
Abstract
Sugarcane is the most important sugar and biofuel crop. MADS-box genes encode transcription factors that are involved in developmental control and signal transduction in plants. Systematic analyses of MADS-box genes have been reported in many plant species, but its identification and characterization were not possible until a reference genome of autotetraploid wild type sugarcane specie, Saccharum spontaneum is available recently. We identified 182 MADS-box sequences in the S. spontaneum genome, which were annotated into 63 genes, including 6 (9.5%) genes with four alleles, 21 (33.3%) with three, 29 (46%) with two, 7 (11.1%) with one allele. Paralogs (tandem duplication and disperse duplicated) were also identified and characterized. These MADS-box genes were divided into two groups; Type-I (21 Mα, 4 Mβ, 4 Mγ) and Type-II (32 MIKCc, 2 MIKC*) through phylogenetic analysis with orthologs in Arabidopsis and sorghum. Structural diversity and distribution of motifs were studied in detail. Chromosomal localizations revealed that S. spontaneum MADS-box genes were randomly distributed across eight homologous chromosome groups. The expression profiles of these MADS-box genes were analyzed in leaves, roots, stem sections and after hormones treatment. Important alleles based on promoter analysis and expression variations were dissected. qRT-PCR analysis was performed to verify the expression pattern of pivotal S. spontaneum MADS-box genes and suggested that flower timing genes (SOC1 and SVP) may regulate vegetative development.
Similar content being viewed by others


Introduction
MADS-box is one of the extensively studied transcription factor families that plays a vital role in diverse biological functions1. This gene family is categorized into two groups; Type-I (Mα, Mβ, Mγ) and Type-II (MIKCc and MIKC*)2. MADS-box family genes are characterized by the most conserved DNA binding domain, MADS-domain of about 60 amino acids in length at the N-terminal region and involved in DNA binding to their target genes based on consensus sequence CC(A/T)6GG (CArG box)3. In addition, type-II lineage contains three additional domains, (1) I-domain (intervening), for DNA-binding specificity and dimer formation, positioned between the M and K domain, (2) K-domain (keratin), characterized by coil-coil structure and responsible for protein–protein interactions and (3) C-domain (C-terminus), is the trans-activation domain and the least conserved domain among all4. On the other hand, MIKC*-type-II proteins have less-conserved K domain and a larger I domain as compared to MIKCc type-II proteins5. In Arabidopsis, the MIKCc proteins have been further divided into 12 subfamilies2, 6. Unlike type-II, type-I MADS-box proteins are simpler in structure as they lack the K domain and considered the more ancient proteins shared by plants and animals7, 8. In most of the plant species, type-I MADS-box genes have a faster birth and death rate than type-II genes due to weaker purifying selection and higher prevalence of segmental gene duplications9. To date, limited knowledge is available regarding type-I genes functions in plants.
Most of the evidences suggested that MADS-box genes are involved in many significant developmental and physiological processes, such as the gametophyte cell division, floral transition, regulation of floral organs and fruit development and ripening10,11,12,13,14. However, these genes were initially recognized as floral organ identity genes in Arabidopsis and antirrhinum. Modified from the original ABC model15, the ABCDE model that determines the identity of floral organs has been presented. Different floral organs identities are controlled by various combinations of types of genes, A + E (sepals), A + B + E (petals), B + C + E (stamens), C + E (carpels) and D + E (ovules)16. In Arabidopsis, genes belong to these functional classes were AP1 (APETALA1) in class A, PI (PISTILATA) and AP3 (APETALA3) in class B, AG (AGAMOUS) in class C, STK/AGL11 (SEEDSTICK/AGAMOUS-LIKE 11) and SHP (SHATTERPROOF) in class D and SEP(SEPALLATA) genes in class E16,17,18.
Four MADS sub-families, FLC, AGL24, SVP and SCO1 have been found as floral regulators. AGL24 and SCO1 act as floral promoters, whereas SVP and FLC functioned as flowering inhibitors19. To date, limited information about MADS-box transcription family is available for vegetative organs development (stem and root). Given its important roles, this gene family has been characterized in many plant species. However, no significant information is available regarding this family in important subtropical and tropical crop sugarcane.
Sugarcane (Saccharum spp.) is a complex polyploid (2n = 4x–16x = 32–128)20,21,22 and the most important sugar crop in the world’s economy. In the Saccharum complex, alot of variations in phenotypic traits exist23, 24. Genomic variation within Saccharum complex can provide understanding about polyploidization, adaptation, stress tolerance, sugar and biomass accumulation25. Though insight into their allelic variation has been demanding and, it is critical to identify the alleles important in controlling these traits in sugarcane. Recent generation of reference genome of wild type sugarcane, Saccharum spontaneum Ap85-441 (n = 4x = 32) has made it possible to explore allelic variations in MADS-box gene family26. S. spontaneum is autopolypoid with excellent stress tolerance27. Modern sugarcane cultivars are allopolyploids and interspecific hybrids mostly derived from crosses between domesticated S. officinarum and wild species S. spontaneum, followed by a series of backcrosses with S. officinarum. Sugarcane stem is the most economically important part used as raw material for sucrose and bagasse for biofuel production28. Although S. spontaneum has low biomass, but its mechanism of stem ripening is similar to that of cultivated sugarcane. In this research, we performed genome-wide identification, classification of MADS-box alleles, their structure, motif analysis, chromosome location of MADS-box genes in S. spontaneum and their expression profiling in vegetative organs, as well as their role in stem ripening.
Material and methods
Plant material
Saccharum spontaneum AP85-441 (n = 4x) tissues and SES208 seedlings were collected from Center for Genomics and Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China. These two S. spontaneum accessions were shared by Hawaii Agriculture Research Center (HARC) sugarcane genome project. No voucher specimen was deposited to a herbarium as these plant materials are readily available at HARC. Roots (R), leaves (L), stem1 (1–5 internodes; immature), stem2 (12–14 internodes; maturing) and stem3 (20–22 internodes; matured) of AP85-441 were collected, separated quickly. Further, SES-208, 35 days old seedling leaves and stems were collected after 24, 48 and 96 h. of ethylene and 48 h of auxin, gibberellin and abscisic acid treatment. Three replicates for each sample were collected and kept at − 80 °C for RNA extraction and expression analysis.
Identification and chromosomal localization of MADS-box genes in S. spontaneum genome
Two methods were applied to identify MADS-box genes in the recently published S. spontaneum genome26. Firstly, HMM (Hidden Markov Model) profiles for previously submitted Arabidopsis MADS-box domains SRF-type-I (PF00319) and MEF2-type-II (PF09047) were obtained from the Pfam database (https://pfam.sanger.ac.uk)29. These profiles were used to discern MADS-box genes in the S. spontaneum genome by using HMMER-3.1b2 software (https://hmmer.janelia.org/)30. All of the proteins with an e-value lower than 0.1 were selected. Secondly, whole MADS-box proteins sequences of Arabidopsis and sorghum were downloaded from TAIR (https://www.arabidopsis.org/) and PlantTFDB (https://planttfdb.cbi.pku.edu.cn/) respectively. BLASTp searches were applied to predict MADS-box genes in the S. spontaneum genome using all Arabidopsis and sorghum MADS-box proteins as queries. Finally, candidate genes were examined for conserved domain by Pfam (https://Pfam.sanger.ac.uk/) and CDD (https://www.ncbi.nlm.nih.gov/cdd/) with threshold e-value < 0.0001. Molecular weight (MW) and Isoelectric point (PI) of all proteins were estimated by using ProtParam (https://web.expasy.org/protparam/). The physical location along chromosomes were determined by BLASTn searches, using all sequences as query against the S. spontaneum draft genome26. MapGene2Chrom web v2 (mg2c.iask.in/)31 online program was used to draw the location of S. spontaneum MADS-box genes on physical map of chromosomes based on their coordinates.
Classification and subcellular localization of MADS-box genes
Saccharum spontaneum MADS-box members were grouped together into gene models based on the phylogenetic relationship with sorghum MADS-box genes. To further classify them into sub-groups, 102 MADS-box genes in Arabidopsis and 65 genes in sorghum were used as reference2, 32. Reference alleles of S. spontaneum MADS-box gene models were aligned to those of Arabidopsis and sorghum by MEGA7-Clustal W option33, and alignment results were used to construct a phylogenetic tree using maximum likelihood method with Jones–Taylor–Thornton (JTT) model. The tree was visualized by iTOL (https://itol.embl.de/) online program34. Subcellular localization of S. spontaneum MADS-box gene model’s representative alleles were predicted by WoLFPSORT (https://www.genscript.com/wolf-psort.html) and CELLO v2.5 (https://cello.life.nctu.edu.tw/).
Comparison of S. spontaneum MADS-box gene structure and conserved motif with sorghum
Coding DNA sequences were used to construct an evolutionary tree for reference alleles of S. spontaneum MADS-box gene models together with sorghum by using Clustal W and maximum likelihood method via MEGA version 733. Intron/Exon map was constructed using gff3 annotation from the published S. spontaneum genome assembly26 by Gene Structure and Display Server 2.0 (gsds.cbi.pku.edu.cn/) online program35.
Moreover, their protein sequences were searched for conserved motifs by MEME 5.1.0 (meme-suite.org/tools/meme) online program with the following parameters: number of motifs-20, optimum width ranges from > 6 to < 200. Motif distribution was built with TBtools software (https://github.com/CJ-Chen/TBtools). Finally, obtained motifs were annotated by using SMARTBLAST-NIH (https://blast.ncbi.nlm.nih.gov/smartblast/) online program. Furthermore, gene structure and motif distribution for all S. spontaneum MADS-box sequences were also obtained using the same methods.
Transcriptome analysis
Total RNA was isolated from the collected samples using the Illumina TruSeq RNA Sample Preparation Kit (RS-122-2001(2), Illumina) following the manufacturer’s protocol. For each sample, three replicates were used. Sequencing was done by Illumina HiSeq2500 at the Center for Genomics and Biotechnology at the Fujian Agriculture and Forestry University. After that, adapter sequences were removed from raw reads using FASTX-toolkit. Sequence quality was examined by FastQC, and low-quality reads were removed. Clean reads were then mapped to the S. spontaneum genome26 by using Tophat v.2.0.10 and transcriptome assembly and DEG’s analysis were conducted using Cufflinks36 with parameters: p value < 0.05 and log2FC > 1 or < − 1. Furthermore transcript abundance was calculated as Fragment per Kilobase Million (FPKM) using stringtie37. Heat map of MADS-box genes were generated based on the FPKM values of gene models (average FPKM of all alleles and paralogs within a gene model).
Cis-element analysis
1.5 kb sequence upstream of ATG (translation initiation site) of differentially expressed genes during stem development were extracted from S. spontaneum genome sequences. Different cis-elements were predicted at PlantPAN (https://plantpan.itps.ncku.edu.tw/) and PlantCARE (https://bioinformatics.psb.ugent.be/webtools/plantcare/html/) and counted. TBtools38 was used to plot the enrichment of these elements.
Quantitative RT-PCR
Same RNA samples sent for RNA-seq were used for expression quantification by qRT-PCR. The first-strand cDNA was synthesized by EasyScript One-Step gDNA Removal and cDNA Synthesis Supermix. For expression quantification, primers for specific genes based on their representative alleles were designed by Primer Premier 5.0 software (Table S6). qRT-PCR was conducted using SYBR Premix Ex Taq (Tli RNaseH Plus) kit (TaKaRa, Japan), in Bio-Rad iQ5 Real-Time PCR System, with profile 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s. GAPDH (glyceraldehyde-3-phosphate-dehydrogenase) gene was chosen as control for data normalization39, 40. For the reliability of results, three technical replicates of each sample were used. Finally, the 2−ΔΔCt method was employed to compute the relative quantitative gene expression41.
Results
Identification, characterization and classification of S. spontaneum MADS-box genes
For identification of MADS-box genes, we used HMM (Hidden Markov Model) and BLASTp searches to explore S. spontaneum genome. Finally, a total of 182 non-redundant protein sequences were retained after conserved domain confirmation. The ORF length of S. spontaneum MADS-box sequences ranged from 174 to 2388 bp, with encoded proteins ranged from 58 to 796aa. These proteins have a predicted PI ranged from 4.4 to 11.13 and molecular weight ranged from 6479.7 to 87,287.16 KDa. (Table S1). Accession number along with sequences of these 182 non-redundant sequences are provided in Table S6.
An un-rooted tree of 182 S. spontaneum MADS-box sequences was constructed to determine the gene models based on 65 sorghum MADS orthologs (same as Fig. S1), as sorghum is one of the closest relative diploid genera of Saccharum42. All the sequences were grouped into 63 gene models with variable number of alleles, paralogs and tandem-duplicates. Alleles were further confirmed by alleles table provided by26. These models were named as Ss-subgroup with number (1–65) in relevance with sorghum orthologs. Of them, six (9.5%) gene models were having four alleles. Twenty-one gene models were found with three alleles, 29 with two and 7 with only one allele. Interestingly, SbMADS 2, 33, 39, 43, 46, 50, 52, 54, 55 have no homolog in S. spontaneum genome. While two copies of SsSOC1-6a/b, SsAG/SHP-10a/b, SsSEP-22a/b, SsMα-48a/b and three copies of SsMβ-64a/b/c were specific in S. spontaneum genome in comparison with sorghum. Summary of 63 genes models along with their corresponding alleles and paralogs are provided in Table S1.
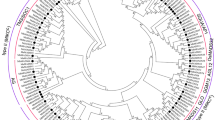
To further clarify the nomenclature, a phylogenetic tree was constructed using representative alleles of S. spontaneum MADS-box gene models with Arabidopsis and sorghum MADS-box protein coding genes. Out of 63, 34 gene models were determined to be type-II MADS-box genes (32 MIKCc, 2 MIKC*), and 29 were confirmed as type-I (21 Mα, 4 Mβ, 4Mγ). Interestingly, we observe species-specific clustering between S. spontaneum and sorghum, which indicated that S. spontaneum is much close to sorghum as they shared a common ancestor about 8 million years ago. We also found one new cluster of 4 gene models (representative alleles Sspon.001C0027130, Sspon.004A0013490, Sspon.006C0021980,Sspon.003B0025330) with no sorghum orthologs, but very close to Arabidopsis Mγ sub-group and were considered as Mγ-like.
Based on the phylogenetic tree analysis, 32 MIKCc were divided into 10 subfamilies. The tree shows that there were three homologs for SVP, AGL12-like, TT16 and SOC1, four for AP3/PI, five for AG/SHP, seven for SEP and one for ANR1. We did not find any FLC/FLM subfamily members in S. spontaneum (Fig. 1).

Phylogenetic relation of MADS-box genes in S.spontaneum, Arabidopsis and sorghum. The phylogenetic tree was constructed using maximum likelihood method. The full-length amino acid sequences were aligned by Clustal-W. The sub-families are presented by different colors.
Chromosomal location of S. spontaneum MADS-box genes
Physical location of 63 genes (182 MADS-box sequences) were mapped onto the homologous chromosomes (Fig. 2). These sequences were randomly distributed among A 36 (~ 20%), B 51 (~ 28%), C 41 (~ 22%) and D 54 (~ 30%) homologous chromosomes, respectively.

Distribution of 182 MADS-box members on homologous chromosomes.
Out of 102 type-II, 32 sequences were distributed in D, while remaining were equally distributed among A(22), B(25) and C(23) homologous chromosomes. Most of these sequences were present on Chr8B (6) and Chr5D (6). Type-I were randomly distributed among A(14), B(26), C(18), and D(22) . Most of these members were mapped on Chr3B (9). Further distribution proportion of MADS-box members in S. spontaneum genome is shown in (Fig. 3).

(A) The percentage of 182 MADS-box members anchored in four homologous groups (A, B, C, and D). (B) Number of different S. spontaneum MADS-box type genes (MIKCc, MIKC*, Mα, Mβ, Mγ) in each homologous group.
Predicted subcellular localization of S. spontaneum MADS-box proteins
Subcellular localization of S. spontaneum MADS-box proteins based on representative alleles of 63 gene models was predicted by two different tools and higher confidence prediction was mentioned in Table 1. Most of the S. spontaneum MADS-box proteins were predicted to be localized in nucleus, while SsAG/SHP-26, SsAGL12-like-23, SsMβ-65 in cytoplasm. SsMα-60, SsMα-63, SsMα-36, SsMα-58, SsMα-44, SsMα-57 were found in chloroplast, SsMα-61, SsMα-59, SsMα-38, SsMβ-64b and SsMγ-like-d in mitochondria. However, SsMβ-64c, SsMγ-like-a, SsMγ-like-b and SsMγ-like-c were predicted in mitochondria and chloroplast. Only one protein SsSVP-29 was predicted in endoplasmic reticulum. This may suggest that type-I genes may require for regulating expression in mitochondria and chloroplast compartments.
Intron–exon structure and conserved motif analysis in comparison with sorghum
Based on the evolutionary relationship of S. spotaneum and sorghum, structural diversity and motif distribution were studied. When compared S. spontaneum MADS-box type II genes structure with sorghum, based on representative allele (Fig. 4A), it was found that both species have similar intron–exon distribution, differ only in the length of exons or introns. The same trend was observed in type-I genes (Fig. 4B) except for SsMα56, 49, 53 and 62, which differ in exon number.

Comparative gene structure analysis of S.spontaneum MADS-box gene models based on representative alleles with sorghum. In exon–intron structure yellow blocks indicate exons, black lines indicate introns (A) Type-II MADS-box genes (B) Type-I MADS-box genes.
We found that all type-I genes have simpler gene structure with exons ranged from 1 to 5, most of them have only one exon, except for SsMα56, 49, 53 and 62. While type-II genes have exons ranged from 1 to 14, most of type-II genes were found with 6–8 exons (Table S1). It was also found that alleles within same gene models were structurally more similar in structure.
Furthermore, we obtained some essential sources of the evolutionary relationship based on similar motif distribution between S. spontaneum and sorghum. (Fig. 5A,B). A total of 20 conserved motifs were identified, which had previously been subjected to SMART annotation. The commonly shared motifs tended to be in the same group suggesting that the same sub-family genes probably had similar functions2, 43. Motif 1 was comprised of approximately 60 amino acids and was the most typical MADS-box domain. Motif 2 represented the K domain, was identified in the majority of type-II proteins. On the other hand, MIKC*-type-II proteins had a less-conserved K domain. This result is consistent with previous studies that showed that the K-box domain was only found in type-II MADS-box genes44. However, in our study, two type-I genes, Sspon.004A0012210, Sspon.004C0005610 also contained a K-box domain. It was also found that alleles of the same gene models have similar motif distribution (Fig. S1).

Comparative motif analysis of S.spontaneum MADS-box proteins based on representative alleles with sorghum. Colors represent the different motifs identified by MEME database with full-length amino acid sequences of SsMADS. Motif 1, 3 and 6 represents MADS-box domain and motif 2 represents K-box domain (A) Type-II MADS-box proteins (B) Type-I MADS-box proteins.
Expression Profile of S. spontaneum MADS-box genes in different tissues
Ripening is a continuous process in sugarcane stem as each internode is an independent growth unit. Sugarcane stem comprised a series of internodes at different physiological stages, i.e., immature (top), at maturation (middle), and mature internodes (bottom)45. To analyze the expression profiles of MADS-box genes, leaves, roots, stem1 (1–5 internodes: immature), stem2 (12–14 internodes: at maturation), stem3 (20–22 internodes: matured) of S. spontaneum variety AP85-441 were sampled and subjected to RNA-seq assays. A summary of RNA-seq mapping reads is provided in Table S2. Out of 63 MADS-box gene models, only 32 gene models showed average expression profiles (FPKM > 0) in at least one tissue tested (Table S2). For AP85, 27 (42.8%) MADS-box gene models were expressed in the leaves, 32 (50.7%) roots, 26 (41.2%) stem1, 26 (41.2%) stem2, 24 (38.03%) stem3, respectively, of which 5 (18.51%), 3 (9.3%) and 4 (15.3%), 5 (19.23%), 5 (20.8%) genes exhibited significant expression levels (FPKM value > 10), of which 1 (20%), 0 (0%), and 2 (50%), 3 (60%), 3 (60%), genes had higher expression levels (FPKM value > 30), respectively.
The most highly expressed gene model SsSVP-4/5was in stem2 that have a FPKM value of 169.81. However, only three (4.34%) gene models (SsSOC1-6a, SsSOC1-6b and SsSVP-4/5) had significant transcript accumulation (FPKM value > 10) in all the five tissues tested, with higher expression in stem2 and stem3. SsAP1/CAL/FUL-9, SVP-29andAGL12-like-21 were also more expressed in stem2 and stem3. These results showed that the SOC1 and SVP MADS-box genes have diverse expression pattern in different tissues and more importantly in stem (Fig. 6, Table S3).

Expression pattern of S.spontaneum MADS-box gene models in different tissues (leaf; root; stem1, 1–5 internodes ; stem2, 12–14 internodes; stem3, 20–22 internodes), after 24 h, 48 h and 96 h of ethylene (ET) and after 48 h. of auxin (IAA), abscisic acid (ABA) and gibberellin (GA) treatment in stem and leaf. The expression value was calculated by fragments per kilobase million (FPKM). The scale represents normalized expression value (log2 (FPKM)).
Overall, type-I gene models maintained either a relatively low transcript level or showed no expression profile in RNA-seq libraries except for SsMα-61. The expression of SsMα-61up-regulated in leaves while down-regulated in both root and stem, indicating that type-I genes are perhaps leaf-specific and have crucial role in leaf development.
Expression profile of S. spontaneum MADS-box genes after hormones treatment
Ethylene plays a crucial role in sucrose accumulation and stem ripening in sugarcane. To investigate the expression patterns of MADS-box genes, two tissues leaf and stem were collected from 35 days old seedling of SES-208 after 24, 48 and 96 h of ethylene treatment. RNA-seq mapping reads summary is provided in Table S2. Nineteen MADS-box genes were expressed in at least one tissue (FPKM > 1). Out of all, SVP and SOC1 gene models (SsSVP-4/5, SsSOC1-6a, SsSOC1-6b SsSVP-17, SsSVP-29) show higher expression in stem as compared to leaf at all time points. The most enriched expression for these genes models was observed at 48 h and 96 h. Besides, some genes, such as SsMa57, SsAG/SHP-10a and SsAG/SHP-10b were expressed much higher in leaf than in stem. Overall, lower or no expression was observed by the type-I gene in both tissues tested (Fig. 6; Table S5). The same expression pattern trend for SVP and SOC1 gene models was observed after IAA, ABA, and GA treatment (48 h) (Fig. 6. Table S5) SVP gene models (SsSVP-17, 29, 4/5) showed higher expression in the stem than in leaves. While SOC1-6b has slightly higher expression in leaves than stem after ABA and GA treatment. SOC1-6a expressed more in leaves than stem after IAA treatment.
Expression pattern of SOC1 and SVP genes after hormone treatment (especially ethylene) was consistent with results mentioned above in different stem sections suggested that MADS-box genes may interact directly or indirectly with hormone signal transduction genes to control stem ripening in sugarcane.
Allelic variations in expression and cis-elements identification of S. spontaneum MADS-box differentially expressed genes
The expression level of alleles of differentially expressed genes of SVP and SOC1 (SsSOC1-6a, SsSOC1-6b; SsSVP-4/5, SsSVP-17, SsSVP-29) also showed differences. In view of the significance of cis-elements in the promoter region on gene regulation, we extracted the 1.5 kb sequence upstream to the start site (ATG) of each gene alleles to obtained cis-elements that were catagories into three groups: growth and development, stress responses, and phytohormone responses by PlantPAN46 and PlantCARE47. For growth and development, light-responsive elements G-box and Sp1 were mostly enriched cis-elements in these promoters (Fig. 7). SsSVP-4/5 alleles Sspon.001C0005280 showed higher expression level in stem2 and 3 with a higher number (31) of light-responsive elements. However, the lowest number (2) of light responsive elements were observed in SsSVP-17 allele Sspon.004B0002060 with no expression in any tissue tested.

Allelic variations of S.spontaneum differentially expressed MADS-box gene models (A) It represents the different cis-elements counts, the intensity of red color shows the enrichment of cis-elements in different alleles of gene models (B) It represents the allelic expression pattern variations in various tissues, and after hormones treatment, (48 h). The scale represents normalized expression value (log2 (FPKM)).
Interestingly, it was observed that alleles with higher expression within a gene model have higher number of light-responsive elements comparing with its partner alleles, For example, SsSOC1-6b alleles showed different expression level (from high to low) within vegetative different stem sections tested: Sspon.001C0001370 > Sspon.001B0000911 > Sspon.001D0001730 > Sspon.001A0001360, with 22, 14,14, 19 light-responsive elements respectively. Alleles of gene model SsSVP4/5 also showed significant difference in expression; Sspon.001C0005280 > Sspon.001D0004841 > Sspon.001B0036011 with 31, 17, 17 light-responsive elements, respectively. However, alleles within SsSOC1-6a, SsSVP-29 have a slight difference in expression and have an almost equal number of light-responsive elements. Moreover, the same allelic expression pattern trend was observed after hormone treatment (48 h) suggested that not all but some alleles within differentially expressed gene models are important in controlling traits in sugarcane. The distribution of hormone-related cis-elements was detected differently and showed in (Fig. 7). The enrichment of promoters containing light-responsive cis-elements in S. spontaneum MADS genes might not be ruled out at this point and it is an interesting question to be addressed in future research, once it is known that light-responsive element is involved in plant growth and development48, 49.
Quantitative real-time PCR (qRT-PCR) analysis of differentially expressed S. spontaneum MADS-box genes
According to the RNA-seq data, S.spontaneum MADS-box genes SsSVP-4/5, SsSOC1-6b, SsSOC1-6a, SsSVP-29 and SsSVP-17 were selected to validate the transcriptome data and subjected to qRT-PCR analysis. After normalization, we found that qRT-PCR results were consistent with the RNA-seq data for all the selected S. spontaneum MADS-box genes in root, leaves, different stem sections, and after ethylene treatment (Fig. 8). These results demonstrated that transcriptome data is appropriate for exploring the expression profiles of MADS-box genes in sugarcane.

Relative expression of S. spontaneum MADS-box gene models by qRT-PCR. Expression pattern of SsSOC1-6a, SsSOC1-6b, SsSVP-4/5, SsSVP-17 and SsSVP-29 are shown in different tissues (leaf, root, stem and under ethylene treatment). Blue bars represents the relative expression in qPCR and orange line represents the expression values (FPKM) in transcriptome for corresponding genes. Correlation coefficient is also given as “r”.
Discussion
In total, 182 MADS-box sequences were identified from the S. spontaneum genome. These were grouped into 63 genes, with four (6, 9.5%) three (21, 33.3%) two (29, 46%) and one (7, 11.1%) alleles. Based on phylogenetic relationships with A. thaliana and S. bicolor type-II genes, S. spontaneum type-II members were further classified into 10 sub-families. Interestingly, no FLC/FLM homologs were found in the S. spontaneum genome. This sub-family has been confirmed in regulating flowering time by autonomous pathways and vernalization50, 51. Similar results have been found in rice, sorghum, maize and cotton in the light of the fact that vernalization is not required for flowering in these species32, 52, 53.
The number of Introns-exon and their arrangement in gene structure, have an influence on alternative gene splicing to a certain extent with the alternation in gene function54. Due to structural complexity of S. spontaneum MADS-box type-II genes, it could be deduced that type-II genes had more complicated and variable functions comparing with type-I genes, that was in accordance with previous reports on soybean55, Arabidopsis2 and Chinese cabbage56. S. spontaneum MADS-box genes belonging to the same sub-families were structurally more similar with some variations in exon numbers, intron/exon length. Same trend has been observed for motif distribution in this study. Further comparison of gene structure and motif distribution of S. spontaneum MADS-boxgenes with sorghum highlighted the conserved evolution between these two species.
In sugarcane, ripening coincides with the process of sucrose accumulation at different physiological stages of stem57. In our study, expression pattern of flowering genes SVP and SOC1 (SsSOC1-6a, SsSOC1-6b, SsSVP-4/5, SsSVP-17, SsSVP-29) had significant transcript accumulation in stem 2 and 3 (maturation) indicate their diverse function in vegetative development. In banana, SOC1, SVP and CAL/FUL/AP1 highly expressed during fruit developmental and ripening processes, indicating their novel roles rather than functioning on flower58. Recent reports on tomato and soybean suggested the fact that MADS-box genes directly regulate a set of genes involved in sugar metabolism, thereby controlling fruit ripening and root development59, 60. In barley, ectopic expression of SVP-BM1 caused floral reversion and inhibited spike development, with florets at the base of the spike replaced by tillers61. While its overexpression produces chimeric floral structure bearing the typical feature of vegetative shoots so important for shoot identity62.
Interestingly, the same gene models of SOC1 and SVP showed higher expression in stem after ethylene and other hormones treatment suggested that these gene models may involve in the hormone signal transduction pathway to regulate vegetative development. Ethylene has a substantial effect on sugarcane maturation, as exogenous application inhibits the growth of young internodes by inducing sucrose accumulation63. In apple, MADS9 was found to act as a trans-activator for ACO1 and ACS1 promoters64. In banana, MaMADS7 (AG-like) interacted with a MaACO1 promoter65. In strawberry, FvSOC1 regulates the differentiation of axillary buds to runners, probably through the activation of gibberellin biosynthetic genes, which plays a central role in the photoperiodic control of both generative and vegetative growth66. Overexpression of KdSOC1 gene alter auxin distribution and accumulation along leaf margin to initiate plantlet formation and distribution, crucial for plasticity during plantlet formation under various environmental conditions67.
Furthermore, allelic variation based on expression data and promoter analysis confirms the fact that not all but some alleles within these genes are important for controlling important vegetative traits in sugarcane. These alleles are enriched with light-responsive elements involved in plant growth and development. So, it can be assumed that flower timing genes (SVP/SOC1), together with some other genes networks, may regulate vegetative and generative development through separate genetic pathways. The importance of these genes in vegetative development might not be ruled out at this point and it is an interesting question to be addressed in future research.
Conclusion
Our study comprehensively analyze MADS-box genes in wildtype sugarcane, S. spontaneum. We identify 182 sequences that are annotated into 63 genes with variable numbers of alleles and paralogs. More importantly, some floral regulator gene models SVP and SOC1 are highly expressed in stem sections, indicate their role in vegetative development. The same MADS-box genes express significantly after hormone treatment, suggesting that SVP and SOC1 could be involved in the hormone signal transduction pathway to regulate stem development. Of course, the more specific functional differentiation of those genes, specifically alleles, needs further study. Genome-based identification, particularly the tissue-specific expression of these genes, provides essential information for understanding the development and transcriptional regulation of the vegetative development and stem ripening in sugarcane and may potentially aid in the understanding of the molecular basis of many agriculturally important sugarcane traits.
Data availability
RNA-seq data was collected from Prof. Ray Ming and Prof. Jisen Zhang of National Sugarcane Engineering Technology Research Center, Fujian Provincial Key Laboratory of Haixia Applied Plant Systems Biology, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China.
References
Messenguy, F. & Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 316, 1–21 (2003).
Par̆enicová, L. et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: New openings to the MADS world. Plant Cell 15, 1538–1551 (2003).
De Bodt, S., Raes, J., Van de Peer, Y. & Theißen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 8, 475–483 (2003).
Kramer, E. M., Dorit, R. L. & Irish, V. F. Molecular evolution of genes controlling petal and stamen development: Duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149, 765–783 (1998).
Adamczyk, B. J. & Fernandez, D. E. MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol. 149, 1713–1723 (2009).
Becker, A. & Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489 (2003).
Alvarez-Buylla, E. R. et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. 97, 5328–5333 (2000).
Kaufmann, K., Melzer, R. & Theißen, G. MIKC-type MADS-domain proteins: structural modularity, protein interactions and network evolution in land plants. Gene 347, 183–198 (2005).
Nam, J. et al. Type I MADS-box genes have experienced faster birth-and-death evolution than type II MADS-box genes in angiosperms. Proc. Natl. Acad. Sci. 101, 1910–1915 (2004).
Ditta, G., Pinyopich, A., Robles, P., Pelaz, S. & Yanofsky, M. F. The SEP4 gene of Arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 14, 1935–1940 (2004).
Fang, S.-C. & Fernandez, D. E. Effect of regulated overexpression of the MADS domain factor AGL15 on flower senescence and fruit maturation. Plant Physiol. 130, 78–89 (2002).
Favaro, R. et al. MADS-box protein complexes control carpel and ovule development in Arabidopsis. Plant Cell 15, 2603–2611 (2003).
Ito, Y. et al. DNA-binding specificity, transcriptional activation potential, and the rin mutation effect for the tomato fruit-ripening regulator RIN. Plant J. 55, 212–223 (2008).
Liljegren, S. J. et al. SHATTERPROOF MADS-box genes control seed dispersal in Arabidopsis. Nature 404, 766 (2000).
Haughn, G. W. & Somerville, C. R. Genetic control of morphogenesis in Arabidopsis. Dev. Genet. 9, 73–89 (1988).
Zahn, L. M., Feng, B. & Ma, H. Beyond the ABC-model: Regulation of floral homeotic genes. Adv. Bot. Res. 44, 163–207 (2006).
Bowman, J. L., Alvarez, J., Weigel, D., Meyerowitz, E. M. & Smyth, D. R. Control of flower development in Arabidopsis thaliana by APETALA1 and interacting genes. Development 119, 721–743 (1993).
Theißen, G. Development of floral organ identity: Stories from the MADS house. Curr. Opin. Plant Biol. 4, 75–85 (2001).
Michaels, S. D. et al. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. 33, 867–874 (2003).
Grivet, L., Glaszmann, J.-C. & D'Hont, A. Molecular evidence for sugarcane evolution and domestication. In IVth Molecular Biology Workshop (CIRAD-CA, Montpellier, France, 2003).
Roach, B. Origin and improvement of the genetic base of sugarcane. Proc. Aust. Soc. Sugar Cane Technol. 11, 34–47 (1989).
D’Hont, A. et al. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharum spp.) by molecular cytogenetics. Mol. General Genet. MGG 250, 405–413 (1996).
Nayak, S. N. et al. Promoting utilization of Saccharum spp. genetic resources through genetic diversity analysis and core collection construction. PLoS ONE 9, e110856 (2014).
Todd, J. et al. Phenotypic characterization of the Miami World Collection of sugarcane (Saccharum spp.) and related grasses for selecting a representative core. Genet. Resources Crop Evolut. 61, 1581–1596 (2014).
Zhang, J. et al. Genome size variation in three Saccharum species. Euphytica 185, 511–519 (2012).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. https://doi.org/10.1038/s41588-018-0237-2 (2018).
Brandes, E. W. & Sartoris, G. B. Sugar-Cane: its origin and improvement. In Yearbook of the United States Department of Agriculture 561–624 (1936).
Bezerra, T. L. & Ragauskas, A. J. A review of sugarcane bagasse for second-generation bioethanol and biopower production. Biofuels Bioprod. Biorefin. 10, 634–647 (2016).
Finn, R. D. et al. Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 (2013).
Finn, R. D., Clements, J. & Eddy, S. R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37 (2011).
Jiangtao, C. et al. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan Hereditas 37, 91–97 (2015).
Zhao, Y. et al. Whole-genome survey and characterization of MADS-box gene family in maize and sorghum. Plant Cell Tissue Organ Culture (PCTOC) 105, 159–173 (2011).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evolut. 33, 1870–1874 (2016).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016).
Guo, A., Zhu, Q., Chen, X. & Luo, J. GSDS: A gene structure display server. Yi chuan Hereditas 29, 1023–1026 (2007).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562 (2012).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Chen, C., Chen, H., He, Y. & Xia, R. TBtools, a Toolkit for Biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv. https://doi.org/10.1101/289660 (2018).
Ling, H., Wu, Q., Guo, J., Xu, L. & Que, Y. Comprehensive selection of reference genes for gene expression normalization in sugarcane by real time quantitative RT-PCR. PLoS ONE 9, e97469 (2014).
Andrade, L. M. et al. Reference genes for normalization of qPCR assays in sugarcane plants under water deficit. Plant methods 13, 28 (2017).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001).
Zhang, Q. et al. Structure, phylogeny, allelic haplotypes and expression of sucrose transporter gene families in Saccharum. BMC Genomics 17, 88. https://doi.org/10.1186/s12864-016-2419-6 (2016).
Song, X. et al. Comprehensive analysis of the flowering genes in Chinese cabbage and examination of evolutionary pattern of CO-like genes in plant kingdom. Sci. Rep. 5, 14631 (2015).
Zhang, L. et al. Genome-wide identification, characterization of the MADS-box gene family in Chinese jujube and their involvement in flower development. Sci. Rep. 7, 1025 (2017).
Botha, F. C. & Black, K. G. Sucrose phosphate synthase and sucrose synthase activity during maturation of internodal tissue in sugarcane. Funct. Plant Biol. 27, 81–85 (2000).
Chow, C.-N. et al. PlantPAN3.0: A new and updated resource for reconstructing transcriptional regulatory networks from ChIP-seq experiments in plants. Nucleic Acids Res. 47, D1155–D1163 (2018).
Lescot, M. et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 30, 325–327 (2002).
Gangappa, S. N. & Botto, J. F. The multifaceted roles of HY5 in plant growth and development. Mol. Plant 9, 1353–1365. https://doi.org/10.1016/j.molp.2016.07.002 (2016).
Gangappa, S. N., Maurya, J. P., Yadav, V. & Chattopadhyay, S. The regulation of the Z- and G-box containing promoters by light signaling components, SPA1 and MYC2, Arabidopsis. PLoS ONE 8, e62194. https://doi.org/10.1371/journal.pone.0062194 (2013).
Helliwell, C. A., Wood, C. C., Robertson, M., Peacock, W. J. & Dennis, E. S. The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 46, 183–192 (2006).
Greb, T. et al. The PHD finger protein VRN5 functions in the epigenetic silencing of Arabidopsis FLC. Curr. Biol. 17, 73–78 (2007).
Arora, R. et al. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8, 242 (2007).
Ren, Z. et al. Genome-wide identification of the MIKC-type MADS-box gene family in Gossypium hirsutum L. unravels their roles in flowering. Front. Plant Sci. 8, 384 (2017).
Tian, Y. et al. Genome-wide identification and analysis of the MADS-box gene family in apple. Gene 555, 277–290 (2015).
Shu, Y., Yu, D., Wang, D., Guo, D. & Guo, C. Genome-wide survey and expression analysis of the MADS-box gene family in soybean. Mol. Biol. Rep. 40, 3901–3911 (2013).
Duan, W. et al. Genome-wide analysis of the MADS-box gene family in Brassica rapa (Chinese cabbage). Mol. Genet. Genomics 290, 239–255 (2015).
Fernandes, A. Refratômetro de campo. Boletim Técnico Copersucar São Paulo 19, 5–12 (1982).
Liu, J. et al. Genome-wide analysis of banana MADS-box family closely related to fruit development and ripening. Sci. Rep. 7, 3467 (2017).
Qin, G. et al. A tomato vacuolar invertase inhibitor mediates sucrose metabolism and influences fruit ripening. Plant Physiol. 172, 1596–1611. https://doi.org/10.1104/pp.16.01269 (2016).
Liu, W. et al. A novel sucrose-regulatory MADS-Box transcription factor GmNMHC5 promotes root development and nodulation in soybean (Glycine max [L.] Merr.). Int J. Mol. Sci. 16, 20657–20673 (2015).
Trevaskis, B. et al. Short vegetative phase-like MADS-box genes inhibit floral meristem identity in barley. Plant Physiol. 143, 225–235 (2007).
Liu, C. et al. Specification of Arabidopsis floral meristem identity by repression of flowering time genes. Development 134, 1901–1910 (2007).
Chong, B. F., Mills, E., Bonnett, G. D. & Gnanasambandam, A. Early exposure to ethylene modifies shoot development and increases sucrose accumulation rate in sugarcane. J. Plant Growth Regul. 29, 149–163 (2010).
Ireland, H. S. et al. Apple SEPALLATA1/2-like genes control fruit flesh development and ripening. Plant J. 73, 1044–1056 (2013).
Liu, J. et al. Role for the banana AGAMOUS-like gene MaMADS7 in regulation of fruit ripening and quality. Physiol. Plant. 155, 217–231 (2015).
Mouhu, K. et al. The Fragaria vesca homolog of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS1 represses flowering and promotes vegetative growth. Plant Cell 25, 3296–3310. https://doi.org/10.1105/tpc.113.115055 (2013).
Zhu, C. et al. Over-expression of KdSOC1 gene affected plantlet morphogenesis in Kalanchoe daigremontiana. Sci. Rep. 7, 5629 (2017).
Funding
Genome sequencing and annotation were supported by startup fund from Fujian Agriculture and Forestry University, the International Consortium for Sugarcane Biotechnology project #35, US DOE DE-SC0010686, and EBI BP2012OO2J17. Publication fee and qPCR experiment were supported by startup fund from Fujian Agriculture and Forestry University.
Author information
Authors and Affiliations
Contributions
M.F. and R.M. conceived the project and designed experiments. M.F., X.Z., and Z.P. carried out the experiment. M.F., J.L., and D.Z. analyzed the data. M.F. wrote the manuscript. R.M. revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fatima, M., Zhang, X., Lin, J. et al. Expression profiling of MADS-box gene family revealed its role in vegetative development and stem ripening in S. spontaneum. Sci Rep 10, 20536 (2020). https://doi.org/10.1038/s41598-020-77375-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-77375-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
