sequenced at 10 × coverage with Illumina paired-end sequencing (HiseqXTEN PE150) and assembled as described in Muqaddasi et al.35. The sequences of the genes present in the interval were mapped to the sequences of the parental lines and then extracted. Mapping was performed with the Geneious mapper set to “Custom sensitivity” based on the “Medium–Low Sensitivity” parameters but with a maximum gap size of 2000 bp to accommodate for missing data regions. Multiple sequence alignments (MSA) for each gene consisting of the sequences of the reference and of the two parental lines were obtained by running the same mapping procedure using the genic sequences of Chinese Spring as reference. Polymorphisms between both parental lines were annotated in the MSAs. To narrow down to potential candidates, the functional annotations of the genes in the interval were inspected which revealed the presence of two CLAVATA3/ESR (CLE)-related proteins, six MADS box transcription factors, and the FLOWERING LOCUS T (VRN-B3 or FT-B1, TraesCS7B02G013100). The potential effect of the non-synonymous substitutions affecting FT-B1 and all other high-confidence genes present within the interval were analyzed with PROVEAN (Protein variation effect analyzer)56. The bread wheat epigenomic map (http://bioinfo.sibs.ac.cn/cs_epigenome)36 was used to identify the location of potential transcription factors in the 1.5 kb region upstream from FT-B1.
Interaction between FT-B1 and VRN-A1
To test the potential interaction between the candidate gene FT-B1 and the vernalization gene VRN-A1, the whole population was genotyped with a Kompetitive Allele Specific PCR (KASP) marker based on the FT-B1 SNP (Supplementary Table S6) and VRN-A1 primers23. All the traits were analyzed with a two-way factorial ANOVA and allele-wise phenotypic distribution was illustrated by plotting the boxplots. The effect of the different alleles at both loci for all traits were evaluated with a Wilcoxon rank-sum test.
Intragenomic, intra- and interspecific analyses
The EnsemblPlant database (https://plants.ensembl.org) was used to identify and recover the protein sequences of all genes classified as paralogs of the three homoeologues (TraesCS7A02G115400, TraesCS7B02G013100, and TraesCS7D02G111600). The sequences were then aligned with MAFFT v7.45057,58 in Geneious. The CerealsDB database (www.cerealsdb.uk.net)59 and the function “Extract genotype for a specific SNP” were used to obtain the genotyping score of the Axiom 820 K array marker AX-94810990 covering the non-synonymous mutation identified in FT-B1. Samples with the ancestral (G) or the derived allele (C) were scored with BB and AA, respectively. Among the 475 genotypes recovered, only the samples identified as varieties, landraces, tetraploid and Aegilops speltoides, according to Winfield et al.37, were kept to evaluate the frequency of the mutation across bread wheat and its progenitors. Interspecific analyses were performed on the set of 377 homolog sequences retrieved by the PROVEAN analysis.
References
Sakuma, S. & Schnurbusch, T. Of floral fortune: Tinkering with the grain yield potential of cereal crops. New Phytol. 225, 1873–1882 (2020).
Gauley, A. & Boden, S. A. Genetic pathways controlling inflorescence architecture and development in wheat and barley. J. Integr. Plant Biol. 61, 296–309 (2019).
Koppolu, R. & Schnurbusch, T. Developmental pathways for shaping spike inflorescence architecture in barley and wheat. J. Integr. Plant Biol. 61, 278–295 (2019).
Guo, Z. et al. Genome-wide association analyses of 54 traits identified multiple loci for the determination of floret fertility in wheat. New Phytol. 214, 257–270 (2017).
Pretini, N. et al. A comprehensive study of spike fruiting efficiency in wheat. Crop Sci. 60, 1541–1555 (2020).
Pretini, N. et al. Identification and validation of QTL for spike fertile floret and fruiting efficiencies in hexaploid wheat (Triticum aestivum L.). Theor. Appl. Genet. 133, 2655–2671 (2020).
Liu, J. et al. A genome-wide association study of wheat spike related traits in China. Front. Plant Sci. 9, 1584. https://doi.org/10.3389/fpls.2018.01584 (2018).
Würschum, T., Leiser, W. L., Langer, S. M., Tucker, M. R. & Longin, C. F. H. Phenotypic and genetic analysis of spike and kernel characteristics in wheat reveals long-term genetic trends of grain yield components. Theor. Appl. Genet. 131, 2071–2084 (2018).
Chen, Z. et al. Pleiotropic QTL influencing spikelet number and heading date in common wheat (Triticum aestivum L.). Theor. Appl. Genet. 133, 1825–1838 (2020).
Debernardi, J. M., Lin, H., Chuck, G., Faris, J. D. & Dubcovsky, J. microRNA172 plays a crucial role in wheat spike morphogenesis and grain threshability. Development 144, 1966–1975 (2017).
Greenwood, J. R., Finnegan, E. J., Watanabe, N., Trevaskis, B. & Swain, S. M. New alleles of the wheat domestication gene Q reveal multiple roles in growth and reproductive development. Development 144, 1959–1965 (2017).
Zhang, B. et al. Novel function of a putative MOC1 ortholog associated with spikelet number per spike in common wheat. Sci. Rep. 5, 12211. https://doi.org/10.1038/srep12211 (2015).
Kuzay, S. et al. Identification of a candidate gene for a QTL for spikelet number per spike on wheat chromosome arm 7AL by high-resolution genetic mapping. Theor. Appl. Genet. 132, 2689–2705 (2019).
Muqaddasi, Q. H. et al. TaAPO-A1, an ortholog of rice ABERRANT PANICLE ORGANIZATION 1, is associated with total spikelet number per spike in elite European hexaploid winter wheat (Triticum aestivum L.) varieties. Sci. Rep. 9, 13853. https://doi.org/10.1038/s41598-019-50331-9 (2019).
Voss-Fels, K. P. et al. High-resolution mapping of rachis nodes per rachis, a critical determinant of grain yield components in wheat. Theor. Appl. Genet. 132, 2707–2719 (2019).
Kardailsky, I. et al. Activation tagging of the floral inducer FT. Science 286, 1962–1965 (1999).
Kobayashi, Y., Kaya, H., Goto, K., Iwabuchi, M. & Araki, T. A pair of related genes with antagonistic roles in mediating flowering signals. Science 286, 1960–1962 (1999).
Corbesier, L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033 (2007).
Kojima, S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell. Physiol. 43, 1096–1105 (2002).
Komiya, R., Ikegami, A., Tamaki, S., Yokoi, S. & Shimamoto, K. Hd3a and RFT1 are essential for flowering in rice. Development 135, 767–774 (2008).
Tsuji, H. et al. Hd3a promotes lateral branching in rice. Plant J. 82, 256–266 (2015).
Yan, L. et al. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 103, 19581–19586 (2006).
Zhang, X. K. et al. Allelic variation at the vernalization genes Vrn-A1, Vrn-B1, Vrn-D1, and Vrn-B3 in Chinese wheat cultivars and their association with growth habit. Crop Sci. 48, 458–470 (2008).
Dixon, L. E. et al. Developmental responses of bread wheat to changes in ambient temperature following deletion of a locus that includes FLOWERING LOCUS T1. Plant Cell Environ. 41, 1715–1725 (2018).
Finnegan, E. J. et al. Zebularine treatment is associated with deletion of FT-B1 leading to an increase in spikelet number in bread wheat. Plant Cell Environ. 41, 1346–1360 (2018).
Dixon, L. E. et al. TEOSINTE BRANCHED 1 regulates inflorescence architecture and development in bread wheat (Triticum aestivum). Plant Cell 30, 563–581 (2018).
Karlgren, A. et al. Evolution of the PEBP gene family in plants: Functional diversification in seed plant evolution. Plant Physiol. 156, 1967–1977 (2011).
Hedman, H., Källman, T. & Lagercrantz, U. Early evolution of the MFT-like gene family in plants. Plant Mol. Biol. 70, 359–369 (2009).
Jin, S., Nasim, Z., Susila, H. & Ahn, J. H. Evolution and functional diversification of FLOWERING LOCUS T/TERMINAL FLOWER 1 family genes in plants. Semin. Cell Dev. Biol. https://doi.org/10.1016/j.semcdb.2020.05.007 (2020).
Hanzawa, Y., Money, T. & Bradley, D. A single amino acid converts a repressor to an activator of flowering. Proc. Natl. Acad. Sci. USA 102, 7748–7753 (2005).
Chardon, F. & Damerval, C. Phylogenomic analysis of the PEBP gene family in cereals. J. Mol. Evol. 61, 579–590 (2005).
Faure, S., Higgins, J., Turner, A. & Laurie, D. A. The FLOWERING LOCUS T-like gene family in barley (Hordeum vulgare). Genetics 176, 599–609 (2007).
Izawa, T. Adaptation of flowering-time by natural and artificial selection in Arabidopsis and rice. J. Exp. Bot. 58, 3091–3097 (2007).
Zheng, X.-M. et al. Evolution of the PEBP gene family and selective signature on FT-like clade. J. Syst. Evol. 54, 502–510 (2016).
Muqaddasi, Q. H., Jayakodi, M., Börner, A. & Röder, M. S. Identification of consistent QTL with large effect on anther extrusion in doubled haploid populations developed from spring wheat accessions in German Federal ex situ Genebank. Theor. Appl. Genet. 132, 3035–3045 (2019).
Li, Z. et al. The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 20, 139. https://doi.org/10.1186/s13059-019-1746-8 (2019).
Winfield, M. O. et al. High-density SNP genotyping array for hexaploid wheat and its secondary and tertiary gene pool. Plant Biotechnol. J. 14, 1195–1206 (2016).
Boden, S. A. et al. Ppd-1 is a key regulator of inflorescence architecture and paired spikelet development in wheat. Nat. Plants 1, 14016. https://doi.org/10.1038/nplants.2014.16 (2015).
Nitcher, R., Pearce, S., Tranquilli, G., Zhang, X. & Dubcovsky, J. Effect of the Hope FT-B1 allele on wheat heading time and yield components. J. Hered. 105, 666–675 (2014).
Guo, Z., Chen, D., Röder, M. S., Ganal, M. W. & Schnurbusch, T. Genetic dissection of pre-anthesis sub-phase durations during the reproductive spike development of wheat. Plant J. 95, 909–918 (2018).
Guo, Z. et al. Genome-wide association analyses of plant growth traits during the stem elongation phase in wheat. Plant Biotechnol. J. 16, 2042–2052 (2018).
Balasubramanian, S., Sureshkumar, S., Lempe, J. & Weigel, D. Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. PLOS Genet. 2, e106. https://doi.org/10.1371/journal.pgen.0020106 (2006).
Hemming, M. N., Walford, S. A., Fieg, S., Dennis, E. S. & Trevaskis, B. Identification of high-temperature-responsive genes in cereals. Plant Physiol. 158, 1439–1450 (2012).
Li, C. et al. Wheat VRN1, FUL2 and FUL3 play critical and redundant roles in spikelet development and spike determinacy. Development 146, dev175398. https://doi.org/10.1242/dev.175398 (2019).
Shaw, L. M. et al. FLOWERING LOCUS T 2 regulates spike development and fertility in temperate cereals. J. Exp. Bot. 70, 193–204 (2019).
Gauley, A. & Boden, S. A. Step-wise increases in FT1 expression regulate seasonal progression of flowering in wheat (Triticum aestivum). New Phytol. https://doi.org/10.1111/nph.16910 (2020).
Babben, S. et al. Association genetics studies on frost tolerance in wheat (Triticum aestivum L.) reveal new highly conserved amino acid substitutions in CBF-A3, CBF-A15, VRN3 and PPD1 genes. BMC Genom. 19, 409. https://doi.org/10.1186/s12864-018-4795-6 (2018).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2020). https://www.R-project.org/.
Soleimani, B. et al. Comparison between core set selection methods using different Illumina marker platforms: A case study of assessment of diversity in wheat. Front. Plant Sci. 11, 1040. https://doi.org/10.3389/fpls.2020.01040 (2020).
Wang, S. et al. Characterization of polyploid wheat genomic diversity using a high-density 90,000 single nucleotide polymorphism array. Plant Biotechnol. J. 12, 787–796 (2014).
Zeng, Z. B. Precision mapping of quantitative trait loci. Genetics 136, 1457–1468 (1994).
Wang, S., Basten, C. J. & Zeng, Z. B. Windows QTL Cartographer 2.5_011 (North Carolina State University, Raleigh, 2012).
Churchill, G. A. & Doerge, R. W. Empirical threshold values for quantitative trait mapping. Genetics 138, 963–971 (1994).
Silva, L. D. C. E., Wang, S. & Zeng, Z.-B. Composite interval mapping and multiple interval mapping: Procedures and guidelines for using Windows QTL cartographer. In Quantitative Trait Loci (QTL): Methods and Protocols (ed. Rifkin, S. A.) 75–119 (Humana Press, Totowa, 2012).
International Wheat Genome Consortium et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 361, eaar7191. https://doi.org/10.1126/science.aar7191 (2018).
Choi, Y. & Chan, A. P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucl. Acids Res. 30, 3059–3066 (2002).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Wilkinson, P. A. et al. CerealsDB 2.0: An integrated resource for plant breeders and scientists. BMC Bioinform. 13, 219. https://doi.org/10.1186/1471-2105-13-219 (2012).
Acknowledgements
This study was funded by internal sources of IPK. We are grateful to Ellen Weiß, Anette Heber, Ute Ostermann, and Sonja Allner for help in data collection. We thank Guy Golan for discussion and comments on the manuscript. We are grateful to the three anonymous reviewers for their critical comments and suggestions.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.S.R. and Q.H.M. conceived, designed, and performed the experiments. J.P. and M.W.G. performed genotyping. J.B. and Q.H.M. analyzed the data. J.B. and M.S.R. contributed to the interpretation of the results and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
On behalf of all authors, Jonathan Brassac states that there is no conflict of interest. J.P. and M.W.G. are members of the company TraitGenetics GmbH. This does, however, in no way limit the availability or sharing of data and materials.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brassac, J., Muqaddasi, Q.H., Plieske, J. et al. Linkage mapping identifies a non-synonymous mutation in FLOWERING LOCUS T (FT-B1) increasing spikelet number per spike. Sci Rep 11, 1585 (2021). https://doi.org/10.1038/s41598-020-80473-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80473-0
This article is cited by
-
Overexpression of the WAPO-A1 gene increases the number of spikelets per spike in bread wheat
Scientific Reports (2022)
-
Identification and validation of a QTL for spikelet number on chromosome arm 6BL of common wheat (Triticum aestivum L.)
Molecular Breeding (2022)
-
Identification and characterization of a natural polymorphism in FT-A2 associated with increased number of grains per spike in wheat
Theoretical and Applied Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
