
Abstract
Inflammation plays central roles in the immune response. Inflammatory response normally requires higher energy and therefore is associated with glucose metabolism. Our recent study demonstrates that lncRNA HOTAIR plays key roles in NF-kB activation, cytokine expression, and inflammation. Here, we investigated if HOTAIR plays any role in the regulation of glucose metabolism in immune cells during inflammation. Our results demonstrate that LPS-induced inflammation induces the expression of glucose transporter isoform 1 (Glut1) which controls the glucose uptake in macrophages. LPS-induced Glut1 expression is regulated via NF-kB activation. Importantly, siRNA-mediated knockdown of HOTAIR suppressed the LPS-induced expression of Glut1 suggesting key roles of HOTAIR in LPS-induced Glut1 expression in macrophage. HOTAIR induces NF-kB activation, which in turn increases Glut1 expression in response to LPS. We also found that HOTAIR regulates glucose uptake in macrophages during LPS-induced inflammation and its knockdown decreases LPS-induced increased glucose uptake. HOTAIR also regulates other upstream regulators of glucose metabolism such as PTEN and HIF1α, suggesting its multimodal functions in glucose metabolism. Overall, our study demonstrated that lncRNA HOTAIR plays key roles in LPS-induced Glut1 expression and glucose uptake by activating NF-kB and hence HOTAIR regulates metabolic programming in immune cells potentially to meet the energy needs during the immune response.
Similar content being viewed by others


Introduction
Inflammation is a biological response of the immune system1,2,3. Inflammatory response is triggered by a variety of factors such as pathogenic infection and exposure to toxic compounds3,4,5,6. Activation of immune response removes injurious and toxic stimuli, helps healing, and thus vital to health4,7,8,9. However, uncontrolled inflammation may result in severe inflammatory diseases10. Macrophages play key roles in provide a first line of defense against pathogens, eliminate the foreign substances and apoptotic cells through phagocytosis, and keep tissues healthy11,12. Pattern recognition factors such as toll-like receptors (TLRs) and others expressed in the macrophage surfaces detect the danger signals in the surrounding environment, activate macrophages, and initiate the signaling cascade. There two major type of macrophage activation process, M1 and M2 types. For example, when macrophages detect inflammatory cytokines such as interferon-gamma (IFNγ) and tumor necrosis factor alpha (TNFα) or foreign material such as lipopolysaccharide (LPS), they become activated and undergo a phenotypic change resulting in activated M1 types macrophage polarization12,13,14. However, when they encounter anti-inflammatory cytokines such as IL-4 and IL-13, macrophages are polarized to M2 (suppressive) state4,15. M1 type activated macrophages secrete proinflammatory inflammatory cytokines and chemokines such as IL-1β, IL-6, TNFα, CXCL1, and others, and induce inflammatory immune responses4,7. However, M2 macrophages secret anti-inflammatory cytokines such as IL-10, IL-18, and suppress inflammation and facilitate tissue repair4,15. Interestingly, M1 macrophages preferentially metabolize glucose as an energy substrate, while M2 macrophages primarily utilize fatty acids to fuel cellular functions16,17. Therefore, glucose metabolism is central to the function of classically activated M1 macrophages and could be a potential target for modifying inflammatory responses4,15,16,17. Studies suggest that proinflammatory immune response in M1 macrophages enhances expression of glucose transporter isoform 1 (Glut1), glucose-6-phosphate dehydrogenase, hexokinase, and increases glucose uptake16,18 and elevated Glut1-driven glucose metabolism drives inflammatory immune responses in macrophage19,20.
Signaling mechanism associated with inflammation and immune response is very complex, follows different pathways and is widely associated with various factors2,21,22,23,24. These include activation of transcription factors such as NF-κB, activator protein 1 (AP1), interferon regulatory factors (IRFs), STAT 1/2 (signal transducer and activator of transcription) and many others18,25,26. These activated transcription factors, in turn, stimulate the expression of proinflammatory cytokines, chemokines, interferons and other pro-inflammatory mediators, and thus propagating cellular inflammation27,28,29. Emerging evidences suggest that, in addition to proteins, noncoding RNAs (ncRNA) play critical roles in variety of cell signaling process including in regulation of gene expression and immune response30,31,32. NcRNAs are recently discovered class of transcripts that coded by the genome, but mostly remain untranslated into proteins33,34,35. Even though, they are untranslated, ncRNAs are functional and integral components of signaling processes36,37,38. Recent studies indicate that ncRNAs play important roles in regulating aerobic glycolysis in cancer cells39,40,41. For instance, miR-199a, miR-138, miR-150 and miR-532-5p inhibit the expression of Glut142,43,44, while miR-130b, miR-19a, miR-19b and miR-301a induce Glut1 expression in epithelial cancer cells31,45. Recently, we discovered that a long noncoding RNA (lncRNA), HOTAIR, is involved in regulation of inflammatory immune responses in macrophages30. HOTAIR is an antisense lncRNA (2.2 kb long) which is well known for its function in gene silencing46,47. HOTAIR navigates polycomb repressive complex 2 (PRC2)mand lysine specific demethylase 1 (LSD1) complexes to their target genes promoters48,49,50. PRC2 and LSD1 recruitment introduce promoter histone H3K27-methylation and H3K4-demethylaytion, respectively and induce gene silencing33,47. HOTAIR is suppresses the tumor suppressor gene expression and it is overexpressed in variety of cancers50,51. Studies from our laboratory show that HOTAIR expression is elevated in breast cancer cells., its gene expression is regulated by estradiol52,53 and dysregulated by estrogenic endocrine disrupting chemicals (EDCs, e.g. BPA and DES)52,54. HOTAIR is an oncogenic lncRNA51,55,56. In addition to its well-worn functions in gene expression, HOTAIR is implicated in protein degradation associated with neuronal function and diseases57,58,59.
Our recent study demonstrated that HOTAIR plays critical roles in regulation of NF-κB activation via degradation of IκBα and regulate expression of cytokines and pro-inflammatory genes regulating inflammation and immune response30. Importantly, activation of immune cells in response to infection or other stressors is a metabolically expensive event31,60,61, and immune cells preferentially meet their energy needs by metabolizing glucose62,63. As HOTAIR plays critical role in NF-κB activation and inflammatory response in macrophage, we hypothesized that HOTAIR plays critical roles in inflammation induced metabolic reprogramming. Here, we explored any function of HOTAIR in glucose metabolism in macrophage during inflammation. Our study demonstrated that HOTAIR plays key roles in regulation of the expression of glucose transporter and glucose uptake, and hence regulates glucose metabolism in macrophages during LPS-induced inflammation and immune response.
Materials and methods
Mouse macrophage cell culture
RAW264.7 cells (mouse macrophages) were procured from American Type Cell Culture Collection. These cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated FBS (Fetal bovine serum), 2 mM l-glutamine, 100 units/mL penicillin and 0.1 mg/mL streptomycin in a humidified incubator with 5% CO2 and 95% air at 37 °C64. Cells were counted to seed 2 × 106 cells in 60 mm cell culture plates. After overnight incubation, macrophages were ready for treatment30,65.
Cell treatment with lipopolysaccharide (LPS)
Macrophage cells were treated with ultrapure E. coli-derived LPS (Invivogen), 1.0 μg/mL, for different time periods. The concentration of LPS has been broadly reported to be used by other laboratories to induce immune and pro-inflammatory response in macrophages30,65. Cells were harvested for the preparation of RNA and protein analysis.
RNA extraction and cDNA synthesis
Total RNA was extracted from the cells using TRIzol (Invitrogen) according to the manufacturer’s instructions65. Briefly RAW264.7 macrophages were harvested with TRIzol, mixed with chloroform and centrifuged at 12,000 rpm for 15 min. The aqueous layer was mixed with equal volume of 100% ethanol and centrifuged at 12,000 rpm for 10 min. After washing the pellet with 70% ethanol, purified RNA was dissolved in 30–50 μL of RNase-free water (Sigma) and quantified using a Nanodrop spectrophotometer. 1 μg of the isolated RNA was reverse transcribed into cDNA using iScript RT-supermix (Bio-Rad)30,65.
Real time PCR
Real-time PCR was done using iTaq Universal SYBR Green Supermix (Bio-Rad), with gene specific PCR primers as listed in Table 1. The CFX96 real-time detection system (Bio-Rad) was used for RT-qPCR. Each experiment was repeated three times with three parallel replicates each time. Expression data were normalized to GAPDH and expressed as 2−ΔCt66,67.
Western blot analyses
The treated macrophage cells were washed with ice-cold PBS and then lysed in cell lysis buffer comprising 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% Igepal CA-630, 0.5% Na-deoxycholate, 2 mM Na3VO4, and complete protease inhibitor cocktail and phosphatase inhibitor cocktail (Roche). The resulting cell lysates were centrifuged for 10 min at 13,000 rpm at 4 °C, and the protein concentrations in the supernatants were determined using a BCA protein assay kit (Pierce)68. 20 μg proteins were loaded onto 10% SDS-PAGE gels, followed by electro-transfer onto PVDF-membrane (Immobilon-P, Millipore). The membranes were blocked in 1 × TBST (0.1% Tween-20, 20 mM Tris–Cl (pH 8.0), and 150 mM NaCl) containing 3% skimmed milk and then incubated with the primary antibodies against IκBα (1:1000 dilution, 4814S, Cell Signaling), Glut 1 (1:1000 dilution, 12939S, Cell Signaling), Phospho-p65 (NF-κB subunit, 1:1000 dilution, 3033S, Cell Signaling), HIF-1α (1:1000 dilution, 14,179, Cell Signaling) and β -actin (1:1000 dilution, A2066, Sigma) overnight at 4 °C. Membranes were washed 3 times (1xTBST), incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature and then washed 3 times (1 × TBST). Lastly, immunoreactive proteins were detected using ECL -super signal west femto substrate reagent (Thermo Scientific)30,68. The amount has been quantified by Image Lab 5.2.1 software.
Chromatin Immunoprecipitation (ChIP) assay
The ChIP assay was done as described earlier30,54. The cells were cross-linked with 1% formaldehyde for 10 min at 37 °C, washed twice with ice-cold PBS and harvested using SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris. HCl, pH 8.1) supplemented with complete protease inhibitor (Roche). Cells were sonicated to shear the chromatin (∼ 200–300 bp range). The fragmented chromatin was pre-cleared with protein G agarose beads (16–266, EMD Millipore) and subjected to immunoprecipitation using antibodies specific to CBP (A22, Santa Cruz Biotechnology, Sc369), Phospho-p65 (3033, Cell Signaling), histone H3K4-trimethyl (07–473, EMD-Millipore), histone acetylation (06–599, EMD-Millipore), RNA Pol II (8WG16, Abcam), and β-actin (A2066, Sigma). Immunoprecipitated chromatin was washed, de-crosslinked and deproteinized at 65 °C in presence of 5 M NaCl followed by incubation with proteinase K (Sigma) at 45 °C for 1 h30,54,69. Purified ChIP DNA fragments were analyzed by semi-quantitative PCR and real-time PCR using primers spanning NF-κB binding sites present in the Glut1 promoter (Table 1).
SiRNA-mediated knockdown of HOTAIR
For the siRNA transfection, RAW 264.7 cells were grown up to 60% confluency in 60 mm culture plates and transfected with HOTAIR-siRNA, a pool of 4 different siRNA constructs (SI05685183, SI05685190, SI05685197, and SI05685204 Qiagen)57 and scramble siRNA (1027310 Qiagen) independently using GenMute siRNA and DNA transfection reagent (SL100568, SignaGen Laboratories) according to the manufacturer’s protocol.A cocktail of transfection reagent and siRNA was made prior to transfection. Initially, 12 μL (12 μg) of GenMute reagent was mixed with 300 μL DMEM (minus FBS and antibiotics) in an eppendorf tube. SiRNA was mixed with 100 μL DMEM (without supplements) in a separate eppendorf. Then the diluted siRNA solution was mixed with diluted GenMute reagents and allowed to stand for 30 min in the dark. In the intervening time, cells were washed twice with supplement-free DMEM and then 1.7 mL of supplement-free DMEM was added to each cell culture plate. Finally, siRNA transfection reagents cocktail was applied to the cell plates, mixed gently and incubated for 48 h. Cells were then stimulated with LPS (1 μg/mL) for specified time period and then harvested for RNA/protein extraction or for ChIP assays30,69.
Antisense-mediated knockdown of HOTAIR
For the antisense transfection, RAW 264.7 cells were grown up to 60% confluency in 60 mm culture plates and transfected with HOTAIR-antisense (HOTAIR-AS) and scramble antisense (Scr-AS, no homology to HOTAIR) oligonucleotides56,57 independently (Table 1) using GenMute siRNA and DNA transfection reagent (SL100568, SignaGen Laboratories) according to the manufacturer’s protocol. A cocktail of transfection reagent and antisense oligonucleotides was made prior to transfection. Initially, 12 μL (12 μg) of GenMute reagent was mixed with 300 μL DMEM (minus FBS and antibiotics) in an eppendorf tube. Antisense oligonucleotides were mixed with 100 μL DMEM (without supplements) in a separate eppendorf. Then the diluted antisense solution was mixed with diluted GenMute reagents and allowed to stand for 30 min in the dark. In the intervening time, cells were washed twice with supplement-free DMEM and then 1.7 mL of supplement-free DMEM was added to each cell culture plate. Finally, antisense transfection reagents cocktail was applied to the cell plates, mixed gently and incubated for 48 h. Cells were then stimulated with LPS (1 μg/mL) for specified time period and then harvested for RNA extraction.
NF-κB inhibition assay
RAW264.7 macrophages (2 × 106) were seeded in 60 mm cell culture plates. After overnight incubation cells were initially treated with IKKβ inhibitor (25 μM, SC-514, Sigma) for 1 h to inhibit NF-kB signaling pathway and then cells were treated with LPS (1 μg/mL) and incubated for additional period of time 4 h70. Cells were harvested, total RNA was isolated using TRIzol reagent, reverse transcribed to cDNA and analyzed by qPCR. Protein was also extracted (SC-514 1 h and additional 1 h LPS treatment) after cell harvesting for Western blot30.
Immunofluorescence microscopy analysis
For immunofluorescence staining of macrophages, cells were seeded on cover slips and fixed in 4% paraformaldehyde (PFA) for 15 min at room temperature, washed with 1X PBS (3 times for 5 min each) and blocked with 1X PBS containing 5% goat normal serum and 0.3% Triton-X100 for 1 h. The cells were then incubated with primary antibody (rabbit anti- Glut 1, 1:200, 12939S, Cell Signaling) overnight at 4 °C. After that the cells were washed 3 times with PBS followed by incubation with anti-rabbit Alexa Fluor 564 (Invitrogen), conjugated secondary antibody for 1 h at room temperature. Finally the cells were washed 3 times with PBS and mounted with mounting media containing DAPI. Images were taken by fluorescence microscope (Nikon ECLIPSE TE2000-U) utilizing a 63 × oil objective lens and quantified by ImageJ software30.
Glucose uptake assay
Glucose uptake into the macrophages was determined using Glucose Uptake Assay Kit (Colorimetric, ab136955, Abcam) as per the manufacturer’s Instructions19,45. RAW264.7 macrophages and/or BMDMs were treated with HOTAIR siRNA and/or HOTAIR-antisense for 48 h and then seeded in 96 well plate in serum free medium. After overnight incubation cells were washed and incubated in 2% Bovine serum albumin (BSA) for 40 min. Then cells were stimulated with insulin (+/−), followed by 2-deoxyglucose addition for 20 min. 2-DG-6-phosphate (2-DG6P) standard curve was prepared. In this assay, the 2-DG6P was oxidized to generate NADPH, which was measured at 412 nm in a microplate reader19,45.
Isolation and culture of primary macrophage (bone marrow derived macrophage, BMDM)
BMDM was isolated from mice bones and cultured as described earlier65,71. Wild-type (C57BL6/J) mice were purchased from Jackson Laboratory. All mice are maintained in a specific pathogen free (SPF) facility at UT Southwestern Medical center. All studies were approved by the Institutional Animal Care and Use Committee (IACUC) and were conducted in accordance with the IACUC guidelines, the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the ARRIVE guidelines. For the isolation of BMDMs, femur and tibia were collected from mouse legs. Using 25G needle bone marrows were flushed out with Iscove's Modified Dulbecco's Medium (IMDM), (12440061, Life technologies) and processed for single cell suspension by passing through 22G needle two times. The suspension was centrifuged at 1000 RPM for 5 min. The pellet was re-suspended with BMDM culture media (L-cell-conditioned IMDM medium supplemented with 15% L929 supernatant, 10% FBS, 1% nonessential amino acid, and 1% penicillin–streptomycin) followed by seeding in three 150 mm culture dishes and cultured for 6 days to differentiate into macrophages, while at day 3, 10 mL fresh BMDM culture media was added into each plate. After day 6, the culture plate was washed with ice cold PBS and using cell scrapper cells were gently scraped with ice cold PBS. The BMDM was centrifuged at 1000 rpm for 5 min and re-suspended into BMDM media. The BMDM was counted and seeded in 6-well (2.5 × 106/well) cell culture plates. After overnight incubation the BMDM was treated with LPS or HOTAIR siRNA and processed for further experiments.
Statistical analysis
Each experiment was done in two or three replicates, and then cells were pooled (and treated as one sample), subjected to RNA extraction, RT-PCR, and ChIP analysis, and each experiment was repeated at least three times (n = 3). The real-time PCR analysis of such samples were done in three replicate reactions and repeated in all three independent experiments (n = 3). Data are presented as means ± SD (as stated in the figure legends). Statistical significance was determined by unpaired Student’s t test (GraphPad Prism 6), and P ≤ 0.05 was considered statistically significant30.
Results
LPS-induces GLUT1 expression in macrophage
Our recent studies demonstrate that lncRNA HOTAIR plays critical roles in inflammation and immune signaling and regulates cytokine expression via regulation of NF-κB activation30. As glucose metabolism is well-known to be elevated during inflammation13,16,72, we investigated if HOTAIR plays any roles in glucose uptake and metabolism. To investigate roles of HOTAIR in glucose metabolism during inflammation, initially, we examined if glucose transporters (Gluts) expression are affected upon lipopolysaccharide (LPS, present in membranes of gram negative bacteria) stimulation. We treated mouse macrophage cells (RAW264.7) with LPS and analyzed its impact on various Gluts expression. Briefly, RNA from the control and LPS-treated macrophages are reverse-transcribed into cDNA and analyzed by qPCR using primers specific to different glucose transporters (Glut1, Glut2, Glut3, Glut4, and Glut5). We also analyzed the expression of well-known inflammatory marker genes such as IL-6 and iNOS, and inflammatory noncoding RNA HOTAIR. As expected, the expression of IL-6, iNOS, and HOTAIR are significantly stimulated (by 9800, 272 and 6 folds, respectively) by LPS-treatment (Fig. 1A–C). Interestingly, expression of Glut1 is also significantly induced by LPS in macrophage, while the expression of other gluts (Glut2, Glut3, Glut4 and Glut5) were nominally affected (Fig. 1D). Time course analysis demonstrates that LPS-induced stimulation of Glut1 is maximum at 4 h (Fig. 1E). Western blot analysis also demonstrates that Glut1 expression is increased at the protein level upon treatment with LPS (Figs. 1F and S1, quantification in Fig. 1G). These observations demonstrate that Glut1, which is the primary glucose transporter expressed in macrophages, is upregulated upon LPS-induced inflammation and is potentially involved in glucose uptake and metabolism during inflammation. Notably, LPS-induced Glut1 expression has reported earlier and our observations are in agreement with the previous studies13.

LPS induces Glut1 expression in macrophages. (A–E) RAW264.7 cells were treated with LPS (1 μg/mL) for 4 h (or different time periods), total RNA was isolated, reverse transcribed to cDNA and the expressions of IL-6, iNOS, HOTAIR, and different glucose transporters (Glut1, Glut2, Glut3, Glut4 and Glut5) were analyzed by RT-qPCR. GAPDH was used as control. The expression (relative to GAPDH) of IL-6, iNOS, HOTAIR, Glut1, Glut2, Glut3, Glut4 and Glut5 are shown in panels (A–D). The expression of Glut1 at different time periods of LPS-treatment is shown in panel (E). (F–G) LPS stimulated protein (1 h LPS-treatment) was extracted from macrophages and analyzed by Western blot using antibodies against Glut1 and β-actin (loading control). Quantifications (using ImageLab5.2.1software) is shown in panel (G). Each experiment was repeated at least with three parallel replicates. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001.
Glut1 expression is regulated by NF-κB during LPS stimulation
The transcription factor NF-κB activation plays central roles in inflammation and immune response7,27,73. NF-κB activation is required for the expression of cytokine and pro-inflammatory genes4,15. Notably, under basal condition (in the absence of inflammation), NF-κB is complexed with IκBα and remains inactive74. However, upon inflammation, IκBα gets phosphorylated by the kinase IKKβ that results in its poly-ubiquitination followed by proteasomal degradation, and hence release of NF-κB (NF-κB activation). Therefore, inhibition of IKKβ would result in inhibition NF-κB activation14,74,75. To investigate if NF-κB activation is associated with LPS-induced Glut1 expression, we treated macrophages with an IKKβ kinase inhibitor (SC-514) and analyzed its impacts on LPS-induced Glut1 expression. Western blot analysis demonstrates that, upon treatment with LPS, the level of IκBα is decreased, while the level phospho-p65 (NF-κB subunit) is increased (compare lanes 1 and 2, Figs. 2A and S2, quantification in panel B). Notably, the LPS-induced IκBα degradation level as well as increased phospho-p65 levels, are reversed upon treatment with IKKβ-inhibitor (SC-514) (compare lanes 1, 2 and 4, Figs. 2A and S2, quantification in panel B). These observations suggest that LPS treatment resulted in NF-κB activation and this is inhibited by SC-514. Interestingly, RT-qPCR analysis demonstrates that LPS-treatment induced the expression of HOTAIR and upon IKKβ-inhibition (SC-514 treatment), the LPS-induced expression of HOTAIR is suppressed (Fig. 2C). Interestingly, the Glut1 expression is increased upon treatment with LPS and this LPS-induced Glut1 expression level is suppressed upon treatment with SC-514 (Fig. 2D). The LPS-induced increase in Glut1 protein level and its decrease upon SC-514-treatement is also evident in the Western blot (compare lanes 1, 2 and 4, Fig. 2A,B). These observations demonstrate that Glut1 expression is augmented upon LPS-induced inflammation and this is potentially regulated via NF-κB activation.

Inhibition of NF-κB downregulates LPS-induced Glut1 expression in macrophages. (A,B) RAW264.7 cells were initially treated with IKKβ-inhibitor SC-514 (for 1 h) and then treated with LPS for additional 1 h (for protein analysis) and 4 h (for RNA analysis). Proteins were analyzed by western blotting using antibodies against phospho-p65 (NF-κB subunit), IκBα, Glut1, and β-actin (loading control) (panel A, quantifications in panel B). (C,D) RNA was isolated from above treated and control cells. Expressions (relative to GAPDH) of HOTAIR, and Glut1 were measured by RT-qPCR. GAPDH was used as control. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001.
HOTAIR regulates LPS-induced Glut1 expression
Our recent studies demonstrated that HOTAIR plays key roles in NF-κB activation, cytokine expression, and inflammation30. Here to investigate if HOTAIR plays any roles in regulation of LPS-induced Glut1 expression, glucose uptake, and metabolism, we knocked down HOTAIR (using siRNA) in macrophages, then treated with LPS, and analyzed its impacts on Glut1 expression (RNA and protein levels). Briefly, RAW264.7 macrophages were transfected with HOTAIR- and scramble siRNAs (48 h) followed by treatment with LPS. RNA and proteins were isolated and analyzed by RT-qPCR and Western blot, respectively. Our RT-qPCR analysis demonstrated that the expression of HOTAIR is increased upon treatment with LPS and this LPS-induced expression of HOTAIR is significantly reduced (56%) upon application of HOTAIR siRNA (Fig. 3A). Scramble siRNA has no significant impact on HOTAIR expression level (Fig. 3A). Interestingly, RT-qPCR analysis also showed that Glut1 expression (mRNA level) is increased upon LPS-treatment and the LPS-induced Glut1 level was decreased upon HOTAIR-knockdown (Fig. 3B). Western blot analysis showed that Glut1 protein level is increased by LPS and that is decreased upon HOTAIR-knockdown (HOTAIR-siRNA treatment, Figs. 3C and S3, quantification in 3D). Western blot analysis also showed that LPS- treatment induced the degradation of IκBα and that is inhibited upon HOTAIR-knockdown (compare lanes 1, 2 and 4, Figs. 3C,D and S3). Concomitantly, the LPS-treatment also increased the phospho-p65 (NF-κB subunit) level and which is decreased under HOTAIR-knockdown conditions (compare lanes 1, 2 and 4, Figs. 3C,D and S3). Taken together, these observations demonstrate that LPS-induced Glut1 expression is regulated by HOTAIR in macrophages and this is mediated via NF-κB activation controlled by HOTAIR under LPS-stimulation.

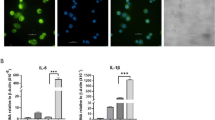
Knockdown of HOTAIR reduces LPS-induced Glut1 expression in macrophages. (A,B) RAW264.7 cells were transfected (48 h) with HOTAIR-siRNA and scramble siRNA followed by treatment with LPS. RNA was analyzed by RT-qPCR for the expression of HOTAIR and Glut1. GAPDH was used as control. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001. (C,D) Proteins from HOTAIR-siRNA and scramble siRNA treatments (48 h) followed by 1 h LPS-treated RAW264.7 cells were analyzed by Western blotting using antibodies against phospho-p65 (NF-κB subunit), IκBα, Glut1, and β-actin (loading control) (panel C). The changes in amounts of NF-κB, IκBα, and Glut1 have been quantified by ImageLab5.2.1 software and shown in panel (D). (E,F) Immunofluorescene analysis: RAW 264.7 cells were transfected with HOTAIR siRNA and scramble siRNA (control) for 48 h and then treated with LPS (1 h), then fixed with paraformaldehyde and immunostained with antibody against Glut1, and counterstained with DAPI. Images were taken by fluorescence microscope (Nikon ECLIPSE TE2000-U) utilizing a 63 × oil objective lens (E) and fluorescence intensity showing the expression of Glut1 was quantified and plotted by ImageJ software (F). Bar is 100 uM. Data represent mean ± SD; *p < 0.05, **p < 0.001. (G,H). RAW264.7 cells were transfected (48 h) with HOTAIR- and scramble antisense oligonucleotide (separately) followed by treatment with LPS. RNA was analyzed by RT-qPCR for the expression of HOTAIR and Glut1. GAPDH was used as control. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001.
To further understand the function of HOTAIR in Glut1 expression, we measured the expression of Glut1 in macrophages by using immunofluorescence assay. We knocked down HOTAIR in RAW264.7 cells by using HOTAIR-siRNA and scramble-siRNA (control) and then treated with LPS. Control and siRNA-treated cells were subjected to immunostaining with Glut1 antibody followed by Alexa Fluor 564 -conjugated secondary antibody and nuclear staining with DNA binding dye DAPI. Cells were then analyzed under a fluorescence microscope (Nikon ECLIPSE TE2000-U)30. We observed that Glut1 expression levels were low in the control cells (in the absence of LPS) and this was increased in upon stimulated with LPS (compare top two panels, Fig. 3E, quantification in panel F). Interestingly, upon HOTAIR-knockdown (HOTAIR-siRNA and LPS treatments), the level of LPS-induced Glut1 expression was decreased relative to LPS alone (Fig. 3E,F). Scramble siRNA has no significant impact on LPS-induced expression of Glut1 (Fig. 3E,F). These results further support our observation that HOTAIR is required for LPS-induced activation of Glut1 in macrophages.
To further address any off-target effects of HOTAIR-siRNA, we have also knocked down HOTAIR using a HOTAIR-specific antisense-oligonucleotide and analyzed its impacts on LPS-induced Glut1 expression. Briefly, RAW264.7 cells were transfected with HOTAIR-antisense and scramble-antisense (separately, as a negative control) for 48, then stimulated with LPS and RNA was analyzed by RT-qPCR. These analysis demonstrated that HOTAIR-antisense specially knocked down HOTAIR level and the knockdown of HOTAIR downregulated the LPS-induced expression of Glut1 (Fig. 3G–H). Our previous studies demonstrated that antisense-mediated knockdown of HOTAIR, down-regulates the LPS-induced increased phospho-NF-κB (phospho-p65) level via inhibition of LPS-induced I-κBα degradation30. Thus, using both siRNA and HOTAIR-antisense mediated knockdown experiments, we demonstrate that HOTAIR regulates Glut1 expression and this is likely via regulation of NF-κB activation. HOTAIR-mediated regulation of Glut1 expression suggests potential roles of HOTAIR in glucose uptake and metabolism under inflammation in immune cells.
HOTAIR regulates LPS induced NF-κB recruitment in the Glut1 promoter
The activation of transcription factors NF-κB plays key roles in the regulation of cytokines and pro-inflammatory genes during inflammation and immune response4,7. Upon inflammation response, the activated NF-κB translocate to the nucleus, binds to the target gene promoters, facilitate activators/coactivators recruitments and that allow chromatin modification and remodeling ultimately resulting in NF-κB target gene activation76,77. As HOTAIR regulates Glut1 expression and it also regulates NF-κB activation upon LPS-stimulation, we examined the LPS-induced enrichment of NF-κB to the Glut1 promoter and if this is regulated via HOTAIR. Glut1 promoter sequence analysis (as described by us previously30,69) revealed the presence of a putative NF-κB binding sites (GGGGATGTCT) upstream of the transcription start site (Fig. 4, top panel, this is in agreement with previous studies78) suggesting its potential regulation by NF-κB. Here, using chromatin immunoprecipitation (ChIP) assay, we have analyzed the NF-κB enrichment at the Glut1 promoter in an LPS-dependent manner and under HOTAIR-knockdown conditions30,69. Briefly, control, scramble-siRNA, and HOTAIR-siRNA-treated RAW264.7 cells were treated with LPS, fixed with formaldehyde, and then subjected to ChIP using antibodies against phosphorylated p65 (NF-κB subunit) and β-actin (control). ChIP DNA fragments were analyzed by qPCR using primers spanning the NF-κB binding site present in the Glut1 promoter. Interestingly, these analyses demonstrated that phospho-p65 (NF-κB) levels were enriched at the Glut1 promoter (NF-κB response element regions) upon treatment with LPS and this LPS-induced NF-κB binding was reduced upon HOTAIR-knockdown (treated with HOTAIR-siRNA followed by LPS) in the Glut1 promoter (Fig. 4, first panel). Scramble siRNA treatment has no significant impact on the LPS-induced enrichment of NF-κB at the Glut1 promoter. Neither LPS nor HOTAIR-knockdown has any significant impact on β-actin (antibody control) enrichment on the Glut1 promoter (Fig. 4, rightmost panel). Notably, along with NF-κB, there are other coactivators which may be associated with LPS-induced Glut1 expression. For example, histone acetyltransferase CBP (CREB binding protein) is known to interact with NF-κB to regulate NF-κB target genes76,77,79. Our ChIP analysis demonstrated that similar to NF-κB, the level of CBP was also enriched at Glut1 promoter and this LPS-induced CBP enrichment was alleviated upon knockdown of HOTAIR (Fig. 4). These observations suggest that HOTAIR plays key roles in LPS-induced NF-κB activation and hence the enrichment of NF-κB and its coactivators such CBP at the Glut1 promoter to regulate its expression in an LPS-dependent manner.

Knockdown of HOTAIR reduces the LPS-dependent recruitment of NF-κB on Glut1 promoter. RAW264.7 macrophage cells were transfected with HOTAIR and scramble-siRNA, then treated with LPS (1.5 h). Cells were then fixed with formaldehyde and subjected to ChIP assay using antibodies specific to phospho-p65, CBP, histone acetylation, H3K4m3, RNA pol II and β-actin (control). The immunoprecipitated DNA fragments were analyzed by qPCR using primers specific to the NF-κB binding regions on Glut1 promoter. The location of NF-κB binding site at the Glut1 promoter is shown in the top panel. Each experiment was repeated at least thrice (n = 3). Data represent mean ± SD; *p < 0.05, **p < 0.001.
Along with activators and coactivators, histone H3K4-trimethylation and histone acetylation are also well known chromatin modifications associated with gene activation80,81,82. Our ChIP analysis demonstrated that levels of H3K4-trimethylation and histone acetylation as well as the level of RNA polymerase II (RNA pol II) were enriched at the Glut1 promoter in the presence of LPS and this was decreased upon HOTAIR knockdown (Fig. 4). This observation suggested that HOTAIR is required for promoter activation (H3K4-methylation and histone acetylation) of Glut1 and this is mediated via activation of NF-κB followed by recruitment of NF-κB and its coregulators including histone methyl-transferases and histone acetyl-transferases at the target gene promoters. The ChIP analysis demonstrated that LPS-induced expression of Glut1 is regulated via transcription factors NF-κB, CBP, and other coactivators and this is regulated by HOTAIR via regulation of NF-κB activation.
HOTAIR regulates glucose uptake under LPS-simulation
As HOTAIR regulated LPS-induced Glut1 expression in macrophage, we investigated if HOTAIR regulates the level of glucose uptake in macrophages during LPS stimulation and inflammation. Notably, Glut1 is a cytosolic protein and this translocates to the cell membrane and thus allows glucose uptake during glucose metabolism83,84,85. It is well-recognized that the level of glucose uptake and metabolism is increased during inflammation and this helps cells to tackle critical inflammatory situation17,20. Here, our studies demonstrate that Glut1 expression is increased upon LPS-stimulation in macrophage and this is regulated by HOTAIR. Therefore, we hypothesize that under LPS-stimulation condition, the level of glucose uptake will increase and this might be regulated via HOTAIR. To test this hypothesis, we measured the level of uptake of glucose (using a commercial kit) in macrophage cells under LPS treatment and in the presence and absence of HOTAIR-knockdowns. Briefly, RAW264.7 macrophages were treated with HOTAIR and scramble siRNAs for 48 h and then stimulated with insulin followed by 2-deoxyglucose (2-DG, a modified glucose) treatment. 2-DG is taken up by the cells by glucose transporter (Glut1 in our case) and gets phosphorylated to 2-DG6P. Once up taken by the cells, 2-DG cannot be further metabolized in glycolysis pathway and therefore will be accumulated inside the cells19,45. Accumulation of 2-DG6P is proportional to glucose uptake by the cells. The accumulated 2-DG6P level was determined by colorimetric reactions (as recommended by the kit)19,45. Interestingly, our results demonstrated that the level of glucose uptake is increased (10 folds) in macrophages upon treatment with LPS and the LPS-induced increased glucose uptake level is significantly reduced upon HOTAIR knockdown (HOTAIR-siRNA treatment) (Fig. 5A). Scramble-siRNA has no significant impacts on LPS-induced glucose uptake level. These observations demonstrate that HOTAIR regulates the LPS-induced glucose uptake during inflammation.

Knockdown of HOTAIR reduces the uptake of glucose in macrophages. RAW264.7 macrophages were treated with HOTAIR siRNA, scramble-siRNA, HOTAIR antisense or scramble-antisense oligonucleotides, separately, for 48 h and after overnight incubation, cells were stimulated with insulin (+/−), followed by 2-deoxyglucose addition for 20 min. 2-DG6P was oxidized and that generates NADPH, which was quantified calorimetrically (measured at 412 nm in a microplate reader). Panels A and B show the glucose uptake analysis under siRNA and antisense-meditated HOTAIR knockdown respectively. Each experiment was repeated at least thrice (n = 3). Data represent mean ± SD; *p < 0.05, **p < 0.001.
To further confirm the effects of HOTAIR on glucose uptake, we knocked down HOTAIR levels using HOTAIR-specific antisense oligonucleotide (a scramble antisense as used in parallel as a negative control) in RAW264.7 cells, then stimulated with insulin followed by 2-deoxyglucose (2-DG, a modified glucose) treatment and then measured the glucose uptake efficiency using similar experiments as described above (Fig. 5B), These results also demonstrated that antisense mediated knockdown of HOTAIR also downregulated the LPS-induced glucose uptake in macrophage (Fig. 5B). This observation in combination with the siRNA-mediated HOTAIR knockdown and glucose uptateke analysis results demonstrated that HOTAIR plays key roles in glucose uptake under inflammatory condition and this is likely mediated via induction of LPS-induced Glut1 expression.
HOTAIR regulates upstream-regulators of glucose metabolism under LPS-simulation
Glucose metabolism is a complex signaling process and there are many upstream signaling cascades that contribute to Glut expression, glucose uptake, and metabolism. Here we demonstrated that HOTAIR plays critical roles in the regulation of Glut1 expression and glucose uptake into macrophages under inflammation. We showed that HOTAIR activates NF-κB which contributes to Glut1 expression and glucose uptake. Notably, HOTAIR down regulates PTEN, which blocks the activation of PI3K/AKT signaling86. PTEN is a tumor suppressor which regulates cell growth and cell apoptosis87. Its often deleted, mutated and aberrantly expressed in a variety of cancer88. Studies have shown that HOTAIR inhibits PTEN gene expression by via methylation89. As PTEN is an upstream regulator of PI3K/AKT signaling which plays major roles in glucose metabolism, and PTEN, is regulated by HOTAIR in cancer cells, we examined if PTEN expression under LPS-stimulation in macrophage and if this is regulated by HOTAIR. RNA samples from the macrophages treated with LPS in the absence and presence of HOTAIR-knockdowns were analyzed by qPCR using PTEN primers. Interestingly, PTEN expression was down-regulated in macrophage upon LPS-stimulation and this was alleviated upon HOTAIR-knockdown (see Fig. 6A,B), suggesting HOTAIR mediated repression of PTEN gene expression.

HOTAIR regulates upstream-regulators of glucose metabolism under LPS-simulation. (A,C): RAW264.7 cells were treated with LPS for varying times. RNA was analyzed by RT-qPCR for the expression of PTEN and HIF-1α. GAPDH was used as control. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001. (B,D): RAW264.7 cells were transfected (48 h) with HOTAIR-siRNA and scramble siRNA followed by treatment with LPS. RNA was analyzed by RT-qPCR for the expression of PTEN (panel B) and HIF-1α (Panel D). GAPDH was used as control. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001. (E,F) Proteins from HOTAIR-siRNA and scramble siRNA treatments (48 h) followed by 1 h LPS-treated RAW264.7 cells were analyzed by Western blotting using antibodies against HIF-1α and β-actin (loading control). The changes in amounts of HIF-1α have been quantified by ImageLab5.2.1 software and shown in panel F.
Additionally, PTEN is known to block the activation of PI3K/AKT signaling and AKT activation stabilizes HIF-1α, which is involved in the induction of Glut1 expression. Glut1 is one of the HIF-1α target genes. PI3K/AKT signaling can also lead to activation of mTOR, which plays role in the glucose metabolism. As PTEN is repressed by HOTAIR under LPS-stimulation of macrophages, we also analyzed the HIF1α expression levels under LPS-stimulation and in the absence and presence of HOTAIR knockdowns. Here our analysis showed that, HIF-1α expression is elevated in macrophages upon LPS-treatment and this is decreased upon HOTAIR knockdown, both at the mRNA and proteins levels, suggesting critical roles of HOTAIR in HIF-1α expression during inflammatory response (Figs. 6C–F and S4). Thus, as HIF-1α is an upstream regulator of Glut1 and HOTAIR regulates HIF-1α, HOTAIR mediated regulation of Glut1 may follow diverse mechanism.
HOTAIR regulates LPS-induced Glut1 expression and glucose uptake in primary bone marrow derived macrophages (BMDM)
We investigated further the importance of HOTAIR in Glut1 regulation and glucose uptakes in primary macrophages (bone marrow derived macrophage, BMDM). Briefly, BMDM cells were isolated from mouse bone marrow and maintained as described by us previously30,65,71. BMDMs were treated with LPS (1 µg/mL, for 4 h) in the presence and absence of HOTAIR-siRNA and scramble-siRNA transfections. RNA was analyzed by RT-qPCR. Interestingly, the level of Glut1 as well as HOTAIR expression was induced upon treatment with LPS in BMDMs (Fig. 7A,B). Application of HOTAIR siRNA resulted in significant knockdown of LPS-induced HOTAIR expression level (Fig. 7A) and this also resulted in a decrease in LPS-induced Glut1 expression (Fig. 7B). Scramble siRNA has no significant impacts on LPS-induced HOTAIR or Glut1 expression (Fig. 7A,B). These observations demonstrated that Glut1 expression is induced upon LPS-stimulation in primary macrophages and this is regulated via lncRNA HOTAIR.

Glut1 expression is induced by LPS in primary macrophages (Bone marrow derived macrophages, BMDM). (A,B) BMDM cells were treated by HOTAIR siRNA and scramble siRNA followed by LPS treatment and RNA was extracted. The expression of HOTAIR and Glut1 was measured by real time PCR. Data represent mean ± SD (n = 3); *p < 0.05, **p < 0.001, ***p < 0.0001. (C,D) BMDM cells were treated by HOTAIR siRNA and scramble siRNA and treated with LPS. Proteins were analyzed by Western blotting using antibodies against phospho-p65 (NF-κB subunit), IκBα, Glut1, and β-actin (loading control). The changes in amounts of NF-κB, IκBα, and Glut1 have been quantified by ImageLab5.2.1 software is shown in panel (D). (E) BMDM cells were treated with HOTAIR and scramble-siRNA for 48 h and after overnight incubation, cells were stimulated with insulin (+/−), followed by 2-deoxyglucose addition for 20 min. 2-DG6P was oxidized and that generates NADPH, which was quantified calorimetrically (measured at 412 nm in a microplate reader). Each experiment was repeated at least thrice (n = 3). Data represent mean ± SD; *p < 0.05, **p < 0.001.
HOTAIR-knocked down and LPS-treated BMDM cells were also analyzed by Western blot to understand the function of HOTAIR for Glut1 expression and NF-κB activation. Interestingly, we found that IκBα protein levels were higher in the control BMDM (no LPS) and this was decreased upon treatment with LPS (compare lanes 1 and 2, Figs. 7C and S5). The level of NF-κB (phospo-p65) protein was increased upon treatment with LPS (compare lanes 1 and 2, Figs. 7C and S5). Interestingly, upon HOTAIR-knockdown, the level of LPS-induced decrease in IκBα protein level was rescued significantly while LPS-induced phospho-p65 NF-κB protein level was decreased (compare lanes 1, 2 and 4, Figs. 6C and S5). Scramble-siRNA has no significant impacts on LPS-induced expression of IκBα and NF-κB (phospho-p65) protein levels. Importantly, Glut1 protein level is also induced by LPS and this was reduced upon treatment with HOTAIR-knockdown (HOTAIR-siRNA treatment, Fig. 7C). Quantification of Western blot showing the expression of IκBα, NF-κB (phospho-p65) and Glut1 protein levels are shown in Fig. 7D.
We also performed the glucose uptake assay (using a commercial kit) in BMDMs under LPS treatment and in the presence or absence of HOTAIR-knockdowns, using similar procedure as described in Fig. 5. Briefly, BMDMs were treated with HOTAIR and scramble siRNAs (48 h), then stimulated with insulin followed by treatment with 2-deoxyglucose (2-DG) which (once uptaken) gets phosphorylated to 2-DG6P and accumulated inside cells19,45. The accumulated 2-DG6P level was determined by colorimetric reactions (as recommended by the kit)19,45. Our results demonstrated that the level of glucose uptake is increased in BMDM upon treatment with LPS and the LPS-induced increased glucose uptake level is significantly reduced upon HOTAIR knockdown (HOTAIR-siRNA treatment) (Fig. 7E). Scramble siRNA has no significant impacts on LPS-induced glucose uptake level. These observations based on primary macrophage analysis further demonstrate that HOTAIR plays a critical role in NF-κB activation, Glut1expression, and glucose uptake during inflammation and immune response in macrophages.
Discussion
Inflammation is the intrinsic immune response of the body towards invading pathogens90,91,92. The inflammatory response causes activation of immune cells such as leukocytes (aka white blood cells), T cells, and B cells, and induces production of inflammatory mediators such as cytokines and chemokines to fight against injury or infection21,93,94. However, uncontrolled and continued inflammation drives the development of many human diseases, including metabolic diseases, obesity, diabetes, autoimmune disorders, neurological disorder, and cancer16,42. The signaling process associated with inflammation and immune response is very complex. Our understanding of inflammation and immune response are largely limited to genomic markers and proteins-based factors that are involved in transcription, phosphorylation, ubiquitination, and protein–protein interactions93,95,96. Even with the huge amount research and options of available therapeutics, many inflammatory diseases still cannot be treated effectively2,80,97. Therefore, understanding the detailed signaling mechanism associated inflammation and immune response is critical for developing effective therapies. Emerging evidences suggest that noncoding RNAs (ncRNAs), play critical roles in various cellular and physiological process including in gene expression, cell differentiation, development, and their dysregulation contributes to critical human disease98,99,100. NcRNAs are recently discovered highly heterogeneous group of transcripts that are coded by the genome, transcribed, but mostly remain untranslated33,35,52. Previously, ncRNAs were considered to be a consequence of transcriptional noise. Recent studies, however, suggest that ncRNAs have distinct cellular functions and are involved in different biological processes101,102,103. In a recent study, we discovered that long noncoding RNA HOTAIR plays critical roles in NF-κB activation, cytokine regulation, inflammation, and immune response30.
Activation of immune cells in response to infection or other stressors is a metabolically expensive event16,31,61,104. The immune cells preferentially meet their energy needs by metabolizing glucose61,62,105. Furthermore, glucose metabolism is involved in mounting inflammatory immune responses18,63,106. Interestingly, the inflammatory response induces expression of glucose metabolizing enzymes and related factors such as Glut1, glucose-6-phosphate dehydrogenase, hexokinase, and increase glycolysis107,108. Therefore, we investigated if HOTAIR is involved in inflammation induced metabolic reprogramming. Our studies demonstrated that, along with proinflammatory genes such as IL-6 and iNOS, the inflammatory lncRNA HOTAIR and Glut1 expression are induced in macrophages upon LPS-stimulation and Glut1 is the major glucose transporter in expressed macrophage and potentially associated with glucose uptake and metabolism during inflammation.
NF-κB activation plays a central role in immune response and inflammation27,28,109. NF-κB activation induces expression of NF-κB regulated cytokines and pro-inflammatory genes90,93. Though it is well known that activation of immune cells in response to infection or other stressors is a metabolically expensive event and metabolizing glucose is a major source of energy to meet the energy requirement of inflammatory response, the detailed signaling mechanism by which NF-κB activation is integrated to glucose metabolism remains elusive. Our mechanistic studies demonstrated that the inhibition of NF-κB (via inhibition of IKKβ kinase by SC-514) significantly reduces LSP-induced Glut1 expression, suggesting potential roles of NF-κB in Glut1 expression during inflammation.
Our recent studies also demonstrated that lncRNA HOTAIR regulates NF-κB activation via degradation of IκBα30. As LPS-induced Glut1 expression is regulated via NF-κB activation which is critically regulated via HOTAIR, we explored the potential role of HOTAIR in LPS-induced Glut1 expression. Indeed our studies demonstrated that siRNA-mediated knockdown of HOTAIR suppressed the LPS-induced expression of Glut1 in macrophage. This is likely because of reduced NF-κB activation under HOTAIR knockdown condition. Importantly, NF-κB activation regulates cytokines and pro-inflammatory genes expression via binding to its target gene promoters, followed by recruitment of activators, coactivators, chromatin modification and remodeling110,111,112. Typical NF-κB regulated genes (such as cytokines) promoters contain binding sites for transcription factor NF-κB and upon inflammation, activated NF-κB gets recruited to the target gene promoters which aids the recruitment of activators, coactivators and chromatin modifiers, resulting in their gene activation76,110,113. Our analysis demonstrated that similar to well-known NF-κB target genes (such as, IL-6, iNOS, etc.), the Glut1 promoter has NF-κB binding site close to the transcription start site. Our ChIP analysis indeed demonstrated that upon LPS-stimulation, NF-κB is activated and is enriched at the Glut1 promoter (NF-κB response element). Along with NF-κB, histone acetyl-transferase and NF-κB associated activator, CBP, is enriched at the Glut1 promoter in response to LPS-treatment76,77. Additionally, gene activation associated chromatin modifications such as histone H3K4-trimethylation and histone acetylation levels are also increased at the Glut1 promoter in an LPS-dependent manner. Importantly, these LPS-induced enrichments in NF-κB, CBP, H3K4-trimethyl, histone acetylation, and RNA polymerase II levels at the Glut1 promoter, were reduced upon knockdown of HOTAIR. These observations further demonstrate critical roles of HOTAIR in LPS-induced Glut1 expression. HOTAIR promotes IκBα degradation which makes NF-κB free from its inhibitory effect and induces availability of activated NF-κB for binding to its target genes promoter such as Glut1 promoter. Thus, our observations suggest further about the crucial roles of HOTAIR in LPS-induced activation of NF-κB and Glut1 expression under inflammation.
As HOTAIR controls the expression glucose transporter Glut1 under LPS-stimulation and Glut1 being the major glucose transporter in macrophage, we explored if HOTAIR also controls the glucose uptake in macrophages during inflammatory responses. Our studies demonstrate that glucose uptake efficiency is increased in macrophages upon LPS stimulation and this is suppressed under HOTAIR-knockdown condition. These observations suggest that HOTAIR is a critical player in Glut1 expression and glucose uptake and potentially regulate glucose metabolism during inflammation. Importantly, glucose metabolism and signaling is complex and involves many upstream regulators and signaling cascade. For example, PI3K/AKT signaling activation is critical to glucose metabolism and PI3K/AKT activation is blocked by PTEN86, which acts as a tumor suppressor to regulate cell growth and cell apoptosis87. In this study, we demonstrated that HOTAIR suppresses PTEN gene expression during LPS-induced inflammation in macrophage. By performing RNA pulldown assay, Zhang et al., have demonstrated that HOTAIR negatively regulate PTEN via directly binding with it114. They also showed that HOTAIR activates the PI3K/Akt pathway to promote Endometrium Cancer progression by suppressing PTEN in vivo114. Furthermore, AKT activation has been reported to stabilize HIF-1α, which is involved in the induction of Glut1 expression115. Indeed, Glut1 is one of the HIF-1α target genes116. In this study we showed that HOTAIR induces HIF-1α expression. Our observation also suggests that HIF-1α is involved in Glut1 expression. PI3K/AKT signaling can also lead to activation of mTOR, which also plays role in the glucose metabolism. Our studies demonstrated that HOTAIR is also involved in the down regulation of PTEN and increase of HIF-1α.
Beyond the studies in cultured macrophages (RAW264.7), we extended our studies to primary cells, BMDMs. Studies in BMDMs also further supported our observations that HOTAIR plays critical roles in NF-κB activation, glucose transporter (Glut1) expression and glucose update during LPS-stimulation, suggesting it potential roles in metabolic reprogramming during inflammation and immune response. Notably, a recent study reported that HOTAIR promotes glycolysis in hepatocytes and this further supports our observation of about the potential roles of HOTAIR in regulation of glucose metabolism117. A model, showing the potential roles of HOTAIR in regulation of glucose uptake via inducing Glut1 expression through IκBα degradation and NF-κB activation, its translocation into the nucleus, and binding to the Glut1 promoter is shown in Fig. 8. Importantly, glucose metabolism and inflammation in macrophages are closely associated with variety chronic metabolic disorder such as obesity and diabetes118,119. The signaling mechanism linking inflammation with metabolic disorders are complex and identifying novel regulatory mechanism for the inflammation associated signaling is critical towards understanding the pathogenesis of metabolic disorders and also for developing novel therapeutics. Notably, independent studies from other laboratories also demonstrated that Glut1 expression is controlled by NF-κB activation. For example, in liver cancer, Glut1 expression is induced by increased NF-κB which is mediated via oncogenic factor LAMTOR578. Another reported that transcription factor Sp1 coordinates with NF-κB and regulates Glut1 expression in response to reactive oxygen specific via activation of MAPK dependent pathways120,121. Therefore, our studies showing the critical roles of lncRNA HOTAIR in NF-κB activation Glut1 expression and glucose uptake under LPS-induced inflammation in macrophages reveals a novel signaling pathway by which is lncRNAs may influence glucose metabolism and may induce metabolic reprogramming in macrophages during imflammation and immune response and therefore, this may serve as a novel therapeutic avenue in the treatment of inflammatory, immune, and metabolic disease. Our studies further suggest that long noncoding RNA are integral components of immune signaling and inflammation and there are novel therapeutic targets for immune and inflammatory diseases.

Proposed model for HOTAIR mediated regulation of glucose metabolism during inflammation. When TLR4 senses LPS, NF-κB is activated that induces HOTAIR expression. In turn, HOTAIR facilitates IκBα degradation and enhances NF-κB activation, its nuclear translocation, and binding to NF-κB regulated Glut1 gene promoter inducing its expressions. Overexpressed Glut1 is translocated to membrane, allows increased glucose uptake and glucose metabolism, during inflammation and immune response. PTEN is repressed by HOTAIR under LPS-stimulation. Thereby, HOTAIR relieved PTEN mediated suppression of HIF-1α. Thus HOTAIR mediated repression of PTEN and induction of HIF-1α under LPS-stimulation, contribute increased Glut1 expression and glucose metabolism.
References
Chen, L. et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218. https://doi.org/10.18632/oncotarget.23208 (2018).
Nathan, C. & Ding, A. Nonresolving inflammation. Cell 140, 871–882. https://doi.org/10.1016/j.cell.2010.02.029 (2010).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820. https://doi.org/10.1016/j.cell.2010.01.022 (2010).
Palsson-McDermott, E. M. & O’Neill, L. A. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113, 153–162. https://doi.org/10.1111/j.1365-2567.2004.01976.x (2004).
Rubartelli, A. & Lotze, M. T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28, 429–436. https://doi.org/10.1016/j.it.2007.08.004 (2007).
Lotze, M. T., Deisseroth, A. & Rubartelli, A. Damage associated molecular pattern molecules. Clin. Immunol. 124, 1–4. https://doi.org/10.1016/j.clim.2007.02.006 (2007).
Lu, Y. C., Yeh, W. C. & Ohashi, P. S. LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. https://doi.org/10.1016/j.cyto.2008.01.006 (2008).
Jabbour, H. N., Sales, K. J., Catalano, R. D. & Norman, J. E. Inflammatory pathways in female reproductive health and disease. Reproduction 138, 903–919. https://doi.org/10.1530/REP-09-0247 (2009).
Girard, S. et al. Role of perinatal inflammation in cerebral palsy. Pediatr. Neurol. 40, 168–174. https://doi.org/10.1016/j.pediatrneurol.2008.09.016 (2009).
Libby, P. Inflammatory mechanisms: The molecular basis of inflammation and disease. Nutr. Rev. 65, S140-146. https://doi.org/10.1111/j.1753-4887.2007.tb00352.x (2007).
Auffray, C., Sieweke, M. H. & Geissmann, F. Blood monocytes: Development, heterogeneity, and relationship with dendritic cells. Annu. Rev. Immunol. 27, 669–692. https://doi.org/10.1146/annurev.immunol.021908.132557 (2009).
Espinosa, V. & Rivera, A. First line of defense: Innate cell-mediated control of pulmonary aspergillosis. Front. Microbiol. 7, 272. https://doi.org/10.3389/fmicb.2016.00272 (2016).
Fukuzumi, M., Shinomiya, H., Shimizu, Y., Ohishi, K. & Utsumi, S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect. Immun. 64, 108–112. https://doi.org/10.1128/IAI.64.1.108-112.1996 (1996).
Jeong, S. J., Pise-Masison, C. A., Radonovich, M. F., Park, H. U. & Brady, J. N. A novel NF-kappaB pathway involving IKKbeta and p65/RelA Ser-536 phosphorylation results in p53 Inhibition in the absence of NF-kappaB transcriptional activity. J. Biol. Chem. 280, 10326–10332. https://doi.org/10.1074/jbc.M412643200 (2005).
Schwandner, R., Dziarski, R., Wesche, H., Rothe, M. & Kirschning, C. J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274, 17406–17409. https://doi.org/10.1074/jbc.274.25.17406 (1999).
Freemerman, A. J. et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289, 7884–7896. https://doi.org/10.1074/jbc.M113.522037 (2014).
Godoy, A. et al. Differential subcellular distribution of glucose transporters GLUT1-6 and GLUT9 in human cancer: Ultrastructural localization of GLUT1 and GLUT5 in breast tumor tissues. J. Cell. Physiol. 207, 614–627. https://doi.org/10.1002/jcp.20606 (2006).
Kominsky, D. J., Campbell, E. L. & Colgan, S. P. Metabolic shifts in immunity and inflammation. J. Immunol. 184, 4062–4068. https://doi.org/10.4049/jimmunol.0903002 (2010).
Ieronymaki, E. et al. Insulin resistance in macrophages alters their metabolism and promotes an M2-like phenotype. J. Immunol. 202, 1786–1797. https://doi.org/10.4049/jimmunol.1800065 (2019).
Ozbudak, I. H. et al. Glucose transporter-1 in pulmonary neuroendocrine carcinomas: Expression and survival analysis. Mod. Pathol. 22, 633–638. https://doi.org/10.1038/modpathol.2009.6 (2009).
Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 140, 771–776. https://doi.org/10.1016/j.cell.2010.03.006 (2010).
Kamruzzaman, M. et al. Quorum-regulated biofilms enhance the development of conditionally viable, environmental Vibrio cholerae. Proc. Natl. Acad. Sci. USA 107, 1588–1593. https://doi.org/10.1073/pnas.0913404107 (2010).
Newton, K. & Dixit, V. M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 4, 3. https://doi.org/10.1101/cshperspect.a006049 (2012).
Alvarez-Errico, D., Lessmann, E. & Rivera, J. Adapters in the organization of mast cell signaling. Immunol. Rev. 232, 195–217. https://doi.org/10.1111/j.1600-065X.2009.00834.x (2009).
Hadjicharalambous, M. R. & Lindsay, M. A. Long non-coding RNAs and the innate immune response. Noncoding RNA 5, 34. https://doi.org/10.3390/ncrna5020034 (2019).
Heward, J. A. & Lindsay, M. A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 35, 408–419. https://doi.org/10.1016/j.it.2014.07.005 (2014).
Liu, T., Zhang, L., Joo, D. & Sun, S. C. NF-kappaB signaling in inflammation. Signal. Transduct. Target. Ther. 2, 1. https://doi.org/10.1038/sigtrans.2017.23 (2017).
Sun, S. C., Chang, J. H. & Jin, J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 34, 282–289. https://doi.org/10.1016/j.it.2013.01.004 (2013).
Zhang, H. & Sun, S. C. NF-kappaB in inflammation and renal diseases. Cell Biosci. 5, 63. https://doi.org/10.1186/s13578-015-0056-4 (2015).
Obaid, M. et al. LncRNA HOTAIR regulates lipopolysaccharide-induced cytokine expression and inflammatory response in macrophages. Sci. Rep. 8, 15670. https://doi.org/10.1038/s41598-018-33722-2 (2018).
Serghiou, S., Kyriakopoulou, A. & Ioannidis, J. P. Long noncoding RNAs as novel predictors of survival in human cancer: A systematic review and meta-analysis. Mol. Cancer 15, 50. https://doi.org/10.1186/s12943-016-0535-1 (2016).
Haemmig, S., Simion, V. & Feinberg, M. W. Long non-coding RNAs in vascular inflammation. Front. Cardiovasc. Med. 5, 22. https://doi.org/10.3389/fcvm.2018.00022 (2018).
Bhan, A. & Mandal, S. S. LncRNA HOTAIR: A master regulator of chromatin dynamics and cancer. Biochim. Biophys. Acta 151–164, 2015. https://doi.org/10.1016/j.bbcan.2015.07.001 (1856).
Bhan, A. & Mandal, S. S. Long noncoding RNAs: Emerging stars in gene regulation, epigenetics and human disease. ChemMedChem 9, 1932–1956. https://doi.org/10.1002/cmdc.201300534 (2014).
Bhan, A., Soleimani, M. & Mandal, S. S. Long noncoding RNA and cancer: A new paradigm. Cancer Res. 77, 3965–3981. https://doi.org/10.1158/0008-5472.CAN-16-2634 (2017).
Xu, J. & Cao, X. Long noncoding RNAs in the metabolic control of inflammation and immune disorders. Cell. Mol. Immunol. 16, 1–5. https://doi.org/10.1038/s41423-018-0042-y (2019).
Chew, C. L., Conos, S. A., Unal, B. & Tergaonkar, V. Noncoding RNAs: Master regulators of inflammatory signaling. Trends Mol. Med. 24, 66–84. https://doi.org/10.1016/j.molmed.2017.11.003 (2018).
Hung, C. L. et al. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 111, 18697–18702. https://doi.org/10.1073/pnas.1415669112 (2014).
Fan, C. et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol. Cancer 16, 130. https://doi.org/10.1186/s12943-017-0699-3 (2017).
Zhang, P., Cao, L., Fan, P., Mei, Y. & Wu, M. LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep. 17, 1204–1220. https://doi.org/10.15252/embr.201642067 (2016).
Gong, Z. et al. Long non-coding RNAs in cancer. Sci. China Life Sci. 55, 1120–1124. https://doi.org/10.1007/s11427-012-4413-9 (2012).
Esteves, J. V. et al. Diabetes modulates MicroRNAs 29b–3p, 29c–3p, 199a–5p and 532–3p expression in muscle: Possible role in GLUT4 and HK2 repression. Front. Endocrinol. 9, 536. https://doi.org/10.3389/fendo.2018.00536 (2018).
Yuan, G., Zhao, Y., Wu, D. & Gao, C. Mir-150 up-regulates glut1 and increases glycolysis in osteosarcoma cells. Asian Pac. J. Cancer Prev. 18, 1127–1131. https://doi.org/10.22034/APJCP.2017.18.4.1127 (2017).
Zhu, H., Xue, H., Jin, Q. H., Guo, J. & Chen, Y. D. MiR-138 protects cardiac cells against hypoxia through modulation of glucose metabolism by targetting pyruvate dehydrogenase kinase 1. Biosci. Rep. 37, 1. https://doi.org/10.1042/BSR20170296 (2017).
Wang, L. et al. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol. Neurodegener. 14, 2. https://doi.org/10.1186/s13024-019-0305-9 (2019).
Elsayed, E. T., Salem, P. E., Darwish, A. M. & Fayed, H. M. Plasma long non-coding RNA HOTAIR as a potential biomarker for gastric cancer. Int. J. Biol. Markers https://doi.org/10.1177/1724600818760244 (2018).
Lan, F. et al. A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449, 689–694. https://doi.org/10.1038/nature06192 (2007).
Jene-Sanz, A. et al. Expression of polycomb targets predicts breast cancer prognosis. Mol. Cell Biol. 33, 3951–3961. https://doi.org/10.1128/MCB.00426-13 (2013).
Tsai, M. C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. https://doi.org/10.1126/science.1192002 (2010).
Xu, G. H. et al. Up-regulation of SR-BI promotes progression and serves as a prognostic biomarker in clear cell renal cell carcinoma. BMC Cancer 18, 88. https://doi.org/10.1186/s12885-017-3761-z (2018).
Kogo, R. et al. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 71, 6320–6326. https://doi.org/10.1158/0008-5472.CAN-11-1021 (2011).
Bhan, A. et al. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J. Mol. Biol. 425, 3707–3722. https://doi.org/10.1016/j.jmb.2013.01.022 (2013).
Bhan, A. et al. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J. Mol. Biol. 426, 3426–3441. https://doi.org/10.1016/j.jmb.2014.07.025 (2014).
Bhan, A. et al. Bisphenol-A and diethylstilbestrol exposure induces the expression of breast cancer associated long noncoding RNA HOTAIR in vitro and in vivo. J. Steroid. Biochem. Mol. Biol. 141, 160–170. https://doi.org/10.1016/j.jsbmb.2014.02.002 (2014).
Tang, Q. & Hann, S. S. HOTAIR: An oncogenic long non-coding RNA in human cancer. Cell Physiol. Biochem. 47, 893–913. https://doi.org/10.1159/000490131 (2018).
Hajjari, M. & Salavaty, A. HOTAIR: An oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 12, 1–9. https://doi.org/10.7497/j.issn.2095-3941.2015.0006 (2015).
Li, L. et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 5, 3–12. https://doi.org/10.1016/j.celrep.2013.09.003 (2013).
Milhem, M. M. et al. Correlation of MTDH/AEG-1 and HOTAIR expression with metastasis and response to treatment in sarcoma patients. J. Cancer Sci. Ther. S5, 1–10 (2011).
Wu, Y. et al. Long noncoding RNA HOTAIR involvement in cancer. Tumour Biol. 35, 9531–9538. https://doi.org/10.1007/s13277-014-2523-7 (2014).
Suzuki, N. et al. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416, 750–756. https://doi.org/10.1038/nature736 (2002).
Ganeshan, K. & Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 32, 609–634. https://doi.org/10.1146/annurev-immunol-032713-120236 (2014).
Vats, D. et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. https://doi.org/10.1016/j.cmet.2006.05.011 (2006).
Delmastro-Greenwood, M. M. & Piganelli, J. D. Changing the energy of an immune response. Am. J. Clin. Exp. Immunol. 2, 30–54 (2013).
Udden, S. M., Waliullah, S., Harris, M. & Zaki, H. The ex vivo colon organ culture and its use in antimicrobial host defense studies. J. Vis. Exp. https://doi.org/10.3791/55347 (2017).
Udden, S. M. N. et al. NOD2 suppresses colorectal tumorigenesis via downregulation of the TLR pathways. Cell Rep. 19, 2756–2770. https://doi.org/10.1016/j.celrep.2017.05.084 (2017).
Sugden, K., Tichopad, A., Khan, N., Craig, I. W. & D’Souza, U. M. Genes within the serotonergic system are differentially expressed in human brain. BMC Neurosci. 10, 50. https://doi.org/10.1186/1471-2202-10-50 (2009).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 (2008).
Udden, S. M., Morita-Fujimura, Y., Satake, M. & Ikawa, S. c-ABL tyrosine kinase modulates p53-dependent p21 induction and ensuing cell fate decision in response to DNA damage. Cell Signal. 26, 444–452. https://doi.org/10.1016/j.cellsig.2013.10.005 (2014).
Ansari, K. I., Kasiri, S. & Mandal, S. S. Histone methylase MLL1 has critical roles in tumor growth and angiogenesis and its knockdown suppresses tumor growth in vivo. Oncogene 32, 3359–3370. https://doi.org/10.1038/onc.2012.352 (2013).
Chen, C. et al. KH-type splicing regulatory protein mediate inflammatory response in gastric epithelial cells induced by lipopolysaccharide. Cell. Biol. Int. 41, 871–878. https://doi.org/10.1002/cbin.10804 (2017).
Zhang, X., Goncalves, R. & Mosser, D. M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 14, 11. https://doi.org/10.1002/0471142735.im1401s83 (2008).
Stunault, M. I., Bories, G., Guinamard, R. R. & Ivanov, S. Metabolism plays a key role during macrophage activation. Mediators Inflamm. 2018, 2426138. https://doi.org/10.1155/2018/2426138 (2018).
Oeckinghaus, A. & Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 1, a000034. https://doi.org/10.1101/cshperspect.a000034 (2009).
Hochrainer, K., Racchumi, G. & Anrather, J. Site-specific phosphorylation of the p65 protein subunit mediates selective gene expression by differential NF-kappaB and RNA polymerase II promoter recruitment. J. Biol. Chem. 288, 285–293. https://doi.org/10.1074/jbc.M112.385625 (2013).
Kishore, N. et al. A selective IKK-2 inhibitor blocks NF-kappa B-dependent gene expression in interleukin-1 beta-stimulated synovial fibroblasts. J. Biol. Chem. 278, 32861–32871. https://doi.org/10.1074/jbc.M211439200 (2003).
Mukherjee, S. P. et al. Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-kappaB-driven transcription. PLoS Biol. 11, e1001647. https://doi.org/10.1371/journal.pbio.1001647 (2013).
Shenkar, R., Yum, H. K., Arcaroli, J., Kupfner, J. & Abraham, E. Interactions between CBP, NF-kappaB, and CREB in the lungs after hemorrhage and endotoxemia. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L418-426. https://doi.org/10.1152/ajplung.2001.281.2.L418 (2001).
Zhou, J., Li, Y., Li, D., Liu, Z. & Zhang, J. Oncoprotein LAMTOR5 activates GLUT1 via upregulating NF-kappaB in liver cancer. Open Med. 14, 264–270. https://doi.org/10.1515/med-2019-0022 (2019).
vander Berghe, N., Cool, R. H. & Wittinghofer, A. Discriminatory residues in Ras and Rap for guanine nucleotide exchange factor recognition. J. Biol. Chem. 274, 11078–11085. https://doi.org/10.1074/jbc.274.16.11078 (1999).
Cui, H., Banerjee, S., Guo, S., Xie, N. & Liu, G. IFN regulatory factor 2 inhibits expression of glycolytic genes and lipopolysaccharide-induced proinflammatory responses in macrophages. J. Immunol. 200, 3218–3230. https://doi.org/10.4049/jimmunol.1701571 (2018).
Gupta, R. A. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. https://doi.org/10.1038/nature08975 (2010).
Schappe, M. S. et al. Chanzyme TRPM7 mediates the Ca(2+) influx essential for lipopolysaccharide-induced toll-like receptor 4 endocytosis and macrophage activation. Immunity 48, 59–74. https://doi.org/10.1016/j.immuni.2017.11.026 (2018).
Blodgett, D. M., De Zutter, J. K., Levine, K. B., Karim, P. & Carruthers, A. Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 130, 157–168. https://doi.org/10.1085/jgp.200709818 (2007).
Navale, A. M. & Paranjape, A. N. Glucose transporters: Physiological and pathological roles. Biophys. Rev. 8, 5–9. https://doi.org/10.1007/s12551-015-0186-2 (2016).
Banerjee, S. K., McGaffin, K. R., Pastor-Soler, N. M. & Ahmad, F. SGLT1 is a novel cardiac glucose transporter that is perturbed in disease states. Cardiovasc. Res. 84, 111–118. https://doi.org/10.1093/cvr/cvp190 (2009).
Zundel, W. et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14, 391–396 (2000).
Li, A., Qiu, M., Zhou, H., Wang, T. & Guo, W. PTEN, insulin resistance and cancer. Curr. Pharm. Des. 23, 3667–3676. https://doi.org/10.2174/1381612823666170704124611 (2017).
Ortega-Molina, A. & Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab 24, 184–189. https://doi.org/10.1016/j.tem.2012.11.002 (2013).
Yu, F., Chen, B., Dong, P. & Zheng, J. HOTAIR Epigenetically modulates PTEN expression via microRNA-29b: A novel mechanism in regulation of liver fibrosis. Mol. Ther. 25, 205–217. https://doi.org/10.1016/j.ymthe.2016.10.015 (2017).
Cekici, A., Kantarci, A., Hasturk, H. & Van Dyke, T. E. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000(64), 57–80. https://doi.org/10.1111/prd.12002 (2014).
Hiemstra, P. S. The role of epithelial beta-defensins and cathelicidins in host defense of the lung. Exp. Lung Res. 33, 537–542. https://doi.org/10.1080/01902140701756687 (2007).
Conner, J. M. & Varon, S. Characterization of antibodies to nerve growth factor: Assay-dependent variability in the cross-reactivity with other neurotrophins. J. Neurosci. Methods 65, 93–99. https://doi.org/10.1016/0165-0270(95)00151-4 (1996).
Chaplin, D. D. Overview of the immune response. J. Allergy Clin. Immunol. 125, S3-23. https://doi.org/10.1016/j.jaci.2009.12.980 (2010).
Kopp, E. B. & Medzhitov, R. Infection and inflammation in somatic maintenance, growth and longevity. Evol. Appl. 2, 132–141. https://doi.org/10.1111/j.1752-4571.2008.00062.x (2009).
Lee, M. J. & Yaffe, M. B. Protein regulation in signal transduction. Cold Spring Harb. Perspect. Biol. 8, 005918. https://doi.org/10.1101/cshperspect.a005918 (2016).
Ambros, V. MicroRNAs and developmental timing. Curr. Opin. Genet. Dev. 21, 511–517. https://doi.org/10.1016/j.gde.2011.04.003 (2011).
Tabas, I. & Glass, C. K. Anti-inflammatory therapy in chronic disease: Challenges and opportunities. Science 339, 166–172. https://doi.org/10.1126/science.1230720 (2013).
Atianand, M. K., Caffrey, D. R. & Fitzgerald, K. A. Immunobiology of long noncoding RNAs. Annu. Rev. Immunol. 35, 177–198. https://doi.org/10.1146/annurev-immunol-041015-055459 (2017).
Wang, K. C. & Chang, H. Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell. 43, 904–914. https://doi.org/10.1016/j.molcel.2011.08.018 (2011).
Hu, G. et al. Molecular mechanisms of long noncoding RNAs and their role in disease pathogenesis. Oncotarget 9, 18648–18663. https://doi.org/10.18632/oncotarget.24307 (2018).
Chen, S. et al. LncRNAs and their role in cancer stem cells. Oncotarget 8, 110685–110692. https://doi.org/10.18632/oncotarget.22161 (2017).
Iyer, M. K. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47, 199–208. https://doi.org/10.1038/ng.3192 (2015).
Anastasiadou, E., Jacob, L. S. & Slack, F. J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18. https://doi.org/10.1038/nrc.2017.99 (2018).
Johnson, A. R., Milner, J. J. & Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 249, 218–238. https://doi.org/10.1111/j.1600-065X.2012.01151.x (2012).
Fox, C. J., Hammerman, P. S. & Thompson, C. B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 5, 844–852. https://doi.org/10.1038/nri1710 (2005).
Hockel, M. & Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 93, 266–276. https://doi.org/10.1093/jnci/93.4.266 (2001).
Kavanagh Williamson, M. et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses 10, 114. https://doi.org/10.3390/v10030114 (2018).
Singh, D. et al. Overexpression of hypoxia-inducible factor and metabolic pathways: Possible targets of cancer. Cell. Biosci. 7, 62. https://doi.org/10.1186/s13578-017-0190-2 (2017).
Tak, P. P. & Firestein, G. S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Invest. 107, 7–11. https://doi.org/10.1172/JCI11830 (2001).
Bhatt, D. & Ghosh, S. Regulation of the NF-kappaB-mediated transcription of inflammatory genes. Front. Immunol. 5, 71. https://doi.org/10.3389/fimmu.2014.00071 (2014).
Ahmed, A. U., Williams, B. R. & Hannigan, G. E. Transcriptional activation of inflammatory genes: Mechanistic insight into selectivity and diversity. Biomolecules 5, 3087–3111. https://doi.org/10.3390/biom5043087 (2015).
Mulero, M. C., Wang, V. Y., Huxford, T. & Ghosh, G. Genome reading by the NF-kappaB transcription factors. Nucleic Acids Res 47, 9967–9989. https://doi.org/10.1093/nar/gkz739 (2019).
Hayden, M. S. & Ghosh, S. Shared principles in NF-kappaB signaling. Cell 132, 344–362. https://doi.org/10.1016/j.cell.2008.01.020 (2008).
Li, D. et al. Long intergenic noncoding RNA HOTAIR is overexpressed and regulates PTEN methylation in laryngeal squamous cell carcinoma. Am. J. Pathol. 182, 64–70. https://doi.org/10.1016/j.ajpath.2012.08.042 (2013).
Hayashi, M. et al. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1alpha under hypoxic conditions in trophoblast-derived cells. J. Endocrinol. 183, 145–154. https://doi.org/10.1677/joe.1.05599 (2004).
Wood, S. M. et al. Selection and analysis of a mutant cell line defective in the hypoxia-inducible factor-1 alpha-subunit (HIF-1alpha). Characterization of hif-1alpha-dependent and -independent hypoxia-inducible gene expression. J. Biol. Chem. 273, 8360–8368. https://doi.org/10.1074/jbc.273.14.8360 (1998).
Wei, S. et al. Promotion of glycolysis by HOTAIR through GLUT1 upregulation via mTOR signaling. Oncol. Rep. 38, 1902–1908. https://doi.org/10.3892/or.2017.5840 (2017).
Saltiel, A. R. & Olefsky, J. M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 127, 1–4. https://doi.org/10.1172/JCI92035 (2017).
Jung, U. J. & Choi, M. S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 15, 6184–6223. https://doi.org/10.3390/ijms15046184 (2014).
Kao, Y. S. & Fong, J. C. Endothelin-1 induces glut1 transcription through enhanced interaction between Sp1 and NF-kappaB transcription factors. Cell. Signal. 20, 771–778. https://doi.org/10.1016/j.cellsig.2007.12.012 (2008).
Kao, Y. S. & Fong, J. C. Endothelin-1 induction of Glut1 transcription in 3T3-L1 adipocytes involves distinct PKCepsilon- and p42/p44 MAPK-dependent pathways. Biochim. Biophys. Acta 1780, 154–159. https://doi.org/10.1016/j.bbagen.2007.11.013 (2008).
Acknowledgements
We thank all the Mandal lab members for helpful discussions. Research in Mandal laboratory is supported by grants from National Institute of Health 1R15HL142032.
Author information
Authors and Affiliations
Contributions
M.O. performed all the experiments included in the manuscript, analyzed the data. N.U. isolated the BMDM. P.A. supervised the animal experiments related to BMDM isolation, S.S.M. supervised the overall projects, designed experiments, coordinated the data analysis, and written the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Obaid, M., Udden, S.M.N., Alluri, P. et al. LncRNA HOTAIR regulates glucose transporter Glut1 expression and glucose uptake in macrophages during inflammation. Sci Rep 11, 232 (2021). https://doi.org/10.1038/s41598-020-80291-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80291-4
This article is cited by
-
Novel long non-coding RNAs associated with inflammation and macrophage activation in human
Scientific Reports (2023)
-
Myeloid cell-targeted therapies for solid tumours
Nature Reviews Immunology (2023)
-
Diagnostic significance of serum lncRNA HOTAIR and its predictive value for the development of chronic complications in patients with type 2 diabetes mellitus
Diabetology & Metabolic Syndrome (2021)
-
Silica-coated magnetic-nanoparticle-induced cytotoxicity is reduced in microglia by glutathione and citrate identified using integrated omics
Particle and Fibre Toxicology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
