
Abstract
Alcohol use disorder exhausts substantial social and economic costs, with recent dramatic increases in female problem drinking. Thus, it is critically important to understand signaling differences underlying alcohol consumption across the sexes. Orexin-1 receptors (Ox1Rs) can strongly promote motivated behavior, and we previously identified Ox1Rs within nucleus accumbens shell (shell) as crucial for driving binge intake in higher-drinking male mice. Here, shell Ox1R inhibition did not alter female mouse alcohol drinking, unlike in males. Also, lower dose systemic Ox1R inhibition reduced compulsion-like alcohol intake in both sexes, indicating that female Ox1Rs can drive some aspects of pathological consumption, and higher doses of systemic Ox1R inhibition (which might have more off-target effects) reduced binge drinking in both sexes. In contrast to shell Ox1Rs, inhibiting shell calcium-permeable AMPA receptors (CP-AMPARs) strongly reduced alcohol drinking in both sexes, which was specific to alcohol since this did not reduce saccharin intake in either sex. Our results together suggest that the shell critically regulates binge drinking in both sexes, with shell CP-AMPARs supporting intake in both sexes, while shell Ox1Rs drove drinking only in males. Our findings provide important new information about sex-specific and -general mechanisms that promote binge alcohol intake and possible targeted therapeutic interventions.
Similar content being viewed by others
Introduction
Despite extensive efforts, alcohol use disorder (AUD) remains a significant health problem, with substantial medical, social and economic costs1,2,3,4,5,6. Binge drinking, with high levels of alcohol intake, is an especially problematic and pernicious obstacle to treating AUD. About 3/4th of AUD-related costs are due to individuals who binge6, and reducing binge intake lowers health risks3,5 and relapse1. Also, binging in non-dependent, problem drinkers can promote development of more serious alcohol problems7,8,9. Thus, identifying key mechanisms that drive alcohol binge intake may provide novel and translationally useful therapeutic interventions and reduce the burden of alcohol-related costs, especially given the limited pharmacotherapies for AUD10.
In recent years, the rate of hazardous alcohol drinking in human females has risen dramatically11,12,13, making it essential to understand possible mechanistic differences across the sexes that promote binge intake. Sex differences are known to exist for several addiction-related behaviors14, including where female rodents often drink more alcohol than males14,15. Other aspects of alcohol responding have some divergence. For example compulsion-like responding, where intake persists despite negative consequences16,17, is more similar between sexes in some models15,18, including home-cage limited-access drinking15 used here, but not others19,20 (see also “Discussion”).
In seeking the critical mechanisms that drive binge drinking, we have focused on the nucleus accumbens (NAcb) shell (shell) and the contribution of orexin-1-receptors (Ox1Rs) and AMPA-type glutamate receptors (AMPARs) (reviewed in21,22). The shell is a critical regulator of numerous motivated and addiction-related behaviors22,23,24,25 and compulsion-like behaviors26,27,28, including where shell inhibition reduces alcohol drinking but not sweet fluid intake or locomotor activity29,30,31,32,33,34,35,36. Orexin/hypocretins regulate many adaptive behavioral and physiological responses37,38, and Ox1Rs are of particular interest because they drive responding for high-value natural and drug rewards, including alcohol, with little role for less-motivating substances37,38,39,40,41,42. Indeed, shell Ox1R inhibition decreases binge intake in males, especially higher drinkers29,43 (as seen with systemic block of Ox1Rs44,45,46), and systemic Ox1R inhibition reduces many forms of pathological alcohol intake38,44,45,46,47,48,49,50. In addition, calcium-permeable AMPARs (CP-AMPARs) are observed in the shell after exposure to alcohol51,52,53 and other intoxicants and stress-related conditions (see22). In addition, shell CP-AMPARs are known to promote several addiction-related behaviors (reviewed in22), making it imperative to know whether CP-AMPARs in shell are also important modulators of alcohol binge consumption.
Here, we demonstrate that shell Ox1Rs did not regulate binge alcohol drinking in female C57BL/6 mice, in strong contrast to our previously demonstrated Ox1R regulation of binge intake in male mice29,43. However, lower doses of systemic Ox1R inhibition did reduce compulsion-like alcohol drinking in females, similar to males48, indicating that females have Ox1Rs that can regulate some aspects of pathological alcohol-directed behavior. Furthermore, inhibition of CP-AMPARs in the shell significantly reduced alcohol drinking in both males and females. This was specific for alcohol, as inhibiting shell CP-AMPARs had no impact on saccharin intake. Thus, binge alcohol consumption in females required shell CP-AMPARs, similar to males, but did not involve shell Ox1Rs, very different from males. These findings indicate that complementary but partly separable mechanisms in the shell drive binge alcohol drinking in females versus males, with important implications for differential treatment strategies to counteract alcohol addiction in the sexes.
Material and methods
Limited daily access (LDA) drinking
All procedures followed Guide for Care and Use of Laboratory Animals provided by the NIH, with approval of the UCSF IACUC. Single housed adult C57BL6/J mice (Jackson Labs) drank under a limited daily access (LDA) paradigm, which is a two-bottle choice (2BC) variant of Drinking-in-the-Dark (with 15% alcohol (v/v) versus water), as we29,43,48,54 (see also55) have used previously. At 7–8 weeks of age, mice had a single 24-h 2BC session. Thereafter, mice drank under LDA for 2 h/day, 5 days/week, starting 2.5–3 h into the dark cycle. After ~ 3 week LDA, with handling 2–5 min per day for the week before surgery, mouse cohorts intended for microinjections underwent intra-shell cannulation surgery (details below). After 1-wk recovery and ~ 2-week more LDA, there was a week of LDA with handling: 2–3 days of handling (2–5 min/day), then 2–3 days of handling where the cannula plug was removed and returned, then 1 day with saline injection. Thereafter, we began intracranial injection experiments (details below). These methods and those below were the same as those used in our previous studies of shell Ox1Rs in male mice29,43,48,54. Systemic injection studies occurred with equivalent timing except without surgery. Separate cohorts of mice were used for each experiment, including systemic injections.
Quinine-resistant alcohol drinking was tested by adulterating alcohol with 100 µM quinine, as in previous studies15,48. For saccharin drinking (tested in separate cohorts of mice), the timing and length of the session across days was the same as for alcohol, except mice instead consumed 0.05% saccharin. We have previously used this29,43,48 since mice drink approximately the same volume of this concentration of saccharin as with alcohol. Blood alcohol concentrations were determined exactly as previously described48.
To compare basal drinking levels (determined on vehicle test days) with possible changes in drinking with a pharmacological agent, we used a method as in43. In particular, determining the percent change in drinking after drug exposure (100 × (drug/vehicle) – 100) can result in large difference when drinking levels under vehicle are lower. Thus, we instead used a method involving a log transformation of the change in drinking with drug, log(100 × drug/vehicle). The strengths and weaknesses of these two methods (percent change vs log transformed percent change) are discussed in detail in43.
Surgery and microinjection
Intra-shell cannulation methods were as in29,43. Surgery occurred after 3-week LDA, with 1-wk recovery before resuming LDA. Cannula were implanted targeting shell (AP + 1.5 mm, ML ± 0.5 mm, DV − 4.5 mm). Pharmacological agents were microinjected (0.2 µl/side, 33-gauge needle extending 0.3 mm beyond cannula tip, at a rate of 200nL/min) 30 min before a drinking session. Vehicle or receptor blocker was administered 1-week apart, using a within-subject, Latin-squares, randomized design. Importantly, animals were exposed to each experimental condition twice: conditions (drug vs vehicle) were randomized for one round of injections, then the same schedule was used for a second round. Each animal thus received 5–6 injections, where 5 injections were planned (initial saline during handling and 4 experimental injections) and a 6th occurred if there were problems with injector clogging (which happened infrequently). For each experimental condition from a given animal, drinking data from the two injection days were averaged to give a single intake value for vehicle and a single value for drug. We routinely utilize this approach to reduce variability in drinking measures29,43,48,56,57. Histology was performed to verify the location of cannula placements, and is shown in Suppl. Fig. 1.
Reagents
All intracranial reagents were injected bilaterally in 0.2 µl/side (see above). We utilized SB-334867 (SB) as an Ox1R inhibitor29,48 since it has been used across many studies, with well-established dosages and specific behavioral effects37,38,40,42,58,59,60,61. Also, behaviors inhibited by SB are reduced by other Ox1R inhibitors38. The SB dose used intracranially (3 µg/side) reduces alcohol but not saccharin intake29 without effect in the NAcb Core, nor in the shell at 1 µg/side29,43, and Ox2R inhibition in the shell with an equivalent dose of TCS-OX2-29 has no effect on male binge alcohol intake29. This SB dose also decreases stress but not drug-prime reinstatement of morphine CPP, or locomotor activity, when infused in the shell61, reduces alcohol- but not sucrose-seeking (mPFC58) and decreases alcohol but not saccharin or food intake (icv60). Thus, these findings validate SB as an Ox1R inhibitor with selective impacts on reward-directed behavior. It also would be useful, in future studies, to identify how well systemic SB crosses the blood–brain barrier, the mechanisms of systemic SB first-pass metabolism, and the spread of SB after intracranial injection.
Calcium-permeable AMPARs (CP-AMPARs) were inhibited using 1-naphthylacetyl spermine (NASPM) at 20 µg/side, as used in many studies62,63,64,65. This and higher doses in NAcb decrease cocaine seeking, but have no effect on cocaine self-administration, sucrose seeking62,65 or behavioral flexibility66.
SB and NASPM were purchased from Sigma. Drugs were made fresh for each day of use. NASPM was diluted in 0.9% sterile saline (NaCl) for intracranial injections. For systemic injections, SB at 3 mg/kg was diluted in 2% DMSO and 25% beta-Cyclodextrin (BCD) and SB at 30 mg/kg was diluted in 2% Tween 80 and saline. For intracranial injections, SB was diluted in 100% DMSO: while 100% DMSO is a high dose for intracranial, we29,43 and other groups67,68,69,70 have used this intracranial vehicle and shown that it does not have non-specific effects. Importantly, our studies are performed with a randomized, Latin-squares design, with alcohol drinking on days in between intracranial test sessions. Any possible lingering toxicity of DMSO should impact drinking on subsequent days, but this was not observed (see29). However, others have reported the possibility of DMSO-related damage71,72, and thus, despite within-animal comparisons used here and in our other studies, we cannot rule out the possibility of DMSO-related changes in animals studied here.
Statistics and analyses
The majority of comparisons were within-animal (vehicle vs drug). These were examined using paired t-test for normally distributed data and Wilcoxon matched-pair signed rank test (Wilcoxon) for non-normal data, and between-animal comparisons were examined using t-test, or Mann–Whitney test for non-normal data. We also performed two-way ANOVA with sex between-factor and vehicle/drug within-factor for all conditions where t-tests showed significant differences. In some comparisons, subject’s data were normalized to baseline to compare across sexes. We also analyzed the correlation between basal alcohol drinking levels and impact of a given treatment (determined as log [100 × (intake during drug treatment)/(intake during vehicle)], as in43, where log value of 2 (log [100]) indicates no treatment effect). Statistical comparisons were performed with GraphPad Prism or SPSS. Data are shown as mean ± SEM and scatter of raw data, and, for comparison, box-and-whiskers plots in grey, with bars showing 25%-75% range, whiskers showing 5%-95% range, and median shown by crossbar; some data point values are given in figure legend when much higher than other data.
Results
In agreement with previous findings14,15, female alcohol-only drinking was significantly higher (3.33 ± 0.17 g/kg) than male intake (2.38 ± 0.13 g/kg) (t55 = 3.881, p = 0.0003). In contrast, quinine-resistant consumption did not differ between sexes (females: 1.97 ± 0.21 g/kg; males: 1.99 ± 0.18 g/kg; t44 = 0.0780, p = 0.9369). Both were measured in the week before intracranial injections. Also, in separate groups of mice, we examined blood alcohol concentrations (BACs) achieved in male and female alcohol-drinking mice after a 2 h intake session. BACs strongly correlated with intake in both sexes (Suppl. Fig. 2). Females (n = 12) had an average of 4.02 ± 0.29 g/kg and 107.96 ± 15.62 mg% BAC, with correlation of p = 0.0473 (R2 = 0.3383). Males (n = 10) had an average of 2.68 ± 0.32 g/kg and 99.72 ± 23.44 mg% BAC, with correlation of p = 0.0002 (R2 = 0.8330). Thus, considering 80 mg% to reflect binge-level intake, both sexes on average showed binge-level consumption levels under the LDA alcohol-only intake model used here.
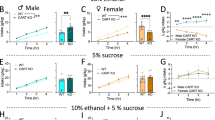
We have previously shown that shell Ox1Rs are critical for promoting binge alcohol drinking in male C57BL6/J mice, especially in higher-drinking subjects29,43, using the broadly utilized Ox1R blocker SB-334867 (SB) at a dose from many other studies29,58,61,73,74,75 (see “Material and methods”). Thus, we examined the impact of this SB dose in the shell on alcohol-only intake in female C57BL6/J mice. Interestingly, and unlike in males29,43, SB inhibition of Ox1Rs in the shell did not alter alcohol-only intake in females (Fig. 1A, n = 12, t11 = 0.1372, p = 0.8934), nor did it alter concurrent water intake (Fig. 1B, p = 0.8457, Wilcoxon). A two-way ANOVA found a significant sex-treatment interaction (F1,41 = 4.516, p = 0.040) but no overall treatment effect (F1,41 = 3.751, p = 0.060); data for male intra-shell SB binge drinking (n = 31) were from Lei et al.43, and the trend for treatment effect may reflect the larger sample size in males vs females.

Ox1R inhibition and regulation of binge and compulsion-like drinking in female C57 mice. (A,B) Intra-shell infusion of the Ox1R inhibitor SB did not alter (A) alcohol-only consumption under LDA (2-h per day drinking), nor (B) did it reduce concurrent water intake. (C,D) Systemic administration of a lower dose of SB (3 mg/kg) significantly reduced quinine-resistant alcohol drinking in both (C) females and (D) males. (E,F) However, lower dose SB did not significantly reduce concurrent water intake in either sex, suggesting a specific effect of systemic SB on compulsion-like alcohol consumption. One data point in (B) for SB was 32.7 ml/kg and in (F) for vehicle was 33.6 ml/kg, not shown on graphs. For Figs. 1, 2, 3 and 4, box-whisker plot (see “Material and methods”) is for the same data shown in adjacent colored bar. Aver-Resist: aversion-resistant drinking; Quin-Alc: quinine (100 µM) in alcohol. ** p < 0.01.
One possibility is that female alcohol intake might more generally occur without the need for Ox1Rs. Thus, we next examined whether quinine-resistant alcohol intake might be inhibited by an Ox1R blocker; we previously showed that a lower dose of SB (3 mg/kg, i.p.), which is low enough to assure specificity for Ox1Rs (see21), reduces quinine-resistant but not alcohol-only intake in male mice48. Also, male and female mouse quinine-resistant drinking under limited access shows similar sensitivity to quinine in alcohol15. Consistent with our previous findings in males48, quinine-resistant alcohol consumption was significantly reduced by systemic injection of 3 mg/kg SB in both females (Fig. 1C, n = 24, t23 = 0.4.524, p = 0.0002) and males (Fig. 1D, n = 22, t21 = 0.2.792, p = 0.0109), with a similar reduction in drinking across the sexes (Mann–Whitney p = 0.4816). A two-way ANOVA found a significant effect of treatment (F1,44 = 24.791, p < 0.001) but no sex-treatment interaction (F1,22 = 0.168, p = 0.684). These results suggest that, even though shell Ox1Rs did not regulate female alcohol-only intake, Ox1Rs can play a role in regulating at least some forms of alcohol drinking in females. In addition, 3 mg/kg systemic SB did not significantly alter concurrent water consumption during compulsion-like intake in females (Fig. 1E, p = 0.8695, Wilcoxon) nor males (Fig. 1F, p = 0.6473, Wilcoxon), indicating a specific effect of systemic SB on aversion-resistant alcohol intake rather than consumption more generally. The trend apparent in Fig. 1F is not significant, and our previous studies show no effect of 3 mg/kg SB on concurrent water drinking during compulsion-like drinking48. Taken together, these findings indicate that some forms of alcohol drinking (aversion-resistant intake) can require Ox1Rs in both females and males, but that shell Ox1Rs were not needed for female binge alcohol-only drinking, unlike in males29,43.
Previous studies have shown that a high systemic dose of SB (30 mg/kg) can reduce alcohol drinking in female mice49, although some have questioned the specificity of this dose (discussed in21). Thus, we examined the impact of this higher SB dose on female and male alcohol-only binge intake. 30 mg/kg systemic SB reduced binge alcohol drinking in both females (Fig. 2A, n = 12, p = 0.0068, Wilcoxon) and males (Fig. 2B, n = 12, p = 0.0122, Wilcoxon). A two-way ANOVA found a significant effect of treatment (F1,22 = 23.546, p < 0.001) but no sex-treatment interaction (F1,22 = 0.292, p = 0.594). However, this higher SB dose did not alter concurrent water intake in either sex (female: Fig. 2C, p = 0.3750: males: Fig. 2D, p = 0.2754; Wilcoxon). The decrease in intake with SB 30 mg/kg did not differ between females and males (female: − 34.9 ± 11%, male: − 26.8 ± 12%, intake level during SB exposure relative to vehicle; p = 0.7553 between sexes Mann–Whitney). Thus, alcohol-only binge intake in females can be regulated by Ox1Rs, in agreement with previous findings49, but our findings above suggest that this did not occur through shell Ox1Rs (Fig. 1A,B), unlike in males29,43.

Higher doses of systemic Ox1R inhibitor reduced alcohol-only drinking in both sexes. (A,B) Higher dose SB (30 mg/kg) given systemically reduced alcohol-only drinking in both (A) females and (B) males. (C,D) Systemic SB at this dose did not reduce concurrent water intake in either sex. *p < 0.05; **p < 0.01.
To examine whether more general shell activity might be critical for female alcohol binging, we examined whether inhibition of calcium-permeable AMPARs (CP-AMPARs) in shell would suppress binge intake. A number of studies have shown that a variety of alcohol drinking models induce expression of CP-AMPARs in the shell22,51,52,53, with unpublished findings suggesting their promotion of alcohol intake22. Thus, we tested whether intra-shell infusion of NASPM (20 µg/side), a widely used and validated CP-AMPAR blocker22 (see “Material and methods”), could reduce binge intake. This intracranial NASPM dose was used in many studies62,63,64,65, and this and higher doses within the NAcb decrease cocaine seeking, but have no effect on cocaine self-administration, sucrose seeking62,65 or behavioral flexibility66. We found that NASPM strongly and significantly reduced alcohol intake in both females (Fig. 3A, n = 12, t11 = 6.344, p < 0.0001) and males (Fig. 3B n = 9, t8 = 4.4, p = 0.0023). In strong contrast, intra-shell NASPM did not reduce concurrent water intake in females (Fig. 3C, p > 0.9999, Wilcoxon) nor males (Fig. 3D, t8 = 0.4294, p = 0.6789), suggesting a specific effect on alcohol consumption. A two-way ANOVA found a significant effect of treatment (F1,19 = 47.063, p < 0.001) and sex-treatment interaction (F1,19 = 11.726, p = 0.003). Also, while there was a trend for a stronger NASPM effect in females for alcohol drinking (female: 62.8 ± 8.3% decrease in intake; male: 42.5 ± 8.8% decrease in intake), this was not significant (p = 0.0955, Mann–Whitney test). Thus, although males but not females required shell Ox1Rs for binge alcohol drinking, shell activity through CP-AMPARs was essential for promoting binge intake in both sexes.

Shell CP-AMPAR inhibition reduced alcohol binging in both sexes. (A,B) Intra-shell infusion of the CP-AMPAR inhibitor NASPM significantly and strongly reduced alcohol-only drinking in both (A) females and (B) males. (C,D) Intra-shell NASPM did not reduce concurrent water intake in either sex. One data point for female concurrent water consumption in (C) was 22.2 ml/kg, not shown on graph. **p < 0.01.
To better understand the specificity of shell CP-AMPAR inhibition on consummatory behavior, we next examined whether NASPM in the shell could alter saccharin intake. However, unlike alcohol drinking, shell NAPSM did not alter saccharin drinking in females (Fig. 4A, n = 9, t8 = 1.677, p = 0.1320) or males (Fig. 4B, n = 9, p = 0.3008, Wilcoxon), nor did it alter concurrent water consumption (females: Fig. 4C, p = 0.8125; males: Fig. 4D, p = 0.1953; Wilcoxon). Together, these results strongly suggest that shell CP-AMPARs strongly and selectively reduced binge alcohol intake in both female and male mice.

Shell CP-AMPAR inhibition did not reduce saccharin intake in either sex. (A,B) Intra-shell infusion of NASPM did not reduce saccharin intake in either sex. (C,D) Intra-shell NASPM did not reduce concurrent water intake in either sex, although a trend is seen in males. One vehicle data point in (C) was 15.7 ml/kg, and one NASPM in (D) was 17.1 ml/kg, not shown on graphs. Saccharin intake was not different across sexes (p > 0.9 Mann–Whitney).
We previously found that shell Ox1Rs are particularly important for driving binge intake in higher-drinking male mice43, suggesting that a lack of overall effect of shell SB in females might mask individual differences. However, there was no significant relation between basal alcohol-only intake level (determined from vehicle injection test days) and the change in drinking with shell SB in females (Fig. 5A, p = 0.6129, R2 = 0.0266). Furthermore, the impact of shell NASPM was not related to basal drinking level in males (p = 0.8147, R2 = 0.0084) or females (p = 0.2739, R2 = 0.0267) (Fig. 5B), suggesting an impact across all individuals (and unlike shell Ox1R inhibition in males which primarily impacts higher drinkers43). We also note that the 3 mg/kg SB impact on quinine-resistant drinking was not related to basal intake level in females (p = 0.5542, R2 = 0.0161) or males (p = 0.1977, R2 = 0.0815) (Fig. 5C), and that there was no relation between basal intake and 30 mg/kg SB effects on alcohol intake in either sex (male: p = 0.0846, R2 = 0.2682; female: p = 0.7817, R2 = 0.0080) (Fig. 5D). Taken together, our results suggest that NASPM in the shell reduced alcohol-only binge drinking across all individuals of both sexes, and that the lack of impact of shell SB on female binge intake was unlikely due to differences in SB effects across individuals.

The impact of Ox1R or CP-AMPAR inhibition was minimally related to basal alcohol intake levels across individual mice. For theses analyses, basal intake in each mouse was determined from vehicle injection days, and the change in drinking with drug was determined by log transforming the percent change in drinking (see “Material and methods”). (A–D) Basal intake was not correlated with change in drinking after (A) shell SB infusion in females, (B) shell NASPM infusion in either sex, (C) systemic 3 mg/kg SB in either sex (quinine-resistant intake), or (D) systemic 30 mg/kg SB infusion in either sex.
Discussion
Binge alcohol drinking, with excessive levels of intake, is a potent and pernicious obstacle to treating AUD, and heavy drinking individuals are responsible for much of the considerable personal and social harm of AUD1,2,3,4,5,6. In addition, problem drinking in females has risen dramatically across recent years11,12,13, and thus it is critical to uncover mechanistic differences across the sexes in order to best develop effective therapies to treat AUD in females versus males. Our previous work has identified the importance of shell Ox1Rs and their role in promoting alcohol intake in higher-drinking male mice29,43. Given this and the observation that female mice drank more than males overall, it was originally predicted that females would be more dependent on shell Ox1Rs. However, here we show that shell Ox1Rs were not needed to promote alcohol-only drinking in female mice. However, aversion-resistant alcohol drinking was similarly reduced by lower doses of systemic Ox1R inhibition in males and females, showing that Ox1Rs can regulate at least some forms of alcohol intake in females. Similarly, higher systemic SB doses also reduced alcohol-only drinking in both sexes (see below). Furthermore, our findings indicate the critical importance of shell CP-AMPARs for supporting binge alcohol intake in both males and females, suggesting that shell mediation of binging was important for alcohol drinking in both sexes, but through complementary mechanisms. Also, this role of shell CP-AMPARs was specific for alcohol, as CP-AMPAR inhibition had no effect on concurrent water intake or saccharin drinking in either sex. Finally, blood alcohol measures confirm that, on average, male and female mice reached binge-level alcohol intake (Suppl. Fig. 2). Taken together, our results suggest that the shell was critical for promoting alcohol-only binge drinking in both sexes (e.g. through CP-AMPARs), but that shell Ox1Rs were important in males but not females (discussed further below).
One central finding of the present study is that shell Ox1Rs were not important for supporting female binge alcohol drinking, while our previous work underscores the critical role of shell Ox1Rs in male alcohol intake29,43. Orexin has long been recognized as an important regulator of alcohol behaviors21,38,39, with full consideration beyond the scope of this work. Previous studies have assessed the relative impact of OxRs across females and males, and the different impact of shell Ox1Rs in females and males could reflect differential function or expression across the sexes. Sex and estrous differences in orexin and OxRs have been observed in the hypothalamus76, but not in cortical and other subcortical areas76,77,78. There are also no sex differences in OxR regulation of morphine mesolimbic activation, sucrose intake, or stress-induced cocaine seeking79,80,81. Further, systemic Ox1R blockers reduce 2-bottle choice alcohol intake in both sexes45,49. However, Ox1R inhibition impacts operant-based alcohol intake in male alcohol-preferring P-rats82 with only a trend in female P-rats49. In outbred rats, Ox1R inhibition reduces alcohol seeking in males38, with effects in females only if alcohol is present83. Considerable future work will be required to disambiguate the number of possible mechanisms that might underlie sex differences in shell Ox1R regulation, including differences in Ox1R expression that might vary across cell types, differing release of orexin, potential orexin interactions with other neuromodulator systems (e.g. 84), and other possibilities. In addition, it would have been useful to examine another behavior in the present studies to confirm that shell SB used in female alcohol-drinking studies was functional. Importantly, shell SB experiments in females were done concurrently with male SB shell experiments43, giving confidence that shell SB was functional and able to modulate alcohol drinking under some conditions (males drinking under DID or intermittent access or LDA, the latter method used for the present studies)43. Nonetheless, it would be valuable in future studies to understand conditions under which female shell Ox1Rs are important for behavior, e.g. reinstatement of morphine CPP61.
We note that shell Ox1Rs were not required for female alcohol-only intake, unlike in males, but systemic inhibition of Ox1Rs utilizing lower doses of SB reduced compulsion-like alcohol drinking in both females and males (for the latter, see also48). In this regard, it is also interesting that, while males and females both exhibit reinstatement for cocaine and sucrose79,81, Ox1R inhibitors reduce both cue- and stress-related reinstatement in males, but only stress- and not cue-induced reinstatement in females79,81. Taken together, these findings might lead to the speculation that Ox1Rs in females are important for more stress-related behaviors (stress-induced reinstatement and compulsion-like intake) but are not involved in more basic behaviors (binge drinking, cued reinstatement), while Ox1Rs in males would be important for a broader range of motivated behaviors. However, across a large sample of mice, we find that shell Ox1Rs are primarily important for alcohol drinking in higher-drinking males, with lesser importance in moderate bingers43, perhaps consistent with previous findings that systemic inhibition of Ox1Rs reduces alcohol drinking in dependent but not non-dependent mice50 and in higher- but not lower-drinking rats and mice44,45,46. However, considerable additional studies would be required to address these sex-, basal-intake-, and challenge-related possibilities. We also note that a higher dose of SB, given systemically, did inhibit alcohol-only consumption in both females and males, in agreement with some previous studies45,49, although some have questioned the specificity of this high dose (see21); the brain site of these effects also remain open, since, in addition to Ox1Rs in the shell and medial prefrontal cortex29, Ox1Rs in central amygdala and ventral tegmental area also promote binge alcohol drinking in mice85. We also note that we did not perform a dose–response of systemic SB here. Our previous work performed a dose–response in male C57 mice for both alcohol-only LDA drinking and aversion-resistant drinking48, and our alcohol-only data in males show a similar dose–response for female C57 mice alcohol-only drinking in Anderson et al.49. Thus, here we used a lower SB concentration (3 mg/kg) for aversion-resistant drinking, which in males impacts quinine-resistant but not alcohol-only drinking48 and does not impact alcohol-only intake in females49. In addition, we tested 30 mg/kg SB for effects on alcohol-only drinking, since this is the only dose to significantly reduce alcohol-only drinking in female C57 mice in49, and this dose is very widely used as an effective dose with some selectivity of behavioral impact21.
As we reviewed elsewhere21, there are two endogenous peptides, orexinA and orexinB, and two orexin receptors, which can mediate orexinergic signaling. Orexin-2-receptors (Ox2Rs) have been related more to sleep, arousal and stress, while Ox1Rs are more related to addiction, reward and motivation. However, there clearly can be crossover, e.g. where Ox2Rs regulate operant responding for alcohol86. We found in mice that Ox2Rs in shell do not contribute to male binge-like alcohol drinking29, while systemic inhibition of Ox2Rs does not regulate male aversion-resistant intake48. Nonetheless, there are clear sex differences in regulation of stress and alcohol behaviors14,87, and thus future studies should examine whether Ox2Rs might play a role in females different from males. Further, while downstream signaling molecules of orexin in neurons are only partially understood, it would be valuable to identify orexin receptor linkage to protein kinase C and other orexin-modulated systems (e.g.88).
While shell Ox1Rs were not needed for female alcohol intake, inhibition of CP-AMPARs in the shell with the widely utilized NASPM significantly and strongly reduced alcohol intake in both males and females. Thus, shell CP-AMPARs were potent promoters of alcohol binging in both sexes. CP-AMPARs in the shell are apparent in relation to various challenges, and inhibition of shell CP-AMPARs can reduce several addiction-related behaviors (reviewed in22). Furthermore, our findings are consistent with previous studies showing51,52,53 and suggesting89,90 that several forms of alcohol exposure lead to the appearance of CP-AMPARs in the shell. It would be interesting in future studies to determine the nature of any molecular changes in CP-AMPARs in female compared to male alcohol-drinking mice. Importantly, while intra-shell inhibition of CP-AMPARs potently reduced binge alcohol intake in both sexes, these effects were specific for alcohol, since intra-shell NASPM did not significantly reduce concurrent water intake during alcohol drinking, nor did it decrease saccharin drinking (tested in a different cohort). Thus, our results support the importance of the shell for promoting binge alcohol drinking across sexes through CP-AMPARs.
We note that we did not assess the possible impact of estrous cycle on drinking in female mice. While hormonal changes can impact drinking levels under some conditions (e.g.91), several studies find that the estrous cycle can have limited influence on addiction-related behavior once established14,18,92,93, including binge-like drinking in female mice91 or compulsion-like behaviors for alcohol19. Thus, the findings of Satta and colleagues91 in particular suggest that female C57 mouse alcohol drinking does not vary across the intact estrous cycle. Nonetheless, future studies could examine how the influence of specific shell receptor types on alcohol drinking might vary across the estrous cycle.
Taken together, our findings here and elsewhere29,43 support the critical importance of the shell in driving binge alcohol drinking in female and male mice. However, there are important similarities and differences in underlying mechanisms, since CP-AMPARs were crucial in both sexes, while shell Ox1Rs are only required in higher-drinking male mice43. However, lower systemic doses of the Ox1R blocker SB inhibited compulsion-like alcohol drinking in both sexes, demonstrating that Ox1Rs can regulate at least some forms of pathological alcohol drinking in females similar to males. Since binge alcohol drinking is a strong contributor to the harms of human drinking1,2,3,4,5,6, and problem drinking in females has risen dramatically across recent years11,12,13, our findings uncovering mechanistic differences across the sexes may help to develop more effective sex-selective and -general therapies to treat AUD.
References
Moos, R. H. & Moos, B. S. Rates and predictors of relapse after natural and treated remission from alcohol use disorders. Addiction 101, 212–222 (2006).
Bouchery, E. E., Harwood, H. J., Sacks, J. J., Simon, C. J. & Brewer, R. D. Economic costs of excessive alcohol consumption in the U.S., 2006. Am. J. Prevent. Med. 41, 516–24 (2011).
Dawson, D. A., Grant, B. F. & Li, T. K. Quantifying the risks associated with exceeding recommended drinking limits. Alcohol Clin. Exp. Res. 29, 902–908 (2005).
Larimer, M. E., Palmer, R. S. & Marlatt, G. A. Relapse prevention. An overview of Marlatt's cognitive-behavioral model. Alcohol Res. Health 23, 151–60 (1999).
Rehm, J. et al. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 373, 2223–2233 (2009).
CDC, Excessive Drinking Costs U.S. $223.5 Billion. 2014, Center for Disease Control: Atlanta, GA.
Esser, M. B. et al. Prevalence of alcohol dependence among US adult drinkers, 2009–2011. Prevent. Chronic Dis. CDC 11, 140329 (2014).
Gowin, J. L., Sloan, M. E., Stangl, B. L., Vatsalya, V. & Ramchandani, V. A. Vulnerability for alcohol use disorder and rate of alcohol consumption. Am. J. Psych. 174, 1094–1101 (2017).
Grant, B. F. et al. Epidemiology of DSM-5 alcohol use disorder: Results from the national epidemiologic survey on alcohol and related conditions III. JAMA Psych. 72, 757–766 (2015).
Spanagel, R. Alcoholism: A systems approach from molecular physiology to addictive behavior. Physiol. Rev. 89, 649–705 (2009).
Grant, B. F. et al. Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: Results from the national epidemiologic survey on alcohol and related conditions. JAMA Psych. 74, 911–923 (2017).
White, A. et al. Converging patterns of alcohol use and related outcomes among females and males in the United States, 2002 to 2012. Alcohol Clin. Exp. Res. 39, 1712–1726 (2015).
Carvalho, A. F., Heilig, M., Perez, A., Probst, C. & Rehm, J. Alcohol use disorders. Lancet 394, 781–792 (2019).
Becker, J. B. & Koob, G. F. Sex differences in animal models: Focus on addiction. Pharmacol Rev 68, 242–263 (2016).
Sneddon, E. A., White, R. D. & Radke, A. K. Sex differences in binge-like and aversion-resistant alcohol drinking in C57BL/6J mice. Alcohol Clin. Exp. Res. 43, 243–249 (2019).
Hopf, F. W. Do specific NMDA receptor subunits act as gateways for addictive behaviors?. Genes Brain Behav. 16, 118–138 (2017).
Hopf, F. W. & Lesscher, H. M. Rodent models for compulsive alcohol intake. Alcohol 48, 253–264 (2014).
Randall, P. A., Stewart, R. T. & Besheer, J. Sex differences in alcohol self-administration and relapse-like behavior in Long-Evans rats. Pharmacol. Biochem. Behav. 156, 1–9 (2017).
Fulenwider, H. D., Nennig, S. E., Price, M. E., Hafeez, H. & Schank, J. R. Sex differences in aversion-resistant ethanol intake in mice. Alcohol. Alcohol 54, 345–352 (2019).
Sneddon, E. A., Ramsey, O. R., Thomas, A. & Radke, A. K. Increased responding for alcohol and resistance to aversion in female mice. Alcohol Clin. Exp. Res. (2020).
Hopf, F. W. Recent perspectives on orexin/hypocretin promotion of addiction-related behaviors. Neuropharm 168, 108013 (2020).
Hopf, F. W. & Mangieri, R. A. Do alcohol-related AMPA-type glutamate receptor adaptations promote intake?. Handb. Exp. Pharmacol. 248, 157–186 (2018).
Chaudhri, N., Sahuque, L. L. & Janak, P. H. Ethanol seeking triggered by environmental context is attenuated by blocking dopamine D1 receptors in the nucleus accumbens core and shell in rats. Psychopharm 207, 303–314 (2009).
Chaudhri, N., Sahuque, L. L., Schairer, W. W. & Janak, P. H. Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharm 35, 783–791 (2010).
Marchant, N. J. et al. Role of ventral subiculum in context-induced relapse to alcohol seeking after punishment-imposed abstinence. J Neurosci 36, 3281–3294 (2016).
Mundt, A. et al. High-frequency stimulation of the nucleus accumbens core and shell reduces quinpirole-induced compulsive checking in rats. Eur. J. Neurosci. 29, 2401–2412 (2009).
Piccoli, L. et al. Role of orexin-1 receptor mechanisms on compulsive food consumption in a model of binge eating in female rats. Neuropsychopharm 37, 1999–2011 (2012).
Barker, J. M., Taylor, J. R. & Chandler, L. J. A unifying model of the role of the infralimbic cortex in extinction and habits. Learn. Mem. 21, 441–448 (2016).
Lei, K. et al. Nucleus accumbens shell and mPFC but not insula orexin-1 receptors promote excessive alcohol drinking. Front. Neurosci. 10, 400 (2016).
Baldo, B. A. & Kelley, A. E. Amylin infusion into rat nucleus accumbens potently depresses motor activity and ingestive behavior. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1232–R1242 (2001).
Kasten, C. R. & Boehm, S. L. Intra-nucleus accumbens shell injections of R(+)- and S(-)-baclofen bidirectionally alter binge-like ethanol, but not saccharin, intake in C57Bl/6J mice. Behav Brain Res. 272, 238–247 (2014).
Lum, E. N., Campbell, R. R., Rostock, C. & Szumlinski, K. K. mGluR1 within the nucleus accumbens regulates alcohol intake in mice under limited-access conditions. Neuropharm 79, 679–687 (2014).
Ramaker, M. J., Strong-Kaufman, M. N., Ford, M. M., Phillips, T. J. & Finn, D. A. Effect of nucleus accumbens shell infusions of ganaxolone or gaboxadol on ethanol consumption in mice. Psychopharm 232, 1415–1426 (2015).
Rewal, M. et al. Alpha4-containing GABAA receptors in the nucleus accumbens mediate moderate intake of alcohol. J. Neurosci. 29, 543–549 (2009).
Stratford, T. R. & Wirtshafter, D. Opposite effects on the ingestion of ethanol and sucrose solutions after injections of muscimol into the nucleus accumbens shell. Behav. Brain. Res. 216, 514–518 (2011).
Wilden, J. A. et al. Reduced ethanol consumption by alcohol-preferring (P) rats following pharmacological silencing and deep brain stimulation of the nucleus accumbens shell. J. Neurosurg. 120, 997–1005 (2014).
Mahler, S. V., Moorman, D. E., Smith, R. J., James, M. H. & Aston-Jones, G. Motivational activation: a unifying hypothesis of orexin/hypocretin function. Nat. Neurosci. 17, 1298–1303 (2014).
Mahler, S. V., Smith, R. J., Moorman, D. E., Sartor, G. C. & Aston-Jones, G. Multiple roles for orexin/hypocretin in addiction. Prog Brain Res. 198, 79–121 (2012).
Lawrence, A. J., Cowen, M. S., Yang, H. J., Chen, F. & Oldfield, B. The orexin system regulates alcohol-seeking in rats. Br. J. Pharmacol. 148, 752–759 (2006).
Baimel, C. et al. Orexin/hypocretin role in reward: implications for opioid and other addictions. Br. J. Pharmacol. 172, 334–348 (2014).
Borgland, S. L. et al. Orexin A/hypocretin-1 selectively promotes motivation for positive reinforcers. J. Neurosci. 29, 11215–11225 (2009).
Cason, A. M. et al. Role of orexin/hypocretin in reward-seeking and addiction: implications for obesity. Physiol. Behav. 100, 419–428 (2010).
Lei, K. et al. Nucleus accumbens shell orexin-1 receptors are critical mediators of binge intake in excessive-drinking individuals. Front. Neurosci. 13, 88 (2019).
Alcaraz-Iborra, M. et al. Different molecular/behavioral endophenotypes in C57BL/6J mice predict the impact of OX1 receptor blockade on binge-like ethanol intake. Front. Behav. Neurosci. 11, 186 (2017).
Moorman, D. E. & Aston-Jones, G. Orexin-1 receptor antagonism decreases ethanol consumption and preference selectively in high-ethanol–preferring Sprague-Dawley rats. Alcohol 43, 379–386 (2009).
Moorman, D. E., James, M. H., Kilroy, E. A. & Aston-Jones, G. Orexin/hypocretin-1 receptor antagonism reduces ethanol self-administration and reinstatement selectively in highly-motivated rats. Brain Res 1654, 34–42 (2017).
Olney, J. J., Navarro, M. & Thiele, T. E. Binge-like consumption of ethanol and other salient reinforcers is blocked by orexin-1 receptor inhibition and leads to a reduction of hypothalamic orexin immunoreactivity. Alcohol Clin. Exp. Res. 39, 21–29 (2015).
Lei, K., Wegner, S. A., Yu, J. H. & Hopf, F. W. Orexin-1 receptor blockade suppresses compulsive-like alcohol drinking in mice. Neuropharm 110, 431–437 (2016).
Anderson, R. I., Becker, H. C., Adams, B. L., Jesudason, C. D. & Rorick-Kehn, L. M. Orexin-1 and orexin-2 receptor antagonists reduce ethanol self-administration in high-drinking rodent models. Front. Neurosci. 8, 33 (2014).
Lopez, M. F., Moorman, D. E., Aston-Jones, G. & Becker, H. C. The highly selective orexin/hypocretin 1 receptor antagonist GSK1059865 potently reduces ethanol drinking in ethanol dependent mice. Brain Res. 1636, 74–80 (2016).
Beckley, J. T., et al. The First Alcohol Drink Triggers mTORC1-Dependent Synaptic Plasticity in Nucleus Accumbens Dopamine D1 Receptor Neurons. J. Neurosci. 36, 701–13. PMC4719011 (2016).
Renteria, R., Buske, T. R. & Morrisett, R. A. Long-term subregion-specific encoding of enhanced ethanol intake by D1DR medium spiny neurons of the nucleus accumbens. Addict. Biol. 23, 689–698 (2017).
Laguesse, S., et al. Prosapip1-dependent synaptic adaptations in the nucleus accumbens drive alcohol intake, seeking, and reward. Neuron 96, 145–159 e8 (2017).
Lei, K., Wegner, S. A., Yu, J. H., Simms, J. A. & Hopf, F. W. A single alcohol drinking session is sufficient to enable subsequent aversion-resistant consumption in mice. Alcohol 55, 9–16 (2016).
Lesscher, H. M., van Kerkhof, L. W. & Vanderschuren, L. J. Inflexible and indifferent alcohol drinking in male mice. Alcohol Clin. Exp. Res. 34, 1219–1225 (2010).
Seif, T. et al. Cortical activation of accumbens hyperpolarization-active NMDARs mediates aversion-resistant alcohol intake. Nat. Neurosci. 16, 1094–1100 (2013).
Seif, T. et al. D-Serine and D-cycloserine reduce compulsive alcohol intake in rats. Neuropsychopharm 40, 2357–2367 (2015).
Brown, R. M. et al. Orexin-1 receptor signalling in the prelimbic cortex and ventral tegmental area regulates cue-induced reinstatement of ethanol-seeking in iP rats. Addict. Biol. 21, 603–612 (2015).
Smart, D. et al. SB-334867-A: the first selective orexin-1 receptor antagonist. Br. J. Pharmacol. 132, 1179–1182 (2001).
Carvajal, F. et al. Orexin receptor 1 signaling contributes to ethanol binge-like drinking: Pharmacological and molecular evidence. Behav. Brain Res. 287, 230–237 (2015).
Qi, K., Wei, C., Li, Y. & Sui, N. Orexin receptors within the nucleus accumbens shell mediate the stress but not drug priming-induced reinstatement of morphine conditioned place preference. Front. Behav. Neurosci. 7, 144 (2013).
Conrad, K. L. et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454, 118–121 (2008).
Russell, S. E. et al. Nucleus accumbens AMPA receptors are necessary for morphine-withdrawal-induced negative-affective states in rats. J. Neurosci. 36, 5748–5762 (2016).
Scheyer, A. F. et al. AMPA receptor plasticity in accumbens core contributes to incubation of methamphetamine craving. Biol. Psych. 80, 661–670 (2016).
White, S. L. et al. A critical role for the GluA1 accessory protein, SAP97 cocaine seeking. Neuropsychopharm 41, 736–750 (2016).
Ding, X. et al. N-methyl-D-aspartate receptor-mediated glutamate transmission in nucleus accumbens plays a more important role than that in dorsal striatum in cognitive flexibility. Front. Behav. Neurosci. 8, 304 (2014).
Simms, J. A., Haass-Koffler, C. L., Bito-Onon, J., Li, R. & Bartlett, S. E. Mifepristone in the central nucleus of the amygdala reduces yohimbine stress-induced reinstatement of ethanol-seeking. Neuropsychopharm 37, 906–918 (2011).
Pierce, R. C., Pierce-Bancroft, A. F. & Prasad, B. M. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/Mitogen-activated protein kinase signal transduction cascade. J. Neurosci. 19, 8685–8695 (1999).
James, M. H. et al. Orexin-1 receptor signalling within the ventral tegmental area, but not the paraventricular thalamus, is critical to regulating cue-induced reinstatement of cocaine-seeking. Int. J. Neuropsychopharmacol. 14, 684–690 (2011).
Vendruscolo, L. F. et al. Glucocorticoid receptor antagonism decreases alcohol seeking in alcohol-dependent individuals. J. Clin. Invest. 125, 3193–3197 (2015).
Poon, A. D. et al. Ca2+/calmodulin-dependent protein kinase II and dimethyl sulfoxide affect the sealing frequencies of transected hippocampal neurons. J. Neurosci. Res. 96, 1208–1222 (2018).
Tamagnini, F., Scullion, S., Brown, J. T. & Randall, A. D. Low concentrations of the solvent dimethyl sulphoxide alter intrinsic excitability properties of cortical and hippocampal pyramidal cells. PLoS ONE 9, e92557 (2014).
Espana, R. A. et al. The hypocretin-orexin system regulates cocaine self-administration via actions on the mesolimbic dopamine system. Eur. J. Neurosci. 31, 336–348 (2010).
Hollander, J. A., Lu, Q., Cameron, M. D., Kamenecka, T. M. & Kenny, P. J. Insular hypocretin transmission regulates nicotine reward. Proc. Natl. Acad. Sci. 105, 19480–19485 (2008).
Plaza-Zabala, A., Flores, A., Maldonado, R. & Berrendero, F. Hypocretin/orexin signaling in the hypothalamic paraventricular nucleus is essential for the expression of nicotine withdrawal. Biol. Psych. 71, 214–223 (2012).
Silveyra, P., Cataldi, N., Lux-Lantos, V. & Libertun, C. Role of orexins in the hypothalamic-pituitary-ovarian relationships. Acta Physiol. 198, 355–360 (2010).
Chen, C. T., Dun, S. L., Kwok, E. H., Dun, N. J. & Chang, J. K. Orexin A-like immunoreactivity in the rat brain. Neurosci. Lett. 260, 161–164 (1999).
Lee, J. S., Lee, E. Y. & Lee, H. S. Hypothalamic, feeding/arousal-related peptidergic projections to the paraventricular thalamic nucleus in the rat. Brain Res. 1598, 97–113 (2015).
Cason, A. M. & Aston-Jones, G. Role of orexin/hypocretin in conditioned sucrose-seeking in female rats. Neuropharm 86, 97–102 (2014).
Narita, M. et al. Direct involvement of orexinergic systems in the activation of the mesolimbic dopamine pathway and related behaviors induced by morphine. J. Neurosci. 26, 398–405 (2006).
Zhou, L. et al. Orexin-1 receptor mediation of cocaine seeking in male and female rats. J. Pharm. Exp. Ther. 340, 801–809 (2012).
Jupp, B., Krivdic, B., Krstew, E. & Lawrence, A. J. The orexin(1) receptor antagonist SB-334867 dissociates the motivational properties of alcohol and sucrose in rats. Brain Res. 1391, 54–59 (2011).
Dhaher, R. et al. The orexin-1 receptor antagonist SB-334867 reduces alcohol relapse drinking, but not alcohol-seeking, in alcohol-preferring (P) rats. J. Addict. Med. 4, 153–159 (2010).
Baimel, C., Lau, B. K., Qiao, M. & Borgland, S. L. Projection-target-defined effects of orexin and dynorphin on VTA dopamine neurons. Cell Rep. 18, 1346–1355 (2017).
Olney, J. J., Navarro, M. & Thiele, T. E. The role of orexin signaling in the ventral tegmental area and central amygdala in modulating binge-like ethanol drinking behavior. Alcohol. Clin. Exp. Res. 41, 551–561 (2017).
Brown, R. M., Khoo, S. Y. & Lawrence, A. J. Central orexin (hypocretin) 2 receptor antagonism reduces ethanol self-administration, but not cue-conditioned ethanol-seeking, in ethanol-preferring rats. Int. J. Neuropsychopharmacol. 16, 2067–2079 (2013).
Bangasser, D. A. & Wicks, B. Sex-specific mechanisms for responding to stress. J. Neurosci. Res. 95, 75–82 (2017).
Borgland, S. L., Taha, S. A., Sarti, F., Fields, H. L. & Bonci, A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron 49, 589–601 (2006).
Neasta, J., Ben Hamida, S., Yowell, Q., Carnicella, S. & Ron, D. Role for mammalian target of rapamycin complex 1 signaling in neuroadaptations underlying alcohol-related disorders. Proc. Natl. Acad. Sci. 107, 20093–8 (2010).
Liu, F. et al. mTORC1-dependent translation of collapsin response mediator protein-2 drives neuroadaptations underlying excessive alcohol-drinking behaviors. Mol. Psych. 22, 89–101 (2017).
Satta, R., Hilderbrand, E. R. & Lasek, A. W. Ovarian hormones contribute to high levels of binge-like drinking by female mice. Alcoh. Clin. Exp. Res. 42, 286–294 (2018).
Perry, A. N., Westenbroek, C. & Becker, J. B. The development of a preference for cocaine over food identifies individual rats with addiction-like behaviors. PLoS ONE 8, e79465 (2013).
Perry, A. N., Westenbroek, C., Jagannathan, L. & Becker, J. B. The roles of dopamine and alpha1-adrenergic receptors in cocaine preferences in female and male rats. Neuropsychopharm 40, 2696–2704 (2015).
Acknowledgements
Supported in part by NIAAA P50 AA017072. Raw data are available from corresponding author.
Author information
Authors and Affiliations
Contributions
C.K., K.L., F.W.H. contributed to conception and design of the study. C.K., K.L., J.Y., V.P., M.W., L.L., L.A., S.G., and F.W.H. contributed to data acquisition, analysis and interpretation. C.K., K.L., F.W.H. contributed to preparation of manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwok, C., Lei, K., Pedrozo, V. et al. Differential importance of nucleus accumbens Ox1Rs and AMPARs for female and male mouse binge alcohol drinking. Sci Rep 11, 231 (2021). https://doi.org/10.1038/s41598-020-79935-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79935-2
This article is cited by
-
Cocaine and amphetamine regulated transcript (CART) mediates sex differences in binge drinking through central taste circuits
Neuropsychopharmacology (2024)
-
Sleep dysregulation in binge eating disorder and “food addiction”: the orexin (hypocretin) system as a potential neurobiological link
Neuropsychopharmacology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
