
Abstract
Arrhythmogenic right ventricular cardiomyopathy (ARVC), with skin manifestations, has been associated with mutations in JUP encoding plakoglobin. Genotype–phenotype correlations regarding the penetrance of cardiac involvement, and age of onset have not been well established. We examined a cohort of 362 families with skin fragility to screen for genetic mutations with next-generation sequencing-based methods. In two unrelated families, a previously unreported biallelic mutation, JUP: c.201delC; p.Ser68Alafs*92, was disclosed. The consequences of this mutation were determined by expression profiling both at tissue and ultrastructural levels, and the patients were evaluated by cardiac and cutaneous work-up. Whole-transcriptome sequencing by RNA-Seq revealed JUP as the most down-regulated gene among 21 skin fragility-associated genes. Immunofluorescence showed the lack of plakoglobin in the epidermis. Two probands, 2.5 and 22-year-old, with the same homozygous mutation, allowed us to study the cross-sectional progression of cardiac involvements in relation to age. The older patient had anterior T wave inversions, prolonged terminal activation duration (TAD), and RV enlargement by echocardiogram, and together with JUP mutation met definite ARVC diagnosis. The younger patient had no evidence of cardiac disease, but met possible ARVC diagnosis with one major criterion (the JUP mutation). In conclusion, we identified the same biallelic homozygous JUP mutation in two unrelated families with skin fragility, but cardiac findings highlighted age-dependent penetrance of ARVC. Thus, young, phenotypically normal patients with biallelic JUP mutations should be monitored for development of ARVC.
Similar content being viewed by others


Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC), which constitutes up to 20% of sudden cardiac death cases in individuals under 30 years of age, is an inherited cardiac disease characterized by progressive fibrofatty replacement of right ventricular myocardium that increases the risk of ventricular tachycardia and early-onset sudden death1. This disease has an autosomal dominant mode of inheritance with considerably reduced penetrance with an estimated prevalence of 1:1000 to 1:5000 in the general population2,3,4. ARVC shows extensive genetic heterogeneity and has been previously associated with 14 different genes. The most common causal genes mutated in about 60% of ARVC patients include PKP2, DSP, DSG2, DSC2, and JUP, which all encode desmosomal protein components. Rare non-desmosomal variants in TMEM43, TTN, TGFB3, RYR2, PLN, LMNA, DES, and CTNNA3 comprise less than 1% of all ARVC cases and have been reported in only a few families or clustering in specific areas, such as TMEM43 in Newfoundland, Canada, and PLN in The Netherlands5,6.
While monoallelic mutations in desmosomal protein-encoding genes, such as DSC2, DSP and JUP, have been associated with isolated (non-syndromic) ARVC, biallelic mutations in these genes have been reported with concomitant phenotypes of ARVC and integumentary abnormalities, such as palmoplantar keratoderma (PPK) and woolly hair and/or alopecia7,8,9,10 (Table 1). The cardiocutaneous syndrome, Naxos disease, results from a two-base pair homozygous deletion in the plakoglobin gene, while its related variant, Carvajal syndrome, which shows greater left ventricular predominance, is due to a mutation in the desmoplakin gene. Furthermore, biallelic JUP mutations were reported in association with skin fragility in the spectrum of epidermolysis bullosa (EB), PPK, woolly hair and/or alopecia, and nail dystrophy. The prevalence of cardiac manifestations in these patients has remained uncertain because ARVC is a progressive disease that often does not become manifest until the 3rd or 4th decade of life. Patients with homozygous mutations, however, present earlier with 100% penetrance by adolescence11. Although the original report of biallelic JUP mutations indicated a complete penetrance in cardiac manifestations, the subsequent studies have reported variable outcomes: among seven patients reported in one study, only one patient progressed to left-sided dilated cardiomyopathy (DCM) by 19 years of age11,12; demise of two patients early after birth has been reported13,14; and several patients presented with neither ARVC nor any other cardiac abnormalities15. Therefore, genotype–phenotype correlations regarding the penetrance, age of onset, and variable phenotypes, specifically cardiac involvement in biallelic loss-of-function (LOF) JUP mutations has not been well established (Table 1).
JUP encodes junction plakoglobin, also known as γ-catenin, an intracellular adapter protein that connects intermediate filaments to cadherins in desmosomes. Cell–cell adhesion complexes are abundant in tissues subjected to high levels of mechanical stress, such as the epidermis and the heart muscle16. In cardiomyocytes, plakoglobin plays a key role not only for anchorage of the myofibrillar apparatus to adherens junctions but also for myofibrillar compliance. Abnormal plakoglobin can cause desmosomal dysfunction, elevated degree of cardiac muscle stiffness, and increased total wall stress17.
As part of an effort to characterize a large cohort of patients with skin fragility disorders in the spectrum of EB patients in Iran, a country consisting of 80 million inhabitants with approximately 38% consanguineous marriages, patients with clinical presentations suggestive of different EB subtypes were subjected to genome-wide sequence analyses. Among the 362 genetically distinct families diagnosed with EB, two patients with a homozygous JUP mutation were found and are reported here.
Methods
Patient data
This study was approved by the Institutional Review Board of the Pasteur Institute of Iran. All subjects and the parents of child-subjects gave written informed consent to participate in this research and to allow their image to be published. All methods were performed in accordance with the relevant guidelines and regulations. A total of 362 patients from Iran were subjected to mutational analysis. The criterion for study inclusion was a tentative diagnosis of EB based on history of neonatal cutaneous blistering suggestive of a genetic skin fragility disorder18,19,20.
Targeted next-generation sequencing panel
We developed a disease-specific, gene-targeted panel for next-generation sequencing to identify pathogenic variants in EB. This panel includes 21 distinct genes, 18 of which have been shown to harbor causative mutations in EB and three of which have been implicated in differential diagnosis of skin fragility disorders18. We tested this panel in a group of patients who are representative of a large multiethnic cohort of EB patients in Iran. Most patients had an undefined EB subtype, and in some cases only archived DNA was available. DNA was extracted from peripheral blood samples, taken from patients, their parents, and other clinically affected and unaffected family members, including siblings (if available), using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA) or the salting-out method. For DNA, target enrichment was performed using the TruSeq Custom Amplicon kit (Illumina Inc., San Diego, CA). DesignStudio (Illumina Inc.) was used for library design. All exons, at least 20 base pairs of the intron at each intron–exon boundary, and up to 50 base pairs of 5′- and 3′-UTRs were targeted. The designed library contained the genes CD151, CDSN, CHST8, COL17A1, COL7A1, DSP, DST, EXPH5, FERMT1, ITGA3, ITGA6, ITGB4, JUP, KRT5, KRT14, LAMA3, LAMB3, LAMC2, PKP1, PLEC, and TGM5, divided into 421 targets covered by 968 amplicon probes which were designed to cover 99% of the targeted bases. A total of 36,724,892 reads were aligned to the human genome, with the mean coverage of the target region being × 42418.
Mutation analysis and transcriptome profiling of JUP gene expression by RNA-Seq
Total RNA was extracted from whole skin biopsy obtained from Patient 2 and from six healthy controls using TRIzol Reagent and quantified on a Nanodrop ND-100 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE), followed by RNA quality assessment on an Agilent 2200 TapeStation (Agilent Technologies, Palo Alto, CA). Multiplexed library construction, workflow analysis, and sequencing runs were performed following standard Illumina protocols (Illumina, Inc., San Diego, CA) using the TruSeq Stranded Total RNA kit with indexes from Set A (Cat #: RS-122-2301). Paired-end (2 × 75) sequencing reads were generated on the Illumina NextSeq 500 platform and stored in FASTQ format. FASTQ quality was checked using FastQC, and the TruSeq RNA adapter sequences were removed by Trimmomatic. Alignment and mapping were performed using STAR-2pass (v. 2.5.3a) (https://github.com/alexdobin/STAR/releases) with the human reference genome (GRCh38/hg38) and GENCODE V27 annotations. In the first pass, an initial alignment was executed, and the splice junction information was collected. This information was then used for the second pass, in which the final alignment was performed. The alignments were sorted, the read group was added, and the corresponding index was created (using Picard tools). The static images of Sashimi plots were generated using the Integrative Genomic Viewer (https://software.broadinstitute.org/software/igv/. R programming language (https://www.r-project.org/) was used to perform the expression analysis and plotting to show the distribution range of expression values of several EB related genes and representative housekeeping genes in the patients versus the six controls21,22.
Variant interpretation
After mapping the RNA-Seq results with human reference genome (GRCh38/hg38), the annotated CSV files of whole transcriptome sequencing from the proband was obtained for further analysis. To find the runs of homozygosity (ROH), we set a threshold of > 2 Mb and selected non-frameshift variants from the CSV file. Further, the sequence variants were filtered from the VCF files for missense, nonsense, and splice site-affecting variants. Indel variants were filtered for exonic in-frame insertions and deletions, frameshift mutations, and gained/lost start or stop codon. Additionally, only variants with total frequencies of < 0.001 or those without frequency data available in Genomes, gnomAD and ExAC databases were examined23.
Homozygosity mapping
The single nucleotide polymorphism variants called from RNA-Seq data were used for homozygosity mapping by PLINK 1.90 beta (http://zzz.bwh.harvard.edu/plink/), and were then overlapped with ROHs in the patient’s genome. Considering the given phenotype, segregation analysis in the family, and the presence of several ROHs in the patient with consanguineous parents enabled us to identify a likely pathogenic homozygous variant in JUP in the proband24.
Transmission electron microscopy
Skin biopsy specimens were cut into small pieces (< 1 mm3) and fixed in half-strength Karnovsky fixative for 4 h at room temperature. After being washed in 0.1 M phosphate buffer (pH 7.4), the samples were immersed in 1.3% aqueous osmium tetroxide (TAAB Laboratories, Berkshire, UK) for 2 h, incubated in 2% uranyl acetate (Bio-Rad, Hertfordshire, UK), dehydrated in a graded ethanol series, and then embedded in epoxy resin using propylene oxide (TAAB Laboratories). Ultra-thin sections were stained with uranyl acetate and lead citrate and examined in a Philips CM10 transmission electron microscope (Philips, Eindhoven, The Netherlands)25.
Immunofluorescence microscopy
Skin sections (5 μm) from biopsy from traumatized area obtained from the patient 1 were air-dried and blocked with diluted normal goat serum (Sigma-Aldrich, Dorset, UK), and then incubated with mouse monoclonal anti-gamma Catenin antibody (Abcam, ab11506) diluted 1:250 in phosphate-buffered saline with 30% w/v bovine serum albumin (Sigma-Aldrich). At next step, after washing in phosphate-buffered saline, we labeled slides with fluorescein isothiocyanate secondary antibodies (Invitrogen, Paisley, UK). The sections were counterstained with DAPI. By excluding the primary antibody, negative controls were also generated for this study. We used the same camera and the exposure time (3 s) for image acquisition of all sections26.
Results
Case report
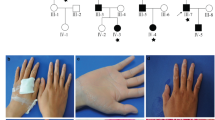
Patient 1 was a 2.5-year-old male born at the 34th gestational week to healthy second cousin once removed consanguineous parents with Iranian-Azeri ancestry (Figs. 1 and 3a). He presented at birth with ulcers and erosions on the perioral and sacral areas. Several days after birth, an ulcer located posterior to the right internal malleolus developed (Fig. 1). At current examination, he presented with scattered erosions and ulcers of the skin, particularly in perioral and axillary areas, as well as plantar fissures; mucosal erosions; palmoplantar keratoderma (PPK); nail dystrophy proceeding to nail loss; and absent scalp hair, eyebrows, and eyelashes (Fig. 1). He exhibited failure to thrive and turned cyanotic while crying.

Clinical findings in patients with JUP-associated spectrum of skin fragility. Patient 1 (upper panel): A 2.5-year-old male manifested with trauma-induced blisters and erosions of the skin; perioral, axillary, plantar, and antecubital fissures; plantar hyperkeratosis; and absence of hair on his scalp. Patient 2 (lower panel): A 22-year-old female showed trauma-induced blisters and erosions; hyperkeratosis of palms, soles, knees and elbows; joint contractures of fingers; and absence of hair on her scalp.
The laboratory tests revealed a normal serum zinc level. The electrocardiogram (EKG) and 24-h Holter monitoring showed sinus tachycardia with inverted T waves in right precordial leads (not shown). Transthoracic echocardiogram displayed normal ventricular ejection fraction, chamber size and motion. The parasternal long and short axis views of right ventricular outflow tract size (PLAX RVOT/BSA; PSAX RVOT/BSA) were normal (12.8 mm/m2 and 12.6 mm/m2, respectively). No cardiac abnormalities were found in the parents. The patient met one major criterion (biallelic JUP mutation) based upon the 2010 modified Task Force Criteria establishing a possible diagnosis of ARVC27.
Patient 2 was an Iranian-Azeri 22-year-old female born at the 33rd week of gestation to healthy second-cousin consanguineous parents (Fig. 3a). At birth, erosions on the scalp, alopecia, absence of eyebrows and eyelashes, dystrophic nails, and left-sided congenital clubfoot were noted. At current examination she presented with manifestations similar to Patient 1, including fragile skin; fissures at skin folds, periorificial areas, and the lateral canthus of the eyes; PPK; hyperkeratotic plaques and follicular hyperkeratosis on the knees and elbows; subungual hyperkeratosis; and nail dystrophy (Fig. 1). Alopecia, sparse curly eyebrows and eyelashes, short stature, low weight, and delay of puberty were also noted. Pruritus and skin fragility had lessened with advancing age. In the family, elective terminations of two subsequent pregnancies had been conducted.
The EKG showed inverted T waves in right precordial leads (V1, V2, and V3) (Fig. 2a) and prolonged terminal activation duration in V1 (80 ms). A 24-h Holter monitoring showed no ventricular ectopy. Echocardiography revealed normal left ventricular ejection fraction (EF), but enlarged RV dimensions. The PLAX RVOT/BSA and PSAX RVOT/BSA were 21 mm/m2 and 24 mm/m2, respectively. Cardiac magnetic resonance imaging (CMRI), with and without Gadolinium, revealed normal myocardial thickness and wall motion, normal chamber size, volume and function, and no delayed myocardial enhancement (Fig. 2b–e). She met at least two major criteria (anterior T wave inversion and the JUP mutation) and one minor criterion (TAD > 55 ms) of the 2019 modified Task Force criteria giving her a definite diagnosis of ARVC27.

EKG, CMRI with Gadolinium findings in Patient 2. (a) EKG shows inverted T waves in leads V1, V2 and V3 (asterisks, major criterion) and a prolonged V3 terminal activation duration (TAD) = 80 ms (minor criterion). Epsilon waves are absent. (There is also a right arm/left arm limb lead reversal). CMRI shows normal myocardial thickness and wall motion along with normal left and right ventricular size, volume, and function (b and c). T2-weighted images (short-TI inversion recovery sequence) in the four-chamber (b) and short-axis (c) views of the ventricles show neither myocardial edema nor inflammation (d and e). Four-chamber (d) and short-axis views (e) of the ventricles in late Gadolinium enhancement sequences show no myocardial enhancement or evidence of myocardial replacement with fibrosis.
Mutation detection
Two different approaches were used for mutation detection. First, DNA-based next-generation sequencing (NGS) utilized a panel consisting of 21 genes associated with different skin fragility syndromes, including EB18. For targeted NGS, DNA was isolated from peripheral blood samples taken from the patients, their parents, and other clinically affected and unaffected family members. For details of the library construction, data capturing and bioinformatics, see the references23,25. The second mutation detection approach utilized RNA-based NGS via RNA-Seq analysis of a whole skin biopsy from Patient 2 as well as six unrelated healthy controls. The resulting data derived from transcriptome-wide variant calling (Fig. 3b), whole-genome homozygosity mapping (Fig. 3c), gene expression profiling visualized by heatmap (Fig. 3e), and gene-specific splicing events depicted by Sashimi plot (Fig. 3f), were elicited by using in-house bioinformatics algorithms22. Data analysis revealed the same homozygous mutation in JUP: [NM_002230], exon 2, c.201delC; p.Ser68Ala*92 in both probands, with their parents being heterozygous for the mutation. This previously unreported mutation was confirmed by bidirectional Sanger sequencing (Fig. 3d). This mutation is not reported in either homozygous or heterozygous state in gnomAD, GME Variome, ExAC, 1000 Genomes, or Iranome. The mutation was predicted to be pathogenic by Annovar, with a CADD score of 46. The 3D structure of junction plakoglobin protein is visualized by PyMol (v.2, Schrodringer, New York, NY), and previously reported mutations in patients without significant clinical cardiac involvement are shown in Fig. 3g28.

Identification of biallelic JUP mutations. (a) Pedigrees of two families with consanguinity. The proband in each family is indicated by an arrowhead. (b) Identification of the mutation in JUP by analysis of annotated variants from RNA-Seq data in Patient 2 using bioinformatics filtering steps indicated. (c) Homozygosity mapping based on RNA-Seq data localized the JUP gene within a homozygosity block of 10.7 Mb on chromosome 17. (d) Sanger sequencing confirmed the presence of the previously unreported homozygous mutation, JUP: NM_002230: exon 2, c.201_201delC, p.Ser68Alafs*92, in both probands. (e) Heatmap visualization of transcriptome analysis revealed markedly reduced level of JUP expression in comparison to the pool of six healthy controls when compared to other genes associated with EB phenotypes and randomly selected housekeeping genes. (f) Sashimi plot of the transcriptome profile of the mutant JUP mRNA revealed normal splicing, indicating lack of exon skipping and/or significant intron retention. (g) Protein visualization of full-length JUP consisting of 745 amino acids. The mutation identified in this study (red) is predicted to result in a frameshift and early truncation of the protein. Two previously reported JUP mutations in patients with similar phenotype are shown (black). The figure was rendered in PyMol (v.2, Schrodinger, New York, NY). For details of WES, homozygosity mapping, RNA sequencing and protein modeling, see PyMol, http://Rymol.org18,24,28,43.
Since ARVC has also been associated with mutations in 13 other genes, besides JUP, including PKP2, DSC2, DSG2, RYR2, SCN5A, TMEM43, DSP, CTNNA3, TGFB3, DES, LMNA, PLN and TTN, the corresponding sequences were individually checked from the RNA-Seq data, and no pathogenic variants were found in these genes.
Immunofluorescent staining
Immunofluorescence mapping using antibodies directed against desmosomal proteins (plakophilin-1, plakoglobin, and desmoplakin I/III) revealed essentially absent staining for plakoglobin in the epidermis of Patient 2 in contrast to pan-epidermal labeling at the periphery of keratinocytes in control skin (Fig. 4).

Immunofluorescence and transmission electron microscopy analysis of skin biopsy from Patient 2. IF staining for junction plakoglobin (encoded by JUP) shows complete absence of the protein in the epidermis of the patient (left lower panel) compared to pan-epidermal keratinocyte cell membrane labeling in an unrelated individual’s healthy skin (left upper panel). Ultrastructurally (right panel), desmosomes appear somewhat small with pinching off within the intercellular desmosomal plaques (arrows) and widening of the intercellular spaces between adjacent keratinocytes (asterisks). For technical details see reference44.
Electron microscopy
Transmission electron microscopy of the skin in Patient 2 revealed intraepidermal blistering and keratinocyte lysis adjacent to desmosomal plaques (Fig. 4). Since the tissue separation is intraepidermal, these findings place these patients into the simplex category of EB.
Discussion
In this study, we reported two patients from unrelated families residing in different parts of Iran harboring a homozygous mutation in the JUP gene. They both presented with skin fragility, PPK, alopecia and nail dystrophy, but ECG abnormalities were present only in the older patient. Neither had significant symptoms or family history for arrhythmia or heart failure. The significant age difference between the two patients provided us the opportunity to follow the progression of cardiac involvement in patients with the same biallelic mutation in JUP with time.
Literature review suggested full penetrance (100%) of ARVC in patients with biallelic JUP mutations, specifically p.Arg265His, p.Glu301Gly, p.Trp680Glyfs*11, and c.2157_2158delTG, presenting with PPK and woolly hair (Table 1)7,11,29,30. However, there was some degree of uncertainty about the cardiac involvement of the patients with biallelic JUP mutations presenting with skin fragility in addition to PPK and woolly hair/alopecia12,13,14,15. A biallelic JUP mutation that resulted in p.Ser24* was primarily associated with skin fragility and ectodermal features without cardiac manifestations in three children from a single family (no. 2 in Table 1)15, although the eldest one manifested with left dilated cardiomyopathy (DCM) at 19 years of age12. However, it was not mentioned whether this patient was screened for mutations in any other ARVC/ALVC or DCM-associated genes. Moreover, DCM can be caused idiopathically or by other causes, such as viral infections. Another biallelic JUP mutation, c.468G > A, a synonymous splice site mutation at the exon3/intron 3 border, was associated with skin fragility, sparse woolly hair, and palmoplantar keratoderma, but no cardiac manifestations were noted in this family with two toddler siblings (Family 1, Table 1)15. Furthermore, biallelic mutations in JUP, p.Gln184* and p.Gln529*, were reported with skin fragility, alopecia, and nail dystrophy; the patients died at 18 and 11 days after birth, respectively13,14. The family of the patient harboring the p.Gln184* mutation consented to post-mortem autopsy, which revealed interventricular septal defect, patchy perivascular fibrosis, and myocyte dropout14. Review of the literature suggested that the nonsense and splice site mutations, located in more proximal exons of junction plakoglobin, including exons 2, 3, 4 and 9, manifested with more severe skin fragility as compared to the frameshift mutations leading to truncated protein in the distal exons, such as exon 12, or missense mutations in the exons 5 that were not associated with skin fragility (Table 1).
Cardiomyopathy in patients with biallelic JUP mutations, characterized by fibrofatty replacement of myocardium with subsequent ventricular dilatation and systolic dysfunction, carries a poor prognosis due to increased risk of ventricular arrhythmias and sudden cardiac death although the patients are often asymptomatic at an early age. Efficient screening of the patients could prevent the catastrophic complications later in life. The current recommended diagnostic approach for ARVC is non-invasive EKG monitoring and cardiac imaging, including echocardiography and CMRI2,31. Since most clinical findings alone are neither sensitive nor specific enough, 2010 Task Force criteria were developed to provide guidance on ARVC diagnosis27. Arrhythmogenic right ventricular cardiomyopathy is now considered within the spectrum of arrhythmogenic cardiomyopathies for which clinically relevant and updated information on genetics and disease mechanism are provided in a 2019 expert consensus statement32,33. The ARVC criteria are composed of six domains including global or regional dysfunction and structural alteration, histological characterization, repolarization abnormalities, depolarization/conduction abnormalities, arrhythmias, and family history27.
In the study by Groeneweg et al., anterior (V1-V3) T wave inversion was found in 74% and prolonged TAD/epsilon waves were seen in 67% and 15% of patients, respectively, with a definite diagnosis of ARVC34. Inverted T waves in right precordial leads in an individual beyond 14 years of age, in the absence of right bundle branch block (RBBB), are found only in 4% of healthy women and 1% of healthy men, thus making it a major criterion with 47% sensitivity and 96% specificity for ARVC35. Furthermore, the pathogenic mutation in JUP is another major criterion, which counts for 0.5 to 2 percent of ARVC patients36.
The reason for the enlarged RV on ECHO but not CMRI in Patient 2 is unclear. However, because echocardiographic imaging of the RV can be limited, we based the absence of structural abnormalities on her CMRI. CMRI plays a major role in the diagnosis of ARVC in early stages. The sensitivity of CMRI is 79–89% for major and 68–78% for minor ARVC criteria while retaining > 95% specificity37. CMRI has some limitations including inter-observer variability in identifying features of ARVC, and it is therefore recommended that CMRI is assessed by an expert specialist familiar with this technique37. Moreover, the sensitivity of CMRI in children is low because arrhythmic presentations precede structural changes38. High negative predictive value of Gadolinium-enhanced CMRI makes it a good choice as a rule-out imaging test for evaluation of structural and functional abnormalities in patients suspected to have ARVC39.
DSP and JUP mutations accounted for 90.8% of the patients displaying cutaneous phenotype with ARVC40. Furthermore, most patients harboring a mutation in ARVC-associated genes present with ARVC by 30 years of age although homozygous patients (e.g., Naxos disease) show 100% penetrance by adolescence42. Long-term cardiac monitoring by EKG, Holter monitoring, echocardiography, and exercise stress test is recommended for the patients harboring mutations in ARVC-associated genes as well as those presenting with PPK and hair shaft anomalies with unknown genotype; however, it was reported that only 2.3% of patients with such cutaneous phenotypes receive relevant cardiac monitoring41,42. In this context, because of age-dependent penetrance and the young age of our patients, further studies and close follow-up are needed, including cardiological monitoring of family members at risk. Such risk can be assessed by mutation analysis of the members of the extended family.
In conclusion, we examined two unrelated patients with skin fragility, separated in age by 20 years, but who harbor the same, previously unreported homozygous JUP mutation. Repolarization/depolarization abnormalities and echocardiographic RV enlargement was seen in the older (definite ARVC) but not the younger (possible ARVC) proband. This cross-sectional observation highlights the age-dependent penetrance of ARVC, the need for close monitoring of patients with pathogenetic JUP mutations; and cascade screening of first degree at risk family members.
Abbreviations
- ALVC:
-
Arrhythmogenic left ventricular cardiomyopathy
- ARVC:
-
Arrhythmogenic right ventricular cardiomyopathy
- CMRI:
-
Cardiac magnetic resonance imaging
- DCM:
-
Dilated cardiomyopathy
- TAD:
-
Terminal activation duration
- EB:
-
Epidermolysis bullosa
- LOF:
-
Loss-of-function
- NGS:
-
Next generation sequencing
- PPK:
-
Palmoplantar keratoderma
- ROH:
-
Runs of homozygosity
References
Corrado, D. et al. Arrhythmogenic right ventricular cardiomyopathy: evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart J. 41, 1414–1429, https://doi.org/10.1093/eurheartj/ehz669 (2019).
Corrado, D., Link, M. S. & Calkins, H. Arrhythmogenic right ventricular cardiomyopathy. N. Engl. J. Med. 376, 1489–1490. https://doi.org/10.1056/NEJMc1701400 (2017).
McKenna, W. J. et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br. Heart J. 71, 215–218. https://doi.org/10.1136/hrt.71.3.215 (1994).
Peters, S. Advances in the diagnostic management of arrhythmogenic right ventricular dysplasia-cardiomyopathy. Int. J. Cardiol. 113, 4–11. https://doi.org/10.1016/j.ijcard.2005.12.015 (2006).
Lazzarini, E. et al. The ARVD/C genetic variants database: 2014 update. Hum. Mutat. 36, 403–410. https://doi.org/10.1002/humu.22765 (2015).
McNally, E., MacLeod, H. & Dellefave-Castillo, L. Arrhythmogenic right ventricular cardiomyopathy. in GeneReviews® [Internet] (eds M. P. Adam, R. A. Pagon et al.). 1993–2020. https://www.ncbi.nlm.nih.gov/books/NBK1131 (University of Washington, Seattle, Seattle (WA), 2005).
McKoy, G. et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet 355, 2119–2124. https://doi.org/10.1016/S0140-6736(00)02379-5 (2000).
Simpson, M. A. et al. Homozygous mutation of desmocollin-2 in arrhythmogenic right ventricular cardiomyopathy with mild palmoplantar keratoderma and woolly hair. Cardiology 113, 28–34. https://doi.org/10.1159/000165696 (2009).
Carvajal-Huerta, L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J. Am. Acad. Dermatol. 39, 418–421. https://doi.org/10.1016/s0190-9622(98)70317-2 (1998).
Smith, E. D. et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 141, 1872–1884. https://doi.org/10.1161/CIRCULATIONAHA.119.044934 (2020).
Protonotarios, N. et al. Genotype-phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J. Am. Coll. Cardiol. 38, 1477–1484. https://doi.org/10.1016/s0735-1097(01)01568-6 (2001).
Boente, M. D. et al. Cardiomyopathy diagnosed in the eldest child harboring p.S24X mutation in JUP. Br. J. Dermatol. 175, 644–646. https://doi.org/10.1111/bjd.14617 (2016).
Pigors, M. et al. Lack of plakoglobin leads to lethal congenital epidermolysis bullosa: a novel clinico-genetic entity. Hum. Mol. Genet. 20, 1811–1819. https://doi.org/10.1093/hmg/ddr064 (2011).
Rotemberg, V. et al. A novel mutation in junctional plakoglobin causing lethal congenital epidermolysis bullosa. J. Pediatr. 191, 266-269e261. https://doi.org/10.1016/j.jpeds.2017.08.029 (2017).
Cabral, R. M. et al. Homozygous mutations in the 5’ region of the JUP gene result in cutaneous disease but normal heart development in children. J. Invest. Dermatol. 130, 1543–1550. https://doi.org/10.1038/jid.2010.7 (2010).
Nekrasova, O. & Green, K. J. Desmosome assembly and dynamics. Trends Cell Biol. 23, 537–546. https://doi.org/10.1016/j.tcb.2013.06.004 (2013).
Isac, C. M. et al. Plakoglobin is essential for myocardial compliance but dispensable for myofibril insertion into adherens junctions. J. Cell Biochem. 72, 8–15 (1999).
Vahidnezhad, H. et al. Multigene next generation sequencing panel identifies pathogenic variants in patients with unknown subtype of epidermolysis bullosa: Subclassification with prognostic implications. J. Invest. Dermatol. 137, 2649–2652 (2017).
Vahidnezhad, H. et al. Dystrophic epidermolysis bullosa: COL7A1 mutation landscape in a multi-ethnic cohort of 152 extended families with high degree of customary consanguineous marriages. J. Invest. Dermatol. 137, 660–669 (2017).
Vahidnezhad, H. et al. Genome-wide single nucleotide polymorphism-based autozygosity mapping facilitates identification of mutations in consanguineous families with epidermolysis bullosa. Exp. Derm. 28, 1118–1121. https://doi.org/10.1111/exd.13501 (2019).
Vahidnezhad, H. et al. Biallelic KRT5 mutations in autosomal recessive epidermolysis bullosa simplex, including a complete human keratin 5 “knock-out”. Matrix Biol. 83, 48–59. https://doi.org/10.1016/j.matbio.2019.07.002 (2019).
Saeidian, A. H., Youssefian, L., Vahidnezhad, H. & Uitto, J. Research techniques made simple: whole-transcriptome sequencing by RNA-Seq for diagnosis of monogenic disorders. J. Invest. Dermatol. 140, 1117–1126 (2020).
Youssefian, L. et al. A novel autosomal recessive GJB2-associated disorder: ichthyosis follicularis, bilateral severe sensorineural hearing loss, and punctate palmoplantar keratoderma. Hum. Mutat. 40, 217–229. https://doi.org/10.1002/humu.23686 (2019).
Youssefian, L. et al. Autosomal recessive congenital ichthyosis: genomic landscape and phenotypic spectrum in a cohort of 125 consanguineous families. Hum. Mutat. 40, 288–298. https://doi.org/10.1002/humu.23695 (2019).
Vahidnezhad, H. et al. Next generation sequencing identifies double homozygous mutations in two distinct genes (EXPH5 and COL17A1) in a patient with concomitant simplex and junctional epidermolysis bullosa. Hum. Mutat. 39, 1349–1354. https://doi.org/10.1002/humu.23592 (2018).
Vahidnezhad, H. et al. Recessive mutation in tetraspanin CD151 causes Kindler syndrome-like epidermolysis bullosa with multi-systemic manifestations including nephropathy. Matrix Biol. https://doi.org/10.1016/j.matbio.2017.11.003 (2017).
Marcus, F. I. et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur. Heart J. 31, 806–814. https://doi.org/10.1093/eurheartj/ehq025 (2010).
Vahidnezhad, H. et al. Molecular dynamics simulation of the consequences of a PYCR1 mutation (p.Ala189Val) in patients with complex connective tissue disorder and severe intellectual disability. J. Invest. Dermatol. 137, 525–528. https://doi.org/10.1016/j.jid.2016.10.007 (2017).
Erken, H. et al. Cardiomyopathy with alopecia and palmoplantar keratoderma (CAPK) is caused by a JUP mutation. Br. J. Dermatol. 165, 917–921. https://doi.org/10.1111/j.1365-2133.2011.10455.x (2011).
Marino, T. C. et al. Novel founder mutation in French-Canadian families with Naxos disease. Clin. Genet. 92, 451–453. https://doi.org/10.1111/cge.12971 (2017).
Savino, K., Bagliani, G., Crusco, F., Padeletti, M. & Lombardi, M. Electrocardiogram and imaging: an integrated approach to arrhythmogenic cardiomyopathies. Card. Electrophysiol. Clin. 10, 413–429. https://doi.org/10.1016/j.ccep.2018.02.011 (2018).
Protonotarios, N. et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia on the basis of the revised diagnostic criteria in affected families with desmosomal mutations. Eur. Heart J. 32, 1097–1104. https://doi.org/10.1093/eurheartj/ehr043 (2011).
Towbin, J. A. et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: executive summary. Heart Rhythm 16, e373–e407. https://doi.org/10.1016/j.hrthm.2019.09.019 (2019).
Groeneweg, J. A. et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ. Cardiovasc. Genet. 8, 437–446. https://doi.org/10.1161/CIRCGENETICS.114.001003 (2015).
Morin, D. P. et al. Usefulness of precordial T-wave inversion to distinguish arrhythmogenic right ventricular cardiomyopathy from idiopathic ventricular tachycardia arising from the right ventricular outflow tract. Am. J. Cardiol. 105, 1821–1824. https://doi.org/10.1016/j.amjcard.2010.01.365 (2010).
Jacob, K. A. et al. Geographical distribution of plakophilin-2 mutation prevalence in patients with arrhythmogenic cardiomyopathy. Neth. Heart J. 20, 234–239. https://doi.org/10.1007/s12471-012-0274-x (2012).
Sen-Chowdhry, S. & McKenna, W. J. Sudden cardiac death in the young: a strategy for prevention by targeted evaluation. Cardiology 105, 196–206. https://doi.org/10.1159/000091640 (2006).
Fogel, M. A., Weinberg, P. M., Harris, M. & Rhodes, L. Usefulness of magnetic resonance imaging for the diagnosis of right ventricular dysplasia in children. Am. J. Cardiol. 97, 1232–1237. https://doi.org/10.1016/j.amjcard.2005.11.045 (2006).
Chellamuthu, S. et al. Is cardiac MRI an effective test for arrhythmogenic right ventricular cardiomyopathy diagnosis?. World J. Cardiol. 6, 675–681. https://doi.org/10.4330/wjc.v6.i7.675 (2014).
Polivka, L., Bodemer, C. & Hadj-Rabia, S. Combination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases. J. Med. Genet. 53, 289–295. https://doi.org/10.1136/jmedgenet-2015-103403 (2016).
Otterspoor, L. C. et al. Arrhythmogenic right ventricular cardiomyopathy: asymptomatic to life threatening as illustrated by the cases of two sisters. Neth. Heart J. 15, 348–353. https://doi.org/10.1007/bf03086013 (2007).
Sattar, Y., Abdullah, H. M., Neisani Samani, E., Myla, M. & Ullah, W. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: an updated review of diagnosis and management. Cureus 11, e5381. https://doi.org/10.7759/cureus.5381 (2019).
Vahidnezhad, H., Youssefian, L., Jazayeri, A. & Uitto, J. Research techniques made simple: genome-wide homozygosity/autozygosity mapping is a powerful tool to identify candidate genes in autosomal recessive genetic diseases. J. Invest. Dermatol. 138, 1893–1900. https://doi.org/10.1016/j.jid.2018.06.170 (2018).
Vahidnezhad, H. et al. Mutations in PLOD3, encoding lysyl hydroxylase 3, cause syndromic recessive dystrophic epidermolyis bullosa-like phenotype with abnormal anchoring fibrils and deficiency in type VII collagen. Matrix Biol. 81, 91–106 (2019).
Acknowledgements
The authors thank Drs. Adam Ertel and Paolo Fortina for assistance in next-generation sequencing. Dr. Sara Pajouhanfar performed genetic counseling, and Dr. Saba Homatash performed echocardiography. Dr. Howard Weitz contributed to the interpretation of cardiac findings. Carol Kelly assisted in manuscript preparation.
Funding
This work was supported by DEBRA International.
Author information
Authors and Affiliations
Contributions
J.U., L.Y., and H.V. conceived the study and initiated this project. H.V. and M.F. interviewed patients and their families and performed genetic counseling. L.Y., A.H.S., M.F., N.M., F.A., S.S., F.R., F.N., L.M., Z.A.S., L.L., A.G., S.Z., A.K., R.T.H., and J.A.M. performed experiments and/or analyzed the data. The manuscript was drafted by M.F., H.V., and J.U. All authors participated in the interpretation of results and critically reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vahidnezhad, H., Youssefian, L., Faghankhani, M. et al. Arrhythmogenic right ventricular cardiomyopathy in patients with biallelic JUP-associated skin fragility. Sci Rep 10, 21622 (2020). https://doi.org/10.1038/s41598-020-78344-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78344-9
This article is cited by
-
Genetic predisposition study of heart failure and its association with cardiomyopathy
The Egyptian Heart Journal (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
