
Abstract
Burkholderia cenocepacia (B. cenocepacia) is an opportunistic bacterium; causing severe life threatening systemic infections in immunocompromised individuals including cystic fibrosis patients. The lack of gasdermin D (GSDMD) protects mice against endotoxin lipopolysaccharide (LPS) shock. On the other hand, GSDMD promotes mice survival in response to certain bacterial infections. However, the role of GSDMD during B. cenocepacia infection is not yet determined. Our in vitro study shows that GSDMD restricts B. cenocepacia replication within macrophages independent of its role in cell death through promoting mitochondrial reactive oxygen species (mROS) production. mROS is known to stimulate autophagy, hence, the inhibition of mROS or the absence of GSDMD during B. cenocepacia infections reduces autophagy which plays a critical role in the restriction of the pathogen. GSDMD promotes inflammation in response to B. cenocepacia through mediating the release of inflammasome dependent cytokine (IL-1β) and an independent one (CXCL1) (KC). Additionally, different B. cenocepacia secretory systems (T3SS, T4SS, and T6SS) contribute to inflammasome activation together with bacterial survival within macrophages. In vivo study confirmed the in vitro findings and showed that GSDMD restricts B. cenocepacia infection and dissemination and stimulates autophagy in response to B. cenocepacia. Nevertheless, GSDMD promotes lung inflammation and necrosis in response to B. cenocepacia without altering mice survival. This study describes the double-edged functions of GSDMD in response to B. cenocepacia infection and shows the importance of GSDMD-mediated mROS in restriction of B. cenocepacia.
Similar content being viewed by others


Introduction
Burkholderia cenocepacia (B. cenocepacia) is an opportunistic aerobic Gram-negative facultative intracellular bacterium widely spread in the environment and can cause severe pulmonary infections in immunocompromised individuals and patients with underlying diseases such as cystic fibrosis (CF) and chronic granulomatous disease1. Treating such infections is challenging due to the evolving antimicrobial resistance of B. cenocepacia2. The infection state can further deteriorate to chronic lung inflammation and a life-threatening systemic infection known as cepacia syndrome, that in turn significantly decreases the patients’ survival1,3,4,5. Discovering alternative treatment targets to alleviate inflammation and resolve B. cenocepacia infections is crucial to the treatment of these severe and resistant infections.
Inflammasome activation is a fundamental mechanism for macrophages to respond to pathogens. The inflammasome promotes the activation and release of pro-inflammatory cytokines interleukin-1beta (IL-1β) and IL-18. This is associated with the formation of pores within the host plasma membrane via the protein Gasdermin D (GSDMD) which often precedes cell death6,7,8,9. GSDMD is expressed as an inactive auto-inhibitory form called GSDMD-full length (GSDMD-FL) which releases its inhibitory C-terminal domain upon activation. The liberated active N-terminal domains (GSDMD-NT) oligomerize and incorporate into the plasma membrane to form transmembrane pores10,11. When the GSDMD pores are excessive and repair mechanisms fail, cell death occurs. GSDMD is activated by caspase1 (CASP1) and/or caspase-11 (CASP11) via the canonical or non-canonical inflammasome pathway, respectively11. Recently, our research team has demonstrated that CASP11 is stimulated upon B. cenocepacia infection which can activate CASP1 in murine macrophages12,13. CASP11 was found to play a critical role in the restriction of many Gram-negative bacteria, including B. cenocepacia, within macrophages12,14. Nonetheless, the role of GSDMD in controlling B. cenocepacia infection is not yet explored.
This study demonstrates that despite lacking a direct bactericidal effect, GSDMD inhibits B. cenocepacia infection, independent from cell death. Instead, GSDMD activation mediates mitochondrial reactive oxygen species (mROS) production, which in turn stimulates the cellular autophagic machinery to degrade the invading bacteria in macrophages. Additionally, different B. cenocepacia secretory systems (T3SS, T4SS, and T6SS) contribute to GSDMD activation and hence bacterial clearance within macrophages. Moreover, GSDMD restricts B. cenocepacia replication while promoting inflammation and neutrophil recruitment in the lungs of infected mice. These results clarify the double-edged functions of GSDMD in response to B. cenocepacia.
Results
CASP11, CASP1, and bacterial secretory systems influence GSDMD cleavage in B. cenocepacia-infected macrophages
GSDMD-FL is inactive and upon cleavage by CASP1 and CASP11, the active NT cleaved form is released (GSDMD-NT)10,11. CASP11 cleaves GSDMD through the non-canonical inflammasome pathway. CASP11 has been shown to restrict Gram-negative bacteria such as B. cenocepacia, B. pseudomallei, Legionella pneumophila and Salmonella Typhimurium as well as Gram-positive bacteria12,14,15,16,17,18,19. In order to investigate whether GSDMD is cleaved during B. cenocepacia infection, bone marrow-derived macrophages from WT, gsdmd−/−, and casp11−/− mice were infected with B. cenocepacia at MOI of 10. At 6 h post-infection, casp11−/− macrophages displayed decreased cleavage of GSDMD-FL via Western blot in cell lysates, cell culture supernatants and cell lysates combined with supernatants (total) (Fig. 1A–C). These data indicate that CASP11 contributes to GSDMD cleavage during B. cenocepacia infection.

CASP11, CASP1, and bacterial secretory systems influence GSDMD cleavage in B. cenocepacia-infected macrophages. (A) Immunoblot analysis of GSDMD, CASP11, CASP1, IL-1β, CASP8, and GAPDH in whole cell lysate of WT, gsdmd−/−, and casp11−/− macrophages infected with B. cenocepacia (B.c.) (MOI10) at 6 h post-infection. (B) Immunoblot analysis of GSDMD, CASP11, CASP1, and IL-1β in cell supernatant (sup) from WT, gsdmd−/− and casp11−/− macrophages treated as in (A). (C) Immunoblot analysis of GSDMD, CASP11, CASP1, IL-1β, CASP8, and GAPDH in total cell lysate and sup from WT, gsdmd−/− , and casp11−/− macrophages treated as in (A). (D) Immunoblot analysis of GSDMD, CASP1, CASP11, IL-1β, and GAPDH in total cell lysate and sup from WT macrophages infected with WT B. cenocepacia, ∆T3SS, ∆T4SS, and ∆T6SS mutants (MOI10) at 6 h post-infection. (A–D) Representative blots from 3 independent experiments.
Previous studies have shown that the presence of cytosolic lipopolysaccharide (LPS) is responsible for the cleavage and auto-activation of CASP11 20,21. Consequently, the liberation of LPS from the phagosome harboring the pathogens is necessary for CASP11 cleavage22. Whether the cleavage of CASP11 post- B. cenocepacia infection is influenced by GSDMD has yet to be determined. Notably, we found that the total (cell lysate and supernatants) amount of cleaved CASP11 in response to B. cenocepacia is reduced in gsdmd−/− macrophages compared to WT macrophages (Fig. 1C). It is important to note that due to the absence of GSDMD pores in gsdmd−/− macrophages, the cleaved form accumulates inside the cell (Fig. 1A). Together, these results indicate that GSDMD contributes to CASP11 cleavage.
In addition to CASP11, GSDMD can also be cleaved by active CASP1 through the canonical inflammasome pathway15. CASP1 is active in casp11−/− and gsdmd−/− macrophages 6 h post- B. cenocepacia infection (Fig. 1A–C). Total amount of cleaved GSDMD is reduced in both casp11−/− and casp1−/− macrophages (Figs. 1C, S1A). This data indicate that during B. cenocepacia infection, GSDMD is cleaved by both CASP11 (non-canonical inflammasome) and CASP1 (canonical inflammasome).
CASP8 can mediate cell death in the absence of GSDMD23,24,25. To test if this is the case during B. cenocepacia infection, we determined whether CASP8 is cleaved at 6 h post- B. cenocepacia infection. Notably, cleaved CASP8 accumulated in gsdmd−/− but not in casp11−/− cell lysates (Fig. 1A). However, we found that total cleaved CASP8 is barely detected in gsdmd−/− and casp11−/− macrophage lysates combined with their supernatants (Fig. 1C).
Bacterial secretory systems (SS) release bacterial virulence factors26. These SS are essential for bacterial persistence within host cells. B. cenocepacia possesses different types (T) of SS including T3SS, T4SS, and T6SS27. The different SS of B. cenocepacia play a role in IL-1β activation13. To determine which SS contributes to GSDMD cleavage and inflammasome activation, WT macrophages were infected with WT B. cenocepacia and different SS mutants for 6 h at MOI of 10. Western blot analysis of the cell lysates combined with the cell culture supernatants (total) demonstrate that infection with SS−/− mutants led to decreased cleavage of GSDMD, CASP1, and IL-1β (Fig. 1D). T3SS−/− and T4SS−/− mutants exerted more cleavage of CASP11 (Fig. 1D). To further test whether different SS play a role in invasion or in the intracellular survival and replication within macrophages, we infected macrophages with the different B. cenocepacia mutants for 0.5, and 6 h then determined intracellular colony forming unit (CFU). Despite having fast growth rates in LB (Fig. S1B,C), all the mutants displayed defective replication within macrophages compared to the parental strain (Fig. S1D,E). This is independent of cell death as they cause less or equivalent cell death compared to WT B. cenocepacia (Fig. S1F). In contrast to the T6SS−/− , which shows increased invasion12, no difference in invasion between WT and T3SS−/− or T4SS−/− could be observed (Fig. S1D). Overall, these results indicate that, both host CASP11 and CASP1 in addition to bacterial T3SS, T4SS, and T6SS contribute to B. cenocepacia-induced GSDMD cleavage within macrophages.
GSDMD contributes to the restriction of B. cenocepacia in vitro
The role of GSDMD during infection differs according to the pathogen. For instance, it enhances Escherichia coli replication28, while it restricts the replication of B. thailandensis and Legionella pneumophila29,30. However, its distinctive role during B. cenocepacia infection is unknown. To unveil this, macrophages from WT, gsdmd−/− , and casp11−/− mice were infected with B. cenocepacia for 0.5, 3, and 6 h and macrophage associated colony forming units (CFU) were determined. Compared to WT macrophages, both gsdmd−/− and casp11−/− macrophages had similar levels of uptake as indicated by similar CFU at 0.5 h, however, they are more permissive to B. cenocepacia at 6 h post-infection (Fig. 2A). Additionally, using red fluorescent B. cenocepacia (K56-2), intracellular bacteria were enumerated after 2 and 6 h (Fig. 2B). Gsdmd−/− macrophages displayed significantly more B. cenocepacia intracellularly when compared to WT at 6 h post-infection (Fig. 2C). These results clearly show that GSDMD can efficiently restrict B. cenocepacia infection within macrophages.

GSDMD contributes to the restriction of B. cenocepacia in vitro. (A) Macrophage associated colony forming unit (CFU) of B. cenocepacia in WT, gsdmd−/−, and casp11−/− macrophages. Data represent mean ± SEM (n = 10). Statistical analysis was performed using two-way ANOVA. (B) Immunofluorescence images of macrophage associated with B. cenocepacia in WT and gsdmd−/− macrophages at 6 h post-infection. Scale bar: 50 µm. (C) Quantification of macrophage associated CFU of B. cenocepacia in WT and gsdmd−/− macrophages at 2 and 6 h post-infection (MOI10). Values are mean ± SEM calculated by scoring 20 randomly chosen fields of view from 7 independent experiments and then normalized to the number of cells. Statistical analysis was performed using two-way ANOVA. (D) Cytokine release from WT, gsdmd−/−, and casp11−/− macrophages infected with B. cenocepacia (MOI10) at 6 h post-infection. Data represent mean ± SEM (n = 20). Statistical analyses were performed using one-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
We showed previously that casp11−/− macrophages release less inflammasome-independent cytokines including KC (CXCL1) in their supernatants 6 h post- B. cenocepacia infection12, yet the mechanism of GSDMD involvement remains unknown. As a pore forming protein, GSDMD may affect the release of IL-1β in addition to inflammasome-independent cytokines. We examined if gsdmd−/− macrophages differentially release cytokines in comparison to WT and casp11−/− macrophages post- B. cenocepacia infection. Indeed, supernatants from gsdmd−/− macrophages elicited significantly less IL-1α, IL-1β, IL-6, and TNF-α when compared to WT and casp11−/− supernatants. Additionally, both casp11−/− and gsdmd−/− macrophages had significantly less CXCL1 release in comparison to WT (Fig. 2D). These results demonstrate that GSDMD can restrict B. cenocepacia and mediate the release of different inflammasome-dependent and -independent, inflammatory cytokines. Together, in the absence of GSDMD, there is more B. cenocepacia yet less cytokine secretion.
GSDMD restricts B. cenocepacia infection irrespective of cell death
Pyroptosis is an inflammatory, programmed cell death associated with excess GSDMD pore formation, and characterized by cytokine efflux, water influx, cell swelling, and finally osmotic lysis31,32. Cell death can deprive the pathogen from the niche required for its persistence and growth. To investigate if GSDMD-mediated restriction of B. cenocepacia is due to its function as an executer of pyroptotic cell death, the release of lactate dehydrogenase (LDH) was measured in supernatants of WT, gsdmd−/− , and casp11−/− macrophages at 6 h post- B. cenocepacia infection. Gsdmd−/− and casp11−/− macrophages exhibited lower levels of LDH released into supernatants when compared to WT (Fig. 3A), indicating that WT macrophage undergo more pyroptotic cell death compared to gsdmd−/− and casp11−/− macrophages. To examine if increased cell death in WT macrophages is responsible for B. cenocepacia restriction and not through other potential functions of GSDMD, glycine was used to protect WT macrophages from cell lysis. Glycine has cytoprotective properties against pyroptosis as it buffers the extracellular media and prevents ion influx, thereby inhibiting swelling and cellular lysis33,34,35,36,37. When treated with glycine, B. cenocepacia growth or gentamicin sensitivity in LB were not affected (Fig. S2A). WT, gsdmd−/− , and casp11−/− macrophages were infected for 6 h in the absence or presence of glycine (5 mM), and the release of LDH and GAPDH were determined. Infected WT macrophages, in the presence of glycine, displayed significantly lower levels of LDH and GAPDH release compared to those not treated with glycine. Thus, in the presence of glycine, there was no significant difference in the LDH and GAPDH release among WT, gsdmd−/− and casp11−/− macrophages (Fig. 3B,C). To determine if protection against pyroptosis affects the production and secretion of inflammatory proteins, WT, gsdmd−/− and casp11−/− macrophages were infected with B. cenocepacia in the absence or presence of glycine. Compared to the non-glycine treated macrophages, WT macrophages displayed decreased CASP1 and IL-1β in the cell supernatant. Conversely, they exhibited more expression of cleaved CASP11, CASP1 and IL-1β in cell lysate in the presence of glycine (Fig. 3C). These results indicated that glycine pre-treatment of macrophages can efficiently protect against pyroptosis during B. cenocepacia infection.

GSDMD restricts B. cenocepacia irrespective of cell death. (A) B. cenocepacia -induced cytotoxicity was calculated by measuring LDH release in supernatants from WT, gsdmd−/−, and casp11−/− macrophages at 6 h post- B. cenocepacia (B.c.) infection with (MOI10) (n = 7). Statistical analysis was performed using one-way ANOVA. (B) B. cenocepacia-induced cytotoxicity was determined as in (A) in presence and absence of glycine (5 mM) (n = 5). Statistical analyses were performed using two-way ANOVA. NS = not statistically significant. (C) Immunoblot analysis in cell supernatant (Sup) showing GAPDH, CASP1, and IL-1β and in whole cell lysate showing GSDMD, CASP1, CASP11, IL-1β, and GAPDH of WT, gsdmd−/−, and casp11−/− macrophages infected with B. cenocepacia (MOI10) at 6 h post-infection in presence and absence of glycine (5 mM). Representative blots from 3 independent experiments. (D) Macrophage associated colony forming unit of B. cenocepacia (MOI10) in WT and gsdmd−/− macrophages at 6 h post-infection in presence of glycine (5 mM). Data represent mean ± SEM (n = 6). Statistical analyses were performed using two-way ANOVA. (E) Cytokine release from WT, gsdmd−/−, and casp11−/− macrophages treated as in (D). Data represent mean ± SEM (n = 20). Statistical analyses were performed using two-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
We then determined if gsdmd−/− macrophages would still be more permissive to B. cenocepacia than WT cells during glycine treatment. Macrophages from WT and gsdmd−/− mice were infected with B. cenocepacia and CFU were determined at 0.5, 3, and 6 h in the presence of glycine. Both WT and gsdmd−/− macrophages exhibited similar invasion of B. cenocepacia. However, gsdmd−/− macrophages were more permissive at 6 h post-infection compared to glycine-protected WT cells (Fig. 3D). These results confirm that GSDMD plays a role in hindering B. cenocepacia replication in addition to its function in mediating cell death.
WT macrophages release more cytokines than gsdmd−/− macrophages (Fig. 2D), however, WT cells exhibited higher levels of cell death during B. cenocepacia infection which may lead to increased inflammatory cytokine release (Fig. 3A). Therefore, we sought to decipher the contribution of GSDMD pore formation or cell lysis to inflammatory cytokine release. To answer this question, we added glycine, which does not block GSDMD pores but does protect against lytic cell death, and collected supernatant 6 h post- B. cenocepacia infection38. Glycine treatment and the prevention of cell lysis reduced the amount of IL-1α, IL-1β, and IL-6 in gsdmd−/− supernatants (Fig. S2B). Glycine-treated gsdmd−/− macrophage supernatants still had significantly less IL-1α, IL-1β, and CXCL1 in comparison to supernatants of WT macrophages, but not TNF-α or IL-6, indicating that the release of these cytokines was not mainly dependent on cell lysis (Fig. 3E). In addition, WT cells exhibited similar amounts of IL-1α, IL-1β, and TNF-α release with and without treatment, but lower levels of CXCL1 and IL-6 in infection supernatants (Fig. S2B). Together, these results confirm that GSDMD promotion of IL-1α, IL-1β, and CXCL1 release is not dependent on cell lysis in vitro.
GSDMD restricts the replication and dissemination of B. cenocepacia in vivo
To test if GSDMD plays a role in B. cenocepacia replication in vivo, WT and gsdmd−/− mice were infected intratracheally with a sub-lethal dose (10 × 106 CFU/mouse) of B. cenocepacia. To verify if equal inoculums of bacteria were delivered to all mice, CFU enumeration was performed on the lung homogenates 4 h post-infection. There was no significant difference in the bacterial loads between WT and gsdmd−/− mice (Fig. 4A). However, after 48 h the bacterial burden was significantly increased in the lung, spleen, and liver of gsdmd−/− mice when compared to the corresponding WT mice (Fig. 4A). This indicates that GSDMD promotes the restriction of B. cenocepacia replication and prevents dissemination in vivo. To determine if the permissiveness of gsdmd−/− is associated with mortality, WT and gsdmd−/− mice were monitored after infection intratracheally with lethal and sub-lethal doses. Interestingly, no significant difference in the survival rate was detected between WT and gsdmd−/− mice in all the indicated doses (Fig. S3A–C), indicating that higher bacterial burden in the absence of GSDMD is not associated with increased mortality.

GSDMD restricts the replication and dissemination of B. cenocepacia in vivo. (A) In vivo CFU from lung, liver, and spleen of WT and gsdmd−/− mice infected with B. cenocepacia (10 × 106 CFU/mouse) at 4 h (n = 10) and 48 h (n = 13). Pooled data from 3 independent experiments are shown as mean ± SEM. Statistical analysis was performed using unpaired two-tailed student’s t-test. (B) Representative 2x (left panel), 4x (middle panel), 20x (right panel) magnification of H&E-stained lung sections from WT (upper panel) and gsdmd−/− (lower panel) mice (n = 5) treated as in (A) for 48 h. Black arrows show the inflammatory cells infiltration. (C) Scatter dot blot showing the difference in the histological stages (parenchymal inflammation, perivascular and peribronchiolar edema, inflammatory cells in blood vessel walls, alveolar macrophages, percentage of affected lung containing neutrophils/neutrophil clusters, percentage of affected lung composed of necrosis/necrotic debris) between lung sections from WT and gsdmd−/− mice (n = 5) treated as in (A) for 48 h. Statistical analysis was performed using Mann–whitney test. 0: Absent; 1: mild; 2: moderate; 3: marked; 4: severe; 5: fully. (D) Cytokine levels at 24 h in the bronchoalveolar lavage fluid (BALF) of WT and gsdmd−/− (n = 20) mice after intratracheally infection with 30 × 106 CFU/mouse. Statistical analysis was performed using two-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
To test if GSDMD plays a role in promoting inflammation in vivo, lung tissues were collected from WT and gsdmd−/− mice infected with 10 × 106 CFU/mouse for 48 h. Hemotoxylin and Eosin-staining was performed to assess morphological and histological changes. Overall, infection of WT lungs was accompanied with severe inflammatory infiltrates and a larger percentage of the lung tissue inflammation when compared to gsdmd−/− lungs (Fig. 4B). Semi-quantitative histopathological scoring demonstrates that the infected WT lung tissues have significantly more areas of inflammation, edema, and necrosis than their gsdmd−/− counterparts. Additionally, they exhibited greater neutrophil infiltration, and clusters mixed with necrotic debris in the parenchyma, affecting alveoli, capillaries and small blood vessels (Fig. 4B,C). Myeloperoxidase (MPO) is one of the degrading enzymes typically produced by neutrophils39,40. To determine if decreased neutrophil infiltration, in the absence of GSDMD, correlates with decreased cytokine and/or MPO release, we collected the bronchoalveolar lavage fluid (BALF) from WT and gsdmd−/− mice infected intratracheally with B. cenocepacia (30 × 106 CFU/mouse) for 24 h and measured various cytokines IL-1α, IL-1β, IL-6, TNF-α, CXCL1 in addition to MPO. There was no significant difference in all the measured cytokines in addition to MPO, except for CXCL1 which was less in gsdmd−/− BALF (Fig. 4D, S3D). Overall, these results show that GSDMD contributes to the restriction of B. cenocepacia replication, lung inflammation, and BALF CXCL1 production in vivo. However, the mechanism of GSDMD-mediated restriction of B. cenocepacia is still unclear.
GSDMD contributes to bactericidal mROS production during B. cenocepacia infection
One of the main challenges to the intracellular lifestyle of B. cenocepacia is the production of host reactive oxygen species (ROS)41,42,43,44, however it is unclear if GSDMD contributes to the production of mROS during B. cenocepacia infection and if it contributes to the restriction of the organism. Therefore, we used the antioxidant N-acetylcysteine (NAC) to determine if decreasing ROS will allow more B. cenocepacia replication within macrophages. Since NAC (3 mM) was found to have a direct effect on B. cenocepacia replication in LB (Fig. S4A), it was added to macrophages post. B. cenocepacia infection. Despite increasing cell death (Fig. S4B), scavenging ROS significantly increased B. cenocepacia replication in all the infected genotypes (Fig. 5A). This indicates that ROS manipulation can significantly affect B. cenocepacia survival within macrophages.


GSDMD contributes to bactericidal mROS production during B. cenocepacia infection. (A) Macrophage associated CFU of B. cenocepacia (B.c.) (MOI10) in WT, gsdmd−/−, and casp11−/− macrophages in presence and absence of N-acetylcysteine (NAC) (3 mM). Data represent mean ± SEM (n = 3). Statistical analysis was performed using two-way ANOVA. (B) Mitosox assay in WT, gsdmd−/−, and casp11−/− macrophages infected with B. cenocepacia (B.c.) (MOI10) at 6 h post-infection in absence or presence of glycine (5 mM). Data represent mean ± SEM (n = 8). Statistical analysis was performed using two-way ANOVA. (C) Fold change comparing the addition of Mitotempo (MT) (20 µM) treatment in macrophage associated CFU of B. cenocepacia (MOI10) in WT, gsdmd−/−, and casp11−/− macrophages at 6 h infection. Data represent mean ± SEM (n = 10). Statistical analysis was performed using two-way ANOVA. (D) TMRM assay in WT and gsdmd−/− macrophages infected with B. cenocepacia (MOI10) at 6 h post-infection. FCCP (uncoupler cause mitochondrial membrane potential (MMP) dissipation) (10 µM) was included as negative control. Fluorescence values were normalized to cell number as measured per well before normalization to the fluorescence of the non-infected wells. Data represent mean ± SEM (n = 12). Statistical analysis was performed using two-way ANOVA. (E) Cytokine release from WT, gsdmd−/−, and casp11−/− macrophages treated as in (C). Data represent mean ± SEM (n = 11). Statistical analysis was performed using paired one-tailed student’s t-test. (F) Immunoblot analysis of GSDMD in isolated phagosomes from WT, gsdmd−/−, and casp11−/− macrophages infected with B. cenocepacia (MOI10) at 6 h post-infection. (G) CFU of B. cenocepacia harvested from WT and gsdmd−/− macrophages and treated with H2O2 for 30 min before plating. Data represent mean ± SEM (n = 3). Statistical analysis was performed using two-way ANOVA. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
GSDMD inserts into mitochondrial membranes, when activated, contributing to mitochondrial ROS (mROS) release which is the major source of ROS in the cell 45. Thus, we postulated that gsdmd−/− macrophages produce less mROS during B. cenocepacia infection, contributing to B. cenocepacia permissiveness. We treated WT and gsdmd−/− macrophages with MitoSOX dye which selectively targets the mitochondria where it is oxidized, producing a highly fluorescent product that is used for mROS measurement. We found that WT and gsdmd−/− macrophages induced comparable mROS during B. cenocepacia infection (Fig. 5B). Yet, the difference in mROS production between the macrophage genotypes could be masked by earlier death of WT macrophages. Therefore, glycine was added and the amount of mROS was determined. Notably, gsdmd−/− and casp11−/− macrophages produced lower mROS in the presence of glycine when compared to non-pyroptotic WT cells (Figs. 5B, S4C). To further confirm if the defective mROS production associated with gsdmd−/− macrophages contributes to permissiveness to B. cenocepacia, Mitotempo (MT), a selective mitochondria-targeted antioxidant, was used46. MT does not induce macrophage death (Fig. S4D) and has no direct bactericidal effect against B. cenocepacia (Fig. S4E). WT, gsdmd−/−, and casp11−/− macrophages were infected with B. cenocepacia for 0.5, 3, and 6 h in the absence and presence of MT (20 µM) then, macrophage associated bacterial CFUs were enumerated. B. cenocepacia replication increased in all the infected macrophage phenotypes, however this increase was more significant in MT-treated WT macrophages compared to gsdmd−/− macrophages at 6 h post-infection (Fig. 5C). Together, these results demonstrate that mROS contributes to the restriction of B. cenocepacia and that WT macrophages produce more mROS than gsdmd−/− and casp11−/− macrophages during B. cenocepacia infection however, the mechanism is unclear.
GSDMD inserts pores in mitochondrial membranes which can increase the production of mROS by disrupting mitochondrial membrane potential (MMP) (ΔΨm)45,47,48,49,50,51,52. To determine if GSDMD disrupts MMP (ΔΨm) during B. cenocepacia. TMRM dye (10 nM) was added 0.5 h before the designated time point (6 h post- B. cenocepacia infection). This dye accumulates in a negatively charged polarized mitochondria giving orange fluorescence. On the other hand, when MMP is disrupted, the dye is dispersed throughout the cell cytosol and fluorescence levels drop dramatically. Accordingly, MMP was found to be higher in gsdmd−/− macrophages during B. cenocepacia infection in comparison to WT counterparts (Fig. 5D). This indicates that activation and cleavage of GSDMD during B. cenocepacia infection leads to disruption of MMP.
Previous studies demonstrated that in human peripheral blood mononuclear cells, mROS promotes production of pro-inflammatory cytokines53,54,55. This led us to explore if defective mROS production in gsdmd−/− macrophages during B. cenocepacia infection contributes to lower cytokine levels (Fig. 3E). By inhibiting mROS via MT, we detected significantly lower pro-inflammatory cytokines release (IL-1α, IL-1β, CXCL1, IL-6 and TNF-α) in the cell culture supernatants of WT murine macrophages 6 h post- B. cenocepacia infection (Fig. 5E). In addition, there was no significant difference in the release of cytokines, except for IL-6, in gsdmd−/− macrophages upon treatment with MT. These data indicate that GSDMD-induced mROS contribute to the secretion of pro-inflammatory cytokines from murine macrophages during B. cenocepacia infection.
GSDMD is recruited to the B. cenocepacia-containing phagosome but does not alter the integrity of the bacterial membrane
To determine if GSDMD co-localizes with B. cenocepacia, we purified phagosomes from B. cenocepacia-infected WT macrophages. We then used Western blot analysis to identify the presence of the cleaved GSDMD on the isolated phagosomes from WT macrophages infected with B. cenocepacia for 6 h (Fig. 5F). We found that cleaved but not full-length GSDMD is present on the phagosomal compartment of the B. cenocepacia-infected WT macrophages56,57. GSDMD was found to have direct bactericidal effect on the membranes of B. thailandensis, by attacking their membrane, thereby making them more vulnerable to H2O2 (2 mM)30,50,58. To test if the presence of GSDMD on the phagosomes containing B. cenocepacia renders the bacterium more susceptible to H2O2, WT and gsdmd−/− macrophages were infected with B. cenocepacia for 6 h in the presence of glycine before treating the harvested intracellular bacteria with different concentration of H2O2 for 30 min (2, 0.75, 0.5 mM), and the number of the surviving bacteria was determined (Fig. 5G). H2O2 significantly decreased the number of bacteria harvested from both WT and gsdmd−/− macrophages, indicating that there is no difference in the H2O2 susceptibility of intracellular B. cenocepacia in the presence of GSDMD. Therefore, we concluded that despite its presence on the phagosomes harboring B. cenocepacia, GSDMD does not have a direct bactericidal effect against B. cenocepacia within macrophages.
Gsdmd −/− mice and their derived macrophages exhibit less autophagosome formation during B. cenocepacia infection
Autophagy is a conserved pathway among eukaryotic cells that degrades cytosolic non-functional organelles, protein aggregates, phagocytosed particles, and intracellular pathogens. It plays an important role in B. cenocepacia clearance from macrophages59. Several studies suggested that GSDMD contributes to the release of mROS which mediates the stimulation of autophagy and bacterial clearance60,61,62. To test this association, WT macrophages were treated with the selective mROS inducer (MitoPQ) and the mROS inhibitor (MT) for 4 h. Autophagy flux was accessed using Bafilomycin A1 (BafA1) which was added 2 h before the designated time point17,63,64. Then, the detection of the cleavage and lipidation of LC3-I into LC3-II was determined. Compared to the non-treated cells, MT treatment led to loss of the autophagic flux in WT macrophages. Accordingly, increasing mROS production by MitoPQ was accompanied by increase of the autophagy flux (Fig. S5A,B). These results demonstrate that mROS positively regulate autophagy in macrophages.
On the other hand, we and others have demonstrated that B. cenocepacia inhibits autophagy regulatory gene expression59,65. Additionally, the reduced activation and release of the pro-inflammatory cytokines negatively regulate autophagy55,66. In gsdmd−/− macrophages, there is higher B. cenocepacia loads along with defective mROS production in addition to defective cytokine release. Together, this led us to hypothesize that these macrophages elicit defective autophagy, which in turn reduces B. cenocepacia clearance. Functional autophagy is depicted by the formation of autophagosomes, called puncta, which can be monitored using confocal microscopy12. To test our hypothesis, WT and gsdmd−/− macrophages were infected with B. cenocepacia and autophagosomes were tracked using an antibody against the autophagy marker LC3. Remarkably, gsdmd−/− macrophages had less B. cenocepacia co-localization with LC3 at 2, and 6 h post-infection compared to WT cells (Fig. 6A,B). Additionally, GSDMD deficiency led to significantly less LC3 puncta formation at 6 h post-infection (Fig. 6C). Furthermore, we quantified the LC3-II level in response to B. cenocepacia via western blot. Compared to corresponding WT macrophages, LC3-II was significantly reduced within gsdmd−/− macrophages (Fig. 6D, S5C). Therefore, GSDMD is required for autophagosome formation during B. cenocepacia infection in vitro.

Gsdmd−/− mice and their macrophages exhibit less autophagosme formation during B. cenocepacia infection. (A) Confocal images of LC3 (green) immunofluorescence assay of B. cenocepacia (B.c.) (red) (MOI10) infected WT and gsdmd−/− macrophages at 6 h post-infection. White arrows points to B. cenocepacia- LC3 co-localization. Scale bar: 10 µM. (B) Quantification of B. cenocepacia co-localized with LC3 in B. cenocepacia infected WT and gsdmd−/− macrophages treated as in (A). Data represent mean ± SEM (n = 5). Statistical analysis was performed using two-way ANOVA. (C) Quantification of total LC3 puncta in B. cenocepacia infected WT and gsdmd−/− macrophages treated as in (A) using ImageJ Software. Values are mean percentage ± SEM calculated by scoring 96 randomly chosen fields of view from 6 independent experiments. Statistical analysis was performed using paired two tailed Student’s t-test.(D) Immunoblot analysis of LC3 in whole cell lysate separated of WT and gsdmd−/− macrophages treated as in (A). (E) Immunoblot analysis of LC3 in lung homogenate separated of WT and gsdmd−/− mice after being infected intratracheally for 4 h by B. cenocepacia (10 × 106 CFU/mouse) (n = 5). (F) Densitometry analysis of immunoblots represented in (E). Data represent mean ± SEM (n = 5). Statistical analysis was performed using unpaired two-tailed student’s t-test *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
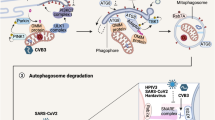
To determine if it plays a similar role in vivo, the lungs of WT and gsdmd−/− mice, infected intratracheally with B. cenocepacia (10 × 106 CFU/mouse) for 4 h, were homogenized and analyzed to determine the amount of LC3II. Compared to corresponding WT lungs, LC3-II was significantly reduced within gsdmd−/− lungs (Fig. 6E,F). Together these data show that during B. cenocepacia infection, autophagy is compromised in the absence of GSDMD both in vitro and in vivo. A schematic illustration of the interplay between GSDMD, mROS, autophagy, cytokines release and B. cenocepacia replication is suggested in (Fig. 7).

Schematic illustration of the interplay between GSDMD, mROS, autophagy, cytokines release and B. cenocepacia restriction. T: type, CASP11: Caspase11, CASP1: Caspase1, GSDMD: Gasdermin D, IL-1β: Interleukin-1β, IL-1α: Interleukin-1α, CXCL1: C-X-C Motif Chemokine Ligand 1, mROS: mitochondrial reactive oxygen species, ΔΨm: mitochondrial membrane potential, Ref: references.
Discussion
B. cenocepacia is a classic opportunistic pathogen characterized by an inherent multidrug resistance67. To this end, better understanding of host–pathogen interaction and the discovery of new host-based approaches that can help control this opportunistic pathogen are clinically exigent. Our latest findings have characterized the crucial role of CASP11 in the restriction of B. cenocepacia12. CASP11 and/or CASP1 can cleave and activate GSDMD11, yet, its role in B. cenocepacia infection is not defined. In this study, we identify the requirements for GSDMD activation in response to B. cenocepacia and the downstream effect on B. cenocepacia growth in vivo and in vitro.
GSDMD inhibits the replication of different bacteria including B. thailandensis19,29,30, but this is not the case for all bacteria. Particularly, GSDMD enhances the replication of other bacteria such as Escherichia coli28. As a pore forming protein that targets cardiolipin, GSDMD inserts into the mitochondrial membrane and disturbs the mitochondrial membrane potential, leading to mROS production during B. cenocepacia infection as reported for other microbes45,68. At present, several prominent reports demonstrated that mitochondria produce less mROS at high membrane potential which corroborates our findings with B. cenocepacia45,47,48,49,50,51,52. Subsequently, mROS can be delivered to bacteria-harboring phagosomes via mitochondrial vesicles contributing to bacterial clearance69,70. The bactericidal effect of mROS against different Gram-negative and Gram-positive bacteria including Salmonella Typhimurium71 and Staphylococcus aureus70, has been described, nevertheless, the contribution of GSDMD was not examined.
On the other hand, mROS were shown to stimulate autophagy, which is an important macrophage defense mechanism against intracellular bacterial pathogens including B. cenocepacia59,65,72. The activation of autophagy promotes the clearance of B. cenocepacia12, Legionella pneumophila18, Listeria monocytogenes73, and Mycobacterium tuberculosis74. Increased mROS production which activates autophagy improves the clearance of Mycobacterium tuberculosis75, Streptococcus pneumoniae76 and Listeria monocytogenes77. Here, we found that autophagy is defective in gsdmd−/− mice and their derived macrophages. Notably, increasing mROS production in macrophages stimulates autophagy and improved B. cenocepacia clearance. Remarkably, phagosomes isolated from B. cenocepacia-infected macrophages harbored cleaved GSDMD within their membranes. It is possible that the formation of GSDMD pore on phagosomal membranes recruits autophagy in an attempt to repair the pore as it happens at the cell membrane78. This will promote uptake by autophagosomes leading to bacterial degradation. Another possibility is that GSDMD pores contribute to the efflux of potassium K + 79 which promotes the activation of autophagy80. It is also possible that GSDMD pore, at the plasma membrane, mediates Ca2 + influx81 which may alter the activation of autophagy82. Therefore, the mechanisms by which GSDMD activation may promote autophagy activity are numerous.
On the other hand, GSDMD can exert direct bactericidal effect against the membranes of few Gram-negative bacteria within macrophages making them more susceptible to the effect of H2O2. These include B. thailandensis30, Salmonella Typhimurium58 and Escherichia coli50. Yet, B. cenocepacia harvested from WT macrophages were not more susceptible to H2O2 when compared to those obtained from gsdmd−/− macrophages. This could be attributed to either the presence of GSDMD on the phagosomal membrane without directly attacking the bacteria within this phagosomes as Liu et al. proposed50 and/or due to the thick polysaccharide on the membrane of B. cenocepacia as Byluand et al. suggested83 for other organisms.
Although gsdmd−/− mice exhibited increased bacterial load in different organs in our study, there was no significant difference in their survival compared to the WT control. Recent reports demonstrated that GSDMD similarly restricted bacteria, but in contrast to our study, GSDMD promoted survival of mice infected with B. thailandensis and Francisella novicida30,84,85. Although, increased bacterial loads are typically associated with decreased survival in mice12,86,87,88, this association is not consistent with inflammation89,90. As such, GSDMD can play a conflicting role during in vivo infections by promoting cytokine production, immune cell recruitment and tissue damage while restricting bacterial infection28. This explains why the absence of GSDMD does not alter the survival of the mice in response to elevated B. cenocepacia loads.
In agreement with this hypothesis, we found that WT lungs exhibited, histologically, more inflammation, neutrophils clustering, and tissue necrosis in comparison to gsdmd−/− lungs. This is consistent with what we found before in casp11−/− lungs in response to B. cenocepacia12. This can be attributed to the difference in cytokine release, especially the release of the pro-inflammatory cytokines which mediate recruitment of immune cells91. The role of CASP11 in the release of pro-inflammatory cytokines and chemokines in bronchoalveolar lavages (BALFs) varies upon infection with different bacterial pathogens. For instance, CASP11 deficiency during B. cenocepacia infection leads to less IL-1β, IL-1α, and CXCL1 production12. On the other hand, in B. thailandensis and Acinetobacter baumannii infection, IL-1β ' secretion in BALF is greatly affected by the lack of CASP1130,92. In a similar fashion, BALF from gsdmd−/− mice contains less IL-18 in response to B. thailandensis30. Here we found that, with the exception of CXCL1, which promotes neutrophil trafficking, IL-1β, IL-1α, IL-6, and TNFα were equivalently released in the lungs of the different mice strains which infected with B. cenocepacia93,94. Hence, it is plausible to conclude that the release of CXCL1 in the BALF during B. cenocepacia infection is GSDMD dependent, and promotes inflammation within the lungs. In vitro, GSDMD controls the release of not only the inflammasome-dependent cytokines (IL-1β and IL-1α) but also the independent one (CXCL1) in response to B. cenocepacia infection. We used glycine to rule out the possibility that cell death contributes to the increase release of cytokines in specific strains. This is in accordance with other studies, which used glycine as a cytoprotective agent, during Candida albicans36,95, Salmonella Typhimurium34,37, and B. thailandensis30 infections. Intracellularly, the lysate of gsdmd−/− macrophages exhibited an accumulation of mature IL-1β. This can be explained by the absence of GSDMD pores and CASP8 activation, which can also cleave proIL-1β96,97. Notably, we found increased CASP8 activation in response to B. cenocepacia infection in gsdmd−/− macrophages as reported in previous studies23,24,25,98. The intracellular accumulation of mature IL-1β in gsdmd−/− macrophages can lead to feedback inhibition of CASP1 cleavage and IL1β processing in response to B. cenocepacia. The lower occurrence of their cleaved forms in the total supernatant and cell lysate of gsdmd−/− macrophages, despite the increased bacterial load, lend weight to this hypothesis of a negative feedback loop, however, it requires further validation. Besides IL-1β, we found less IL-1α in the supernatants of gsdmd−/− macrophages indicating that GSDMD pore also plays a role in IL-1α release. GSDMD activation in response to B. cenocepacia infection may increase CASP11 expression and cleavage by the released IL-1α, IL-1β as we previously reported99. Furthermore, the cytokines released from WT macrophages (IL-1α, IL-1β, and CXCL1) were found to be decreased in WT supernatants after reducing mROS production in response to B. cenocepacia. This emphasizes the role of mROS in bolstering cytokine production53,54,55. Consequently, the aforementioned lower secretion of IL-1α, IL-1β, and CXCL1 in gsdmd−/− macrophages could be attributed to the defective mROS in these cells during B. cenocepacia infection. Additionally, defected autophagy in gsdmd−/− macrophages may contribute in the decreased secretion of cytokines to the extracellular milieu as it has been proposed before100.
It is worth noting that GSDMD cleavage could be downstream of ROS production during canonical inflammasome activation101. On the other hand, in non-canonical inflammasome activation, mROS are believed to be downstream of GSDMD which provides a link by which the non-canonical pathway may activate the canonical inflammasome45. The decreased expression of CASP1 together with defective mROS production in gsdmd−/− macrophages favors the idea that GSDMD cleavage occurs upstream of mROS production during B. cenocepacia infection.
Inflammasome activation can be affected by B. cenocepacia secretory systems (SS) through which, B. cenocepacia is able to secrete effector proteins inside the host cytosol12,56,102. T6SS, but not T3SS, was identified to be responsible in eliciting pyrin-dependent, inflammasome activation within human monocytes and THP-1 cells56. Furthermore, T3SS plays a role in IL-1β maturation13,103, in addition to its role in maintaining B. cenocepacia survival and replication in murine macrophages12. Here, we have demonstrated that, T3SS, T4SS, and T6SS of B. cenocepacia contribute to the cleavage of GSDMD, CASP1, and IL-1β in B. cenocepacia infected macrophages. Moreover, all of these SSs promote intracellular bacterial persistence within murine macrophages. These findings clarify the different activators and factors that affect inflammasome activation in response to B. cenocepacia infection.
Taken together, GSDMD mediates a variety of double-edged functions that may affect bacterial infection and inflammation in a contrasting manner. The recognition that GSDMD exerts cellular functions independently of cell death is gaining momentum and warrants further research.
Future directions
The interplay between mROS, GSDMD and autophagy during different infections may differ which licenses more investigation.
Materials and methods
Bacterial strains
Burkholderia cenocepacia (B. cenocepacia) K56–2 is a clinical isolate obtained from a CF patient who is not directly involved in the study104. Its derivate MH1K is a gentamicin-sensitive strain105. B. cenocepacia K56–2, used in immunofluorescence experiments, is complemented with a plasmid for red fluorescent protein (Ds-Red). B. cenocepacia ΔT3SS, ΔT4SS-1, and ΔT6SS mutants were kindly provided by Dr. Valvano at Queen’s University, Belfast, UK. All bacterial strains were grown overnight in LB medium at 37 °C and 200 rpm as previously described59,64,65,105.
Mice
C57BL/6 wild-type (WT) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Casp11−/− mice were generously given by Dr. Yuan at Harvard Medical School, Boston, MA, USA106. Gsdmd−/− mice were a gift from Dr. Thirumala-Devi Kanneganti at St. Jude Children’s Research Hospital, Memphis, TN, USA. Casp1−/−/casp11Tg mice were kindly provided by Dr. Vishva Dixit at Genentech, San Francisco, CA, USA. All mice were housed in a pathogen-free facility, and experiments were conducted with approval from the Animal Care and Use Committee at the Ohio State University (Columbus, OH, USA) which is accredited by AAALAC International according to guidelines of the Public Health Service as issued in the Guide for the Care and Use of Laboratory Animals (revised 1996).
Bone marrow-derived macrophages
For the generation of primary bone marrow-derived macrophages (BMDMs) from mice, tibias and femurs were flushed with IMDM medium (Thermo Fisher Scientific, 12440053) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, 16000044), 50% L cell-conditioned medium, 0.6 × MEM non-essential amino acids (Thermo Fisher Scientific, 11140050) and 0.1% penicillin and streptomycin (Thermo Fisher Scientific, 15140122). Cells were cultivated at least 6 days at 37 °C in a humidified atmosphere containing 5% CO2 as previously described12,17,19,105,107.
In vitro infection of primary macrophages
Prior to infection, macrophages were cultivated in IMDM medium supplemented with 10% fetal bovine serum for at least 1 h. In vitro infections were performed at MOI10:1, including centrifugation for 5 min at 30×g to synchronize the infection. Cells were infected with B. cenocepacia MH1K for 1 h followed by 30 min incubation with 50 µg/mL gentamicin (Thermo Fisher Scientific, 15750060) to avoid extracellular bacterial replication. To determine the macrophage associated colony forming unit (CFU), macrophages were lysed using 0.1% Triton X-100 (Fisher Scientific, BP151) in PBS at the indicated time points, and serial dilutions of the lysates were incubated on LB agar for 48 h. To protect against cell death, cells were incubated with 5 mM glycine (VWR Life Science, 76201-024) 1 h prior to infection and continuously until the end of the experiment. To inhibit cellular reactive oxygen species (ROS), macrophages were incubated with 3 mM N-Acetyl-l-cysteine (NAC) (ALX-105-005-G005) which was added 1 h post-infection and continuously until the end of the experiment. On the other hand, to specifically inhibit mitochondrial ROS, cells were incubated with 20 µM Mitotempo (Millipore Sigma, SML0737) which was added 1 h prior to infection and continuously until the end of the experiment. Additionally, to specifically induce mitochondrial ROS, cells were incubated with 1 µM MitoPQ (Abcam, ab146819). Furthermore, to inhibit autophagy in order to determine the autophagy flux, the cells were incubated with 100 nM Bafilomycin A1 (Baf A1) (Enzo, BML-CM110-0100) which was added 2 h before designated time point.
In vivo infection
For intratracheal infection, mice were anesthetized with Isoflurane and inoculated with 100 µL of phosphate-buffered saline (PBS; Thermo Fisher Scientific, 14190144) containing 10 × 106 CFU/mouse (organ CFU, histology) unless otherwise noted. To determine the bacterial load in organs, mice were sacrificed at 4 h and 48 h post-infection to collect lung, liver, and spleen for homogenization in PBS as previously described59,107. Data are presented as CFU per gram of organ. For survival experiments, animals were monitored daily for 6–8 days.
Phagosome enrichment
Medium was removed from seeded BMDMs (30 × 106 cells) and replaced with ice-cold PBS. Cells were harvested and washed twice with ice-cold PBS, resuspended in homogenization buffer (250 mM sucrose, 0.5 mM EGTA, 20 mM Hepes/KOH (pH 7.2)), and lysed in a Dura Grind stainless-steel homogenizer (VWR, 22877-280). The resulting cell homogenate was cleared by centrifugation, and organelles were fractionated in a discontinuous sucrose density gradient by ultracentrifugation at 100,000×g. Phagosomes were recovered from the interface of 55% and 65% sucrose layers, separated through a 15% Ficoll cushion at 18,000×g, and concentrated in a final centrifugation step at 18,000×g108,109.
Confocal microscopy
Macrophages were cultured on glass coverslips in 24-well plates and fixed with 4% paraformaldehyde for 20 min at the indicated time points. For permeabilization, cells were treated with ice cold methanol for 10 sec followed by 0.1% Triton X-100 for 20 min before blocking with 5% goat serum (Thermo Fisher Scientific, 16,210,064) in PBS. LC3A/B (Cell Signaling Technology, 4108) was visualized using goat anti-rabbit IgG secondary antibody conjugated to Alexa Fluor 488 (Molecular Probes, A-11008). Nuclei were stained with 1 µg/mL of 4ʹ,6ʹ-diamino-2-phenylindole (DAPI; Molecular Probes, D1306,) in PBS-5% goat serum for 15 min. Images were captured using a laser scanning confocal fluorescence microscope with a 60X objective (Olympus Fluoview FV10i) as previously described19,105. Intensities of LC3I/II were measured using ImageJ Software as previously described12.
Cytokine and MPO analysis
Cytokines in cell culture supernatants were measured by R&D Systems DuoSet ELISA Development Systems (murine IL-1α, DY400, murine IL-1β, DY401, murine IL-6, DY406, murine CXCL1/KC, DY453, murine TNF-α, DY410 and murine Myeloperoxidase (MPO), DY3667) according to the manufacturer’s instructions.
Histological analysis
Lungs were removed from infected mice, and fixed in 10% formalin at room temperature. Sample preparation, processing, hematoxylin & eosin staining, and semi-quantitative slide evaluation using ordinal grading scales was performed by the Histology laboratory within the Department of Veterinary Biosciences at The Ohio State University as previously described59.
Immunoblotting
Protein extraction from macrophages was performed using TRIzol reagent (Thermo Fisher Scientific, 15596026) according to the manufacturer’s instructions. Briefly, after phase separation using chloroform, 100% ethanol was added to the interphase/phenolchloroform layer to precipitate genomic DNA. Subsequently, the phenol-ethanol supernatant was mixed with isopropanol to isolate proteins. The Bradford method was used to determine protein concentrations in the cell lysate. Equal amounts of protein were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane. Membranes were incubated overnight with antibodies against GSDMD (Abcam, ab209845), CASP11 (abcam, ab180673), CASP1 (AdipoGen, AG-20B-0042-C100), murine IL-1b (R&D Systems, AF-401-NA), LC3A/B (Cell Signaling Technology, 12741) and GAPDH (Cell Signaling Technology, 2118). Corresponding secondary antibodies conjugated with horseradish peroxidase in combination with enhanced chemiluminescence reagent (Amersham, RPN2209,) were used to visualize protein bands. Densitometry analyses were performed by normalizing target protein bands to their respective loading control (GAPDH) using ImageJ software as previously described17,19.
LDH assay
LDH release from macrophages infected with B. cenocepacia was measured using the CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega, G7891) according to the manufacturer’s instructions. B. cenocepacia-induced LDH release [%] = ((infected sample-low control)/ (high control-low control))*100 (as described before12).
Mitochondrial ROS assay
To determine mitochondrial superoxide production, macrophages were incubated for 30 min with 2 µM MitoSOX dye (Thermo Fisher Scientific, M36008) diluted in cell imaging solution (formulated as previously described17) at 37 °C in a humidified atmosphere containing 5% CO2. Then cells were washed with PBS and further incubated with cell imaging solution. The fluorescence was read using a SpectraMax i3x microplate reader at 510/580 nm. Fluorescence values are reported as relative to the no treatment.
Mitochondrial membrane potential (MMP) assay
To determine mitochondrial membrane potential, macrophages were incubated for 30 min with 10 nM TMRM dye (Molecular Probes, Invitrogen, UK, T668) diluted in cell imaging solution (formulated as previously described17) at 37 °C in a humidified atmosphere containing 5% CO2. Then cells were washed with PBS and further incubated with cell imaging solution. As a positive control, cells were treated with 10 µM of FCCP (MMP uncoupler) (Millipore Sigma, C2759). The fluorescence was read using a SpectraMax i3x microplate reader at 548/574 nm. Fluorescence values were normalized to cell number as measured per well using a SpectraMax MiniMax 300 Imaging Cytometer, then reported as fold change to the no treatment.
B. cenocepacia susceptibility to H2O2
WT or gsdmd−/− macrophages were infected with B. cenocepacia (MOI10) for 6 h in the presence of glycine (5 mM). The infected cells were washed with PBS twice and lysed in PBS plus 2% saponin plus 15% BSA to harvest intracellular bacteria as previously described30. Lysates were treated for 30 min with PBS, H2O2 (2, 0.75, 0.5 mM) before plating them on LB agar plates30.
Statistical analysis
Data were analyzed using GraphPad Prism 8.3.0. All figures display mean and standard error of the mean (SEM) from at least three independent experiments as indicated in the figures’ legends. Comparisons between groups were conducted with either Student’s t-test, one-way or two-way ANOVA (depending on the data structure) followed by Holm’s adjustment for multiple comparisons as indicated12. Adjusted P < 0.05 was considered statistically significant.
Change history
08 June 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41598-021-92047-9
References
Scoffone, V. C. et al. Burkholderia cenocepacia infections in cystic fibrosis patients: Drug resistance and therapeutic approaches. Front. Microbiol. 8, 1592 (2017).
Scoffone, V. C. et al. Vaccines to Overcome Antibiotic Resistance: The Challenge of Burkholderia cenocepacia. Trends Microbiol. 28, 315–326 (2020).
Rhodes, K. A. & Schweizer, H. P. Antibiotic resistance in Burkholderia species. Drug Resist. Updat. 28, 82–90 (2016).
Dennis, J. J. Burkholderia cenocepacia virulence microevolution in the CF lung: Variations on a theme. Virulence. 8(6), 618–620 (2017).
Hauser, N. & Orsini, J. Cepacia syndrome in a non-cystic fibrosis patient. Case Rep. Infect. Dis. 2015, 537627 (2015).
Estfanous, S. Z. K., Ali, S. A., Seif, S. M., Soror, S. H. A. & Abdelaziz, D. H. A. Inflammasome genes’ polymorphisms in Egyptian chronic hepatitis C patients: Influence on vulnerability to infection and response to treatment. Mediat. Inflamm. 2019, 3273645 (2019).
Swanson, K. V., Deng, M. & Ting, J. P. Y. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 19(8), 477–489 (2019).
Lieberman, J., Wu, H. & Kagan, J. C. Gasdermin D activity in inflammation and host defense. Sci. Immunol. 4(39), 1447 (2019).
Rathkey, J. K. et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 3(26), 2738 (2018).
Aglietti, R. A. & Dueber, E. C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 38(4), 261–271 (2017).
Spel, L. & Martinon, F. Gasdermin D opens the way for NETs. Nat. Rev. Rheumatol. 14(12), 690–692 (2018).
Krause, K. et al. CASP4/caspase-11 promotes autophagosome formation in response to bacterial infection. Autophagy 14(11), 1928–1942 (2018).
Rosales-Reyes, R., Aubert, D. F., Tolman, J. S., Amer, A. O. & Valvano, M. A. Burkholderia cenocepacia type VI secretion system mediates escape of type II secreted proteins into the cytoplasm of infected macrophages. PLoS ONE 7(7), e41726 (2012).
Aachoui, Y. et al. Caspase-11 protects against bacteria that escape the vacuole. Science 339(6122), 975–978 (2013).
Ramos-Junior, E. S. & Morandini, A. C. Gasdermin: A new player to the inflammasome game. Biomed. J. 40(6), 313–316 (2017).
Thurston, T. L. M. et al. Growth inhibition of cytosolic Salmonella by caspase-1 and caspase-11 precedes host cell death. Nat. Commun. 7, 13292 (2016).
Krause, K. et al. Caspase-11 counteracts mitochondrial ROS-mediated clearance of Staphylococcus aureus in macrophages. EMBO Rep. 20(12), e48109 (2019).
Amer, A. O. & Swanson, M. S. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell. Microbiol. 7(6), 765–778 (2005).
Caution, K. et al. Caspase-11 and caspase-1 differentially modulate actin polymerization via RhoA and Slingshot proteins to promote bacterial clearance. Sci. Rep. 5, 18479 (2015).
Shi, J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514(7521), 187–192 (2014).
Ross, C., Chan, A. H., Von Pein, J., Boucher, D. & Schroder, K. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci. Alliance. 1(6), e201800237 (2018).
Mortellaro, A., Diamond, C., Khameneh, H. J. & Brough, D. Novel perspectives on non-canonical inflammasome activation. Immunol. Targets Ther. 4, 131–141 (2015).
Schneider, K. S. et al. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep. 21(13), 3846–3859 (2017).
Sarhan, J. et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 115(46), E10888–E10897 (2018).
Mascarenhas, D. P. A. et al. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog. 13(8), e1006502 (2017).
Green, E. R. & Mecsas, J. Bacterial secretion systems: an overview. Microbiol. Spectr. VMBF-0012-2015 (2016).
Loutet, S. A. & Valvano, M. A. A decade of Burkholderia cenocepacia virulence determinant research. Infect. Immun. 78(10), 4088–4100 (2010).
Kambara, H. et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 22(11), 2924–2936 (2018).
Gonçalves, A. V. et al. Gasdermin-D and Caspase-7 are the key Caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLOS Pathog. 15(6), e1007886 (2019).
Wang, J., Deobald, K. & Re, F. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J. Immunol. 202(12), 3468–3473 (2019).
Frank, D. & Vince, J. E. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 26(1), 99–114 (2019).
Pandeya, A., Li, L., Li, Z. & Wei, Y. Gasdermin D (GSDMD) as a new target for the treatment of infection. Medchemcomm. 10(5), 660–667 (2019).
Weinberg, J. M., Bienholz, A. & Venkatachalam, M. A. The role of glycine in regulated cell death. Cell. Mol. Life Sci. 73(11–12), 2285–2308 (2016).
Fink, S. L. & Cookson, B. T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. J. Immunol. 202(7), 1913–1926 (2006).
Cemma, M. & Brumell, J. H. Bacterial escape artists set afire. Science 339(6122), 912–913 (2013).
Krysan, D. J., Sutterwala, F. S. & Wellington, M. Catching fire: Candida albicans, macrophages, and pyroptosis. PLoS Pathog. 10(6), e1004139 (2014).
Loomis, W. P., den Hartigh, A. B., Cookson, B. T. & Fink, S. L. Diverse small molecules prevent macrophage lysis during pyroptosis. Cell Death Dis. 10(4), 326 (2019).
Evavold, C. L. et al. The pore forming protein gasdermin D regulates interleukin-1 secretion from living macrophages HHS Public Access. Immunity 48(1), 35–44 (2018).
Allen, R. C. & Stephens, J. T. Myeloperoxidase selectively binds and selectively kills microbes. Infect. Immun. 79(1), 474–485 (2011).
Haegens, A. et al. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 182(12), 7990–7996 (2009).
Saini, L. S., Galsworthy, S. B., John, M. A. & Valvano, M. A. Intracellular survival of Burkholderia cepacia complex isolates in the presence of macrophage cell activation. Microbiology 145(12), 3465–3475 (1999).
Keith, K. E. & Valvano, M. A. Characterization of SodC, a periplasmic superoxide dismutase from Burkholderia cenocepacia. Infect. Immun. 75(5), 2451–2460 (2007).
Keith, K. E., Killip, L., He, P., Moran, G. R. & Valvano, M. A. Burkholderia cenocepacia C5424 produces a pigment with antioxidant properties using a homogentisate intermediate. J. Bacteriol. 189(24), 9057–9065 (2007).
Rosales-Reyes, R., Skeldon, A. M., Aubert, D. F. & Valvano, M. A. The Type VI secretion system of Burkholderia cenocepacia affects multiple Rho family GTPases disrupting the actin cytoskeleton and the assembly of NADPH oxidase complex in macrophages. Cell. Microbiol. 14(2), 255–273 (2012).
Platnich, J. M. et al. Shiga toxin/lipopolysaccharide activates caspase-4 and gasdermin D to trigger mitochondrial reactive oxygen species upstream of the NLRP3 inflammasome. Cell Rep. 25(6), 1525-1536.e7 (2018).
Du, K., Farhood, A. & Jaeschke, H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol. 91(2), 761–773 (2017).
Kim, T. S. et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy. 15(8), 1356–1375 (2019).
Wang, J. S., Wu, D., Huang, D. Y. & Lin, W. W. TAK1 inhibition-induced RIP1-dependent apoptosis in murine macrophages relies on constitutive TNF-α signaling and ROS production. J. Biomed. Sci. 22(1), 1–13 (2015).
Rogers, C. et al. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun. 10(1), 1689 (2019).
Liu, X. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535(7610), 153–158 (2016).
Yu, J. et al. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 111(43), 15514–15519 (2014).
Belchamber, K. B. R. et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur. Respir. J. 54(4), 1802244 (2019).
Shi, Q. et al. Mitochondrial ROS activate interleukin-1β expression in allergic rhinitis. Oncol. Lett. 16(3), 3193–3200 (2018).
Bulua, A. C. et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208(3), 519–533 (2011).
Chen, Y., Zhou, Z. & Min, W. Mitochondria, oxidative stress and innate immunity. Frontiers in Physiology. 9, 1487 (2018).
Gavrilin, M. A. et al. Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J. Immunol. 188(7), 3469–3477 (2012).
Lamothe, J., Huynh, K. K., Grinstein, S. & Valvano, M. A. Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell. Microbiol. 9(1), 40–53 (2007).
Jorgensen, I., Zhang, Y., Krantz, B. A. & Miao, E. A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213(10), 2113–2128 (2016).
Abdulrahman, B. A. et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy. 7(11), 1359–1370 (2011).
Kim, J. H. et al. Mitochondrial ROS-derived PTEN oxidation activates PI3K pathway for mTOR-induced myogenic autophagy. Cell Death Differ. 25(11), 1921–1937 (2018).
Kim, S. H. et al. Mitochondrial ROS activates ERK/autophagy pathway as a protected mechanism against deoxypodophyllotoxin-induced apoptosis. Oncotarget 8(67), 111581–111596 (2017).
Filomeni, G., De Zio, D. & Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 22(3), 377–388 (2015).
Tian, L. et al. The cytotoxicity of coxsackievirus B3 is associated with a blockage of autophagic flux mediated by reduced syntaxin 17 expression article. Cell Death Dis. 9(2), 1–12 (2018).
Assani, K., Tazi, M. F., Amer, A. O. & Kopp, B. T. Correction: IFN-γ Stimulates Autophagy-Mediated Clearance of Burkholderia cenocepacia in Human Cystic Fibrosis Macrophages. PLoS ONE 14(2), e0213092 (2019).
Tazi, M. F. et al. Elevated Mirc1/Mir17-92 cluster expression negatively regulates autophagy and CFTR (cystic fibrosis transmembrane conductance regulator) function in CF macrophages. Autophagy. 12(11), 2026–2037 (2016).
Qian, M., Fang, X. & Wang, X. Autophagy and inflammation. Clin. Transl. Med. 6(1), 24 (2017).
Govan, J. R. W. & Deretic, V. Microbial pathogenesis in cystic fibrosis: Mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60(3), 539–574 (1996).
Russo, H. M. et al. Active Caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J. Immunol. 197(4), 1353–1367 (2016).
Geng, J. et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 16(11), 1142–1152 (2015).
Abuaita, B. H., Schultz, T. L. & O’Riordan, M. X. Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe. 24(5), 625-636.e5 (2018).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472(7344), 476–480 (2011).
Sun, Q., Fan, J., Billiar, T. R. & Scott, M. J. Inflammasome and autophagy regulation: A two-way street. Mol. Med. 23, 188–195 (2017).
Mitchell, G. et al. Listeria monocytogenes triggers noncanonical autophagy upon phagocytosis, but avoids subsequent growth-restricting xenophagy. Proc. Natl. Acad. Sci. USA 115(2), E210–E217 (2017).
Ashley, D. et al. Antimycobacterial effects of everolimus in a human granuloma model. J. Clin. Med. 9(7), E2043 (2020).
SanMartín, C. D., Böhme, D. & Rojas-Rivera, D. Calcium & ROS: Two orchestra directors for the requiem of death. Cell Calcium 85, 102113 (2020).
Dong, Y. et al. TLR4 regulates ROS and autophagy to control neutrophil extracellular traps formation against Streptococcus pneumoniae in acute otitis media. Pediatr. Res. (2020) (Under Press).
Sonoda, J. et al. Nuclear receptor ERRα and coactivator PGC-1β are effectors of IFN-γ-induced host defense. Genes Dev. 21(15), 1909–1920 (2007).
Rühl, S. et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362(6417), 956–960 (2018).
Broz, P., Pelegrín, P. & Shao, F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20(3), 143–157 (2020).
Kondratskyi, A., Kondratska, K., Skryma, R., Klionsky, D. J. & Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy. 14(1), 3–21 (2018).
Yang, X. et al. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity 51(6), 983-996.e6 (2019).
Bootman, M. D., Chehab, T., Bultynck, G., Parys, J. B. & Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus?. Cell Calcium 70, 32–46 (2018).
Bylund, J., Burgess, L. A., Cescutti, P., Ernst, R. K. & Speert, D. P. Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J. Biol. Chem. 281(5), 2526–2532 (2006).
Zhu, Q., Zheng, M., Balakrishnan, A., Karki, R. & Kanneganti, T.-D. Gasdermin D promotes AIM2 Inflammasome Activation and is required for host protection against Francisella novicida. J. Immunol. 201(12), 3662–3668 (2018).
Banerjee, I. et al. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 49(3), 413-426.e5 (2018).
Winter, C. et al. Important role for CC chemokine ligand 2-dependent lung mononuclear phagocyte recruitment to inhibit sepsis in mice infected with Streptococcus pneumoniae. J. Immunol. 182(8), 4931–4937 (2009).
Zou, S. et al. Contribution of progranulin to protective lung immunity during bacterial pneumonia. J. Infect. Dis. 215(11), 1764–1773 (2017).
Karlsson, E. A. et al. A perfect storm: Increased colonization and failure of vaccination leads to severe secondary bacterial infection in influenza virus-infected obese mice. MBio 8(5), e00889-e917 (2017).
Kuriakose, T. & Kanneganti, T. D. Gasdermin D flashes an exit signal for IL-1. Immunity 48(1), 1–3 (2018).
Van Der Windt, G. J. W. et al. CD44 deficiency is associated with increased bacterial clearance but enhanced lung inflammation during gram-negative pneumonia. Am. J. Pathol. 177(5), 2483–2494 (2010).
Napier, B. A. et al. Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 213(11), 2365–2382 (2016).
Wang, W. et al. Caspase-11 plays a protective role in pulmonary Acinetobacter baumannii infection. Infect. Immun. 85(10), e00350-e417 (2017).
Sawant, K. V. et al. Chemokine CXCL1 mediated neutrophil recruitment: Role of glycosaminoglycan interactions. Sci. Rep. 6, 33123 (2016).
Carpagnano, G. E. et al. Neutrophilic airways inflammation in lung cancer: The role of exhaled LTB-4 and IL-8. BMC Cancer. 11, 226 (2011).
Wellington, M., Koselny, K., Sutterwala, F. S. & Krysan, D. J. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot. Cell. 13(2), 329–340 (2014).
Chan, A. H. & Schroder, K. Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 217(1), e20190314 (2019).
Shenderov, K. et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1β in Response to TLR4 stimulation through a Caspase-8 and TRIF-dependent pathway. J. Immunol. 192(5), 2029–2033 (2014).
Malireddi, R. K. S., Kesavardhana, S. & Kanneganti, T. D. ZBP1 and TAK1: Master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front. Cell. Infect. Microbiol. 9, 406 (2019).
Caution, K. et al. Caspase-11 mediates neutrophil chemotaxis and extracellular trap formation during acute gouty arthritis through alteration of cofilin phosphorylation. Front. Immunol. 10, 2519 (2019).
Zhang, M., Kenny, S. J., Ge, L., Xu, K. & Schekman, R. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. Elife. 4, e11205 (2015).
Wang, Y. et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 11(12), 1069–1082 (2019).
Green, E. R. & Mecsas, J. Bacterial secretion systems: an overview. Microbiol. Spectr. 4(1), 32 (2016).
Aubert, D. F. et al. A Burkholderia type VI effector deamidates rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe. 19(5), 664–674 (2016).
Holden, M. T. G. et al. The genome of Burkholderia cenocepacia J2315, an epidemic pathogen of cystic fibrosis patients. J. Bacteriol. 191(1), 261–277 (2009).
Abdulrahman, B. A. et al. Depletion of the ubiquitin-binding adaptor molecule SQSTM1/P62 from macrophages harboring cftr ∆f508 Mutation Improves the Delivery of Burkholderia cenocepacia to the Autophagic Machinery. J. Biol. Chem. 288(3), 2049–2058 (2013).
Wang, S. et al. Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92(4), 501–509 (1998).
Akhter, A. et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity 37(1), 35–47 (2012).
Lührmann, A. & Haas, A. A method to purify bacteria-containing phagosomes from infected macrophages. Methods Cell Sci. 22(4), 329–341 (2000).
Magarian Blander, J. & Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440(7085), 808–812 (2006).
Acknowledgements
S.E. and A.B. are supported by are supported by a doctoral fellowship from the Egyptian Bureau of Higher Education. K.H. was supported by a Cure Cystic Fibrosis Columbus training grant and is now supported by a National Institutes of Health T32 training grant. K.K. was supported by a Cystic Fibrosis Foundation Post-doctoral research grant. Studies in the Amer laboratory are supported by NIAID R01 AI24121, NHLBI R01 HL127651-01A1 and C3 Cure Cystic Fibrosis Columbus. The graphical abstract was created with BioRender.com. The authors declare that they have no conflict of interests.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.O.A., S.E. and K.K.; Formal Analysis, S.E.; Investigation, S.E., K.K., M.A., A.A., M.E., K.H., A.B., K.D., C.C., M.A.G., H.H., S.S. and A.O.A.; Resources, A.O.A.; Writing—Original draft, S.E; Writing—Review & Editing, S.E., K.K., M.A., A.A., M.E., K.H., A.B., K.D., C.C., D.B., X.Z., M.A.G., H.H. , S.S. and A.O.A.; In vivo: S.E, M.A., K.C. Phagosome separation: S.E, H.W., T.L.; Statistics: X.Z. and S.E.; Project Administration, A.O.A. and S.E.; Supervision, A.O.A.; Funding Acquisition, A.O.A.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in Figure 6B where were the y-axis numbering “0, 10, 20, 30” was incorrectly given as “0, 1, 2, 3”.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Estfanous, S., Krause, K., Anne, M.N.K. et al. Gasdermin D restricts Burkholderia cenocepacia infection in vitro and in vivo. Sci Rep 11, 855 (2021). https://doi.org/10.1038/s41598-020-79201-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79201-5
This article is cited by
-
DNA hypomethylation promotes the expression of CASPASE-4 which exacerbates inflammation and amyloid-β deposition in Alzheimer’s disease
Alzheimer's Research & Therapy (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
