
Abstract
The vaginal microbiota is thought to play a role in modulating risk of high-risk human papillomavirus (hrHPV) infection. We examined the relationship between the vaginal microbiota and persistent hrHPV infection in HIV-negative and HIV-positive women. We used 16S-rRNA sequencing to characterize the vaginal microbiota of two serial samples taken six months apart from 211 Nigerian women (67%, 142/211 HIV-positive and 33%, 69/211 HIV-negative) and evaluated the association between the vaginal microbiota and persistent hrHPV infection using generalized estimating equation logistic regression models and linear discriminant analysis effect size (LEfSe) algorithm to identify phylotypic biomarkers of persistent hrHPV infection. The high diversity microbiota, Community State Type IV-B, was the most prevalent in both HIV-negative (38% at baseline, 30% at the follow-up visit) and HIV-positive (27% at baseline, 35% at the follow-up visit) women. The relationship between the vaginal microbiota and persistent hrHPV was modified by HIV status. In HIV-negative women, women with Lactobacillus dominant microbiota had lower odds (OR: 0.35, 95% CI 0.14–0.89, p = 0.03) of persistent hrHPV compared to women with Lactobacillus deficient microbiota. While among HIV-positive women, the odds of being persistently infected with hrHPV was higher in women with Lactobacillus dominant microbiota (OR: 1.25, 95% CI 0.73–2.14 p = 0.41). This difference in effect estimates by HIV was statistically significant (p = 0.02). A high diversity vaginal microbial community with paucity of Lactobacillus species was associated with persistent hrHPV infection in HIV-negative women but not in HIV-positive women.
Similar content being viewed by others
Introduction
Cervical cancer is the third commonest cancer among women globally, with 570,000 new cases and 311,400 deaths estimated to have occurred globally in 20181. Persistent infection with one of 12 high risk Human Papillomaviruses (hrHPV) is recognized as a necessary but insufficient cause of cervical intraepithelial neoplasia (CIN) and cervical cancer2. HPV infections are very common3. About 80% of sexually active women are infected with at least one HPV type at some point in their lifetimes3. However, around 90% of infections are cleared within 18 months, as a result of an incompletely understood host cell-mediated immune response4. Persistence of HPV infection leads to the accumulation of mutations in the somatic cellular genome which increases the risk of CIN and cervical cancer5. Interactions between host factors such as the microbiota and viral factors that facilitate persistence and carcinogenesis are not completely understood and remain an active area of research.
There is emerging evidence to suggest that the hosts’ vaginal microbiota play crucial roles in the pathogenesis of HPV infections6,7. The defence mechanisms against colonization of the vagina by pathogenic organisms include the presence of lactic acid producing Lactobacillus species8, an acidic environment9,10 and antimicrobial peptides10,11. Disruptions in these defence mechanisms, as could occur with high diversity vaginal microbiota, may modulate risk of viral infections such as HIV12 and HPV13. High diversity vaginal microbial communities are associated with proinflammatory responses which may directly damage the cervical epithelial barrier, thereby facilitating HPV entry to basal keratinocytes and establishment of infection4,14. The microbiota may also interact with cellular pathways in ways that are not yet clear, to lead to persistent, productive viral infections14,15,16,17.
Most 16S rRNA sequencing studies of the vaginal microbiota and hrHPV infection to date have focused on prevalent hrHPV infection and vaginal microbiome patterns6,13,16,18,19,20,21,22. Collectively, these studies showed increased risk of prevalent HPV infection in women with high diversity vaginal microbiota or with decreased relative abundance of Lactobacillus species13,16,19. Thus far, only one study has investigated the association between the vaginal microbiota and persistent hrHPV21. In that study, vaginal samples collected at one baseline visit were used to characterize the vaginal microbiota, while hrHPV persistence was determined by testing two serial samples taken 12 months apart21. Given that the vaginal microbiota is dynamic, cross-sectional studies of the vaginal microbiota that rely on vaginal sampling at a single time point are inadequate to fully characterize the relationship between the vaginal microbiota and persistent hrHPV23. Therefore, longitudinal studies with repeated vaginal microbiota assessments are warranted. Earlier work by Brotman et al. to address this knowledge gap involved extensive sampling of the vaginal microbiota and repeated HPV testing. However, that study was limited by the lack of data on high risk HPV24.
We previously reported an association between prevalent hrHPV and decreased relative abundance of Lactobacillus species18. In this study, we examine the relationship between the vaginal microbiota measured at two different time points and persistent hrHPV infection in a nested case control study of HIV-negative and HIV-positive Nigerian women.
Results
Study population
From a prospective population—based cohort study of 1020 women, we selected 211 women (94 cases of persistently any HPV infected and 117 controls of persistently HPV negative women) for inclusion in this nested case control study using a 1:1 match for HIV positive women and a 1:2 match for HIV negative women. Details of participant selection are provided in Fig. 1.

Participant flow chart and selection of participants in nested case control analysis.
Table 1 shows the baseline characteristics of the study population stratified by HIV status. Mean (SD) age of participants at enrolment was 38 (8) years while mean age at sexual debut was 20 (3) years. Most participants had more than six years of formal education (88%, 186/211) and were premenopausal (84%, 175/211). More than half of the participants were married (58%, 122/211) and douche regularly (60%, 126/211). The median interval (IQR) between baseline and follow-up visit was 6 (6–8) months.
Of the 117 controls (women who were persistently negative for HPV), 39% (46/117) were HIV negative. Of the 94 cases (women who were persistently positive for any HPV), 24% (23/94) were HIV negative. Among women with persistent any HPV infection, 76% (71/94) had persistent hrHPV infections.
Among cases, the commonest hrHPV types detected were HPV 52 and HPV 35 at both baseline (27%, 25/94 and 23%, 22/94 respectively) and follow up visits (31%, 29/94 and 19%, 18/94 respectively). Details of the frequency distribution of other high risk HPV types identified in women with persistent any HPV infection at baseline and follow up stratified by HIV status are shown in Table 2. In women with persistent hrHPV infection, type specific persistence (same hrHPV type present at baseline and follow up) was commoner (87%, 62/71) than group persistence (13%, 9/71). The commonest hrHPV types in type specific persistence were HPV 52 (31%, 22/71) and HPV 35 (24%, 17/71). Further details of HPV prevalence, persistence and clearance of the parent cohort have been previously published25,26.
There were 422 visits made by the 211 participants selected. Approximately, 84% (353/422) of vaginal microbiota samples taken at these visits were available for 16S rRNA sequence analysis which yielded 12, 967, 629 high quality reads. The mean (SD) number of reads per sample was 36, 021 (14, 375), with a mean (SD) read length of 427 (6) base pairs.
Vaginal microbiome composition in the study population
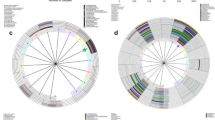
Hierarchical clustering analysis based on species composition and relative abundance identified five major clusters that showed similarities to previously described vaginal microbiome community state types (CST)23. Three of these CSTs were dominated by high proportions of Lactobacillus specie: L. crispatus in CST I, L. gasseri in CST II and L. iners in CST III (Fig. 2). CST I-B had moderately high proportions of Lactobacillus species, most commonly L. crispatus as well as low proportions of pathobionts such as Enterococcus and Streptococcus, which are not commonly seen in CST I. CST IV-B was characterized by greater taxonomic diversity with low proportions of Lactobacillus, pathobionts such as Escherichia, Enterococcus and Streptococcus as well as bacterial vaginosis associated taxa such as Gardnerella, Atopobium, Megasphaera, Sneathia and Prevotella.

Heatmap of the 40 most abundant bacterial taxa in 211 Nigerian women. Hierarchical clustering identified five major clusters. Three of these clusters were dominated by one Lactobacillus species: L.crispatus (CST I), L.gasseri (CST II) and L.iners (CST III). CST I-B and CST IV-B were more diverse with CST I-B having modest proportions of L.crispatus. Ward linkage clustering was used to cluster samples based on their Jensen-Shannon distance calculated in the Vegan package in R66.
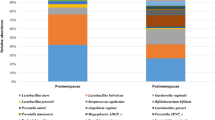
Alpha diversity was significantly lower in CST I compared to CST II (p < 0.001), CST I-B (p < 0.001) and CST IV-B (p < 0.001). There were no significant differences in alpha diversity between CST I and CST III (p = 0.06) (Fig. 3). Lactobacillus species were present in all CSTs (Fig. 4). The median (IQR) relative abundance of Lactobacillus species was 97% (90–98%) in CST I; 75% (50–93%) in CST II; 95% (87–98%) in CST III; 77% (57–95%) in CST I-B and 27% (11–42%) in CST IV-B.

Shannon diversity indices as a measure of alpha diversity by community state types. Compared to CST I, alpha diversity is significantly higher in CST II, CST I-B and CST IV-B.

Relative abundance of Lactobacillus species by Community State Assignments. Lactobacillus species were present in all communities. CST I-B had modest proportions of L.crispatus as well as other Lactobacillus species, most commonly L.rhamnosus. CST IV-B was the most diverse community with higher proportions of non-Lactobacillus species.
In HIV negative women, the point prevalence of CST IV-B was 38% (26/69) at the baseline visit and 30% (21/69) at the follow up visit (Table 3). The difference in proportions (0.12; 95% CI − 0.06–0.30; p = 0.20) was not statistically significant. In HIV positive women, the point prevalence of CST IV-B was 27% (38/142) at the baseline visit and 35% (50/142) at the follow up visit. This CST distribution at the baseline and follow up visit was not statistically significant (difference in proportions = 0.06, 95% CI = − 0.10–0.14, p = 0.75). Of the Lactobacillus species dominant CSTs, CST III was the most prevalent at both baseline (15%, 10/69) and follow up (30%, 21/69) visits in HIV negative women. In HIV positive women, the most prevalent Lactobacillus species dominant CST was CST III at the baseline visit (9%, 13/142) and CST II at the follow up visit (15%, 21/142) (Table 3).
Markov chain transition probabilities indicated substantial variability in inter-CST transitions over the two-time points (baseline and follow-up visits) (Fig. 5). Patterns of transition probabilities were comparable among HIV positive and HIV negative women. In both HIV negative and HIV positive women, the highest inter-state transition probabilities were to the more diverse community (CST IV-B). While the highest self-transition probability, the probability that a CST would be the same at both baseline and follow up, was observed for CST IV-B in both HIV negative and HIV positive women, indicating that CST IV-B was the most stable CST among women in this study.

Markov chain transition probabilities over two time periods, approximately six months apart by HIV status. Arrow weights are proportional to the maximum likelihood estimate of the transition probability between CSTs at the baseline and follow up visits. Node sizes scale with the number of women in each CST. Numeric transition probabilities are shown on arrows for transition probabilities > 0.30. Markov chain schematic transitions were modelled using the markovchain package in R67.
In HIV negative women, transition probabilities from other CSTs to CST IV-B were 0.40 (CST I), 0.67 (CST II), 0.57 (CST III), and 0.33 (CST I-B). There were no transitions from a Lactobacillus dominant CST (CST I, CST II, CST III) to CST I-B, while CST II was the least connected CST. Transitions from CST IV-B were most commonly to CST III (transition probability = 0.55). In HIV positive women, transition probabilities from other CSTs to CST IV-B were 0.18 (CST I), 0.33 (CST II), 0.38 (CST III) and 0.33 (CST I-B). There were no transitions from CST II to CST I-B.
Association between demographic, lifestyle and clinical characteristics and the vaginal microbiome
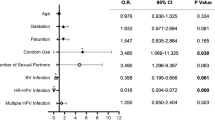
We evaluated potential demographic and behavioural correlates for a Lactobacillus species dominant microbiota and a Lactobacillus crispatus dominant microbiota, treating the vaginal microbiota as a time varying response variable with measurements at two time points – baseline and follow up visits (Table 4). In univariate analyses, being HIV-positive was significantly associated with reduced odds of having a Lactobacillus species dominant microbiome (OR: 0.53, 95% CI 0.34–0.84, p = 0.007) or a L.crispatus dominant microbiome (OR: 0.33, 95% CI 0.15–0.72, p = 0.006). Adjustments for age in multivariable models did not significantly change this relationship. Other than HIV status, there were no other associations between demographic, lifestyle, or clinical characteristics and the vaginal microbiota.
Association between persistent hrHPV infection and vaginal microbiota
A priori, we considered the possibility of HIV modifying the relationship between the vaginal microbiota and persistent hrHPV based on results from previous studies and biological plausibility18, 27. In effect modification analysis, the odds ratio of having persistent hrHPV for women with a Lactobacillus dominant microbiota (> 70%) versus a Lactobacillus depleted microbiota was multiplied by a factor of 3.39 (95% CI 1.18–9.70, p = 0.02) for HIV-positive women as compared to HIV-negative women. Therefore, we present effect estimates by HIV strata.
Among HIV negative women, the odds of persistent hrHPV infection were significantly lower in women whose microbiota were dominated by Lactobacillus species (OR: 0.35, 95% CI: 0.14–0.89, p = 0.03) than women with Lactobacillus depleted microbiota. In HIV positive women, there was no significant association between persistent hrHPV infection and the vaginal microbiota in the Lactobacillus dominant model (OR: 1.25, 95% CI: 0.73–2.14, p = 0.41) (Table 5). There were also no significant associations between the vaginal microbiota and persistent hrHPV in the other models (L.crispatus dominant [> 70%] model and Community State Assignment model) considered for both HIV-negative and HIV-positive women.
In sensitivity analyses varying cut points to dichotomize relative abundance of Lactobacillus species, with values ranging from 50 to 80% in increments of 5%, the association between Lactobacillus dominated microbiota and persistent hrHPV in HIV negative women was insensitive to cut off points between 50 and 80% indicating that results were robust within this range of cut points (Supplementary Table 1). Analysis of characteristics of participants missing microbiome data indicated that missingness at baseline was associated with socioeconomic status; while missingness at follow up was associated with HIV status (Supplementary Table 2). These variables (socioeconomic status, study visit and HIV status) were used as auxiliary variables during multiple imputation for sensitivity analysis. Effect estimates from the imputed data were similar to effect estimates obtained from complete case analysis (Supplementary Table 3).
We used LEfSe modelling to identify specific bacterial taxa that may be differentially abundant in HIV negative women with and without persistent hrHPV; and HIV positive women with and without persistent hrHPV. We included HIV positive women in LEfSe analysis as the broad categories in the regression models may mask differences in non-dominant bacterial taxa. We found that in HIV-negative women, women with persistent hrHPV infection had significant over-representation of Leptotrichia species in their vaginal microbiota compared to HIV-negative women without persistent hrHPV infection (Fig. 6) Conversely, there was significant overrepresentation of Veillonella species in HIV-negative women who were persistently hrHPV negative compared to HIV negative women with persistent hrHPV infection (Fig. 6) . In HIV-positive women, we did not identify any discriminative features between the vaginal microbiota of women with persistent hrHPV infection and women who were persistently HPV negative.

Identified phylotype biomarkers of persistent hrHPV infection among HIV negative women. The phylotype biomarkers are identified as being significantly abundant at alpha value < 0.05. The analysis was run and visualized with the LEfSe program using the Galaxy interface68. The Linear Discriminant Analysis (LDA) score is indicated at the bottom. The greater the LDA score, the more significant the phylotype biomarker is.
Discussion
In this nested case control study of the vaginal microbiota and persistent hrHPV infection in HIV-negative and HIV-positive women, we found that a high relative abundance of Lactobacillus species was associated with reduced odds of persistent hrHPV infection in HIV-negative women but not in HIV-positive women. These results extend findings from our previous study, in which we showed a significant association between a low relative abundance of Lactobacillus species and increased odds of prevalent hrHPV, a more proximal causal intermediate in the relationship between hrHPV and cervical cancer18. Consistent with our previous report on prevalent hrHPV and other studies, we also identified Leptotrichia species to be over-represented in HIV-negative women with persistent hrHPV18,21.
While recent longitudinal studies with repeated vaginal microbiota assessments that use next generation sequencing techniques have shown an association between higher vaginal microbial diversity and prevalent hrHPV or persistent any HPV, our study is the first to show this for persistent hrHPV19,24. In one study conducted among Italian women, Di Paola et. al. provide evidence to show that a high diversity vaginal microbiota, CST IV, at baseline was predictive of future odds of persistent hrHPV, despite not accounting for the temporal dynamics of the vaginal microbiota21. Several other cross-sectional studies have showed that decreased relative abundance of Lactobacillus species is associated with precancerous cervical lesions and cervical cancer28,29,30. Collectively, these results suggest that the vaginal microbiome may play a role in cervical cancer pathogenesis.
It is plausible that the vaginal microbiome modulates risk of HPV acquisition31, although there is limited mechanistic evidence for this relationship. It is also possible, though less probable, that the presence of HPV interacts with the host immune system through complex mechanisms that lead to the presence of immunosuppressive cytokines that favour different vaginal communities17,32,33. Alternatively, sexual activity, a common cofactor for both vaginal microbiota and HPV infection may explain the associations observed.
Consistent with previous studies on the temporal dynamics of the vaginal microbiota, we found that CST IV-B was the most stable CST and when transitions did occur, they were most commonly to the L.iners dominated CST III23. These findings of the stability of CST IV-B may be considered to be qualitatively similar to a longitudinal study among 80 African women which showed that high diversity associated bacteria such as Prevotella bivia and Gadnerella vaginalis were consistently present in 91% and 58% of healthy women with a Nugent score of 0–3 respectively, while L. crispatus was consistently present in only 47%34. These findings may reflect a lack of optimal protection against the growth of strict anaerobes by L. iners or the ability of L.iners to thrive in a range of metabolic stress related conditions that other Lactobacillus species cannot survive in35. Future studies on the functional metabolomics profile associated with L. iners may help to understand the role of this specie in the maintenance and potential restoration of a healthy vaginal microbiota.
We found that HIV-positive women were less likely to have a high relative abundance of Lactobacillus species or L. crispatus compared to HIV-negative women. Microbial diversity has been reported to be an important risk factor for HIV acquisition and viral shedding20,36,37. Recent reports, in which HIV susceptible CD4 + cells are increased in the genital tract in women with high diversity microbial communities, provide further mechanistic evidence for the link between increased risk of HIV acquisition and viral shedding15,16. In addition, it is possible that the lower relative abundance of Lactobacillus species among HIV positive women in our study was due to immunosuppression resulting in subsequent colonization of genital tract by diverse bacteria.
Similar to previous studies, we did not find any significant association between vaginal douching and the vaginal microbiota20,34,38,39. The influence of hygienic practices such as vaginal douching on the microbiota may be mediated by factors such as the type of materials used for douching and timing in the menstrual cycle40,41. In a meta-analysis of intra-vaginal practices and bacterial vaginosis, which included 13 prospective studies in Sub Saharan Africa, vaginal douching with soap, but not other materials, was associated with bacterial vaginosis41. In our study population, majority of women who reported douching regularly, used water to douche, providing some explanation for our findings.
In this analysis, we did not find any association between lifetime number of sexual partners, number of partners in past year, recent sexual activity and the vaginal microbiota. Previous studies have reported reduced Lactobacillus species in women with recent sexual activity23,34. Our inability to detect these relationships may be due to the small sample size of women who reported recent sexual activity in our study.
Among HIV negative women, results from LEfSe analysis indicated that while Leptotrichia species was differentially more abundant in persistent hrHPV infected women, Veillonella species was differentially more abundant in hrHPV negative women. The association between Letptotrichia species and hrHPV infection has been reported in previous studies18,21. Less frequently reported is the observation of a higher abundance of Veillonella in hrHPV negative women. Veillonella are anaerobic, gram negative cocci commonly found in the oral cavity, gut and vaginal tract that play diverse roles in different niches42. The pathogenic role of Veillonella is uncertain and they are currently considered to be of low virulence43,44. While Veillonella species have been associated with more diverse microbial communities, including the formation of biofilms leading to dental plaques in the oral cavity and serious infections such as endocarditis45, they have also been found to be more abundant in HPV negative women compared to HPV positive women13,46, as well as in the oral microbiota of HPV negative infants compared to the oral microbiota of HPV positive infants46. Paradoxically, while they may be less common in HPV positive vaginal microbiota, they are increasingly more abundant in distal end points in the natural history of cervical cancer pathogenesis, such as in precancerous lesions13, Bogert et al. observed that the cytokine response profile of Veillonella species within the gut microbiota is dependent on the combinations of the bacteria species present47. These findings taken together with the paradoxical observations, may highlight the interplay between the integrated responses to multiple representative species within a microbial environment in influencing disease risk and warrant a deeper functional characterization of the vaginal microbiota.
The lack of association between Lactobacillus as a genus and hrHPV negative status in LEfSe analysis is noteworthy, given the strong associations between Lactobacillus species and hrHPV negative status that has been reported in previous studies. However, in this population, it must be noted that the commonest and the most stable microbial communities was the diverse CST IV-B community. This provides credence to current understanding that even in diverse communities, the important catabolic function, of lactic acid production, is conserved among communities despite differences in species composition.
Definitions of HPV persistence vary considerably across studies48. The commonest definitions include HPV positivity measured at two or more time points with an interval of six or more months48. At one extreme of this definition is the requirement for the same HPV genotype to be present at both time points (genotype-specific persistence), and the other extreme is the presence of any HPV at both time points (group persistence). Conceivably, HPV genotype-specific persistence is better suited for identifying women at increased risk of cervical cancer and its precursor lesions, as group persistence could be the result of one HPV type clearing and another being acquired. However, results from a population-based cohort study in Denmark have shown that women with hrHPV group persistence have increased odds of high-grade precancerous lesions that are almost 200-fold higher than observed in women persistently negative for HPV49. Furthermore, in studies investigating the use of group persistence versus genotype specific persistence to inform referral practices, the use of genotype specific persistence did not improve risk stratification over the use of group persistence in women referred for colposcopy indicating that group persistence is a good proxy for measuring type-specific persistence and risk of high grade precancerous lesions50,51.
While our study is the single largest nested case control study of persistent hrHPV and the vaginal microbiota among HIV-negative and HIV-positive women in Africa to date, it has some limitations. As with all case control studies, there is potential for selection bias when the selected cases and controls are systematically different from the target population especially when the selection criteria are associated with both the outcome and the exposure of interest52. In this study, the risk of selection bias is minimized for two reasons. First the parent cohort is a longitudinal population-based cohort; cases and controls selected from this cohort are representative of the source population with regards to exposure—outcome relationships. However, the prevalence of the exposure and outcome may vary substantially between the selected study population and the source population. Therefore, we limit our inferences to associations between exposure and outcome and make no inferences about the generalizability of the prevalence estimates of hrHPV or vaginal microbiota observed in this population. Secondly, while our selection criterion was associated with the outcome (all cases were selected), it was not associated with the exposure of interest. This reduces the risk of bias due to selection.
We included microbiome data from only two time points and did not perform qPCR to determine concentration. The human vaginal microbiota composition can be highly variable over short periods of time due to several factors, therefore sampling over shorter time intervals for extended periods are required. However, we did not find significant transitions of CST-IV to other CSTs, suggesting that additional sampling among women with CST IV may not have changed our results. There was some degree of missingness for vaginal microbiota data which could be a source of bias if differential. However, results from sensitivity analysis where missing vaginal microbiota was imputed based on visit (baseline or follow up), socioeconomic status and HIV status was not markedly different from the results based on complete non-missing data. We assessed HIV status at enrolment into the study but did not repeat HIV testing at follow up. It is possible that some women classified as HIV negative at the beginning of the study seroconverted during follow up, resulting in misclassification. We did not account for the CD4 + counts, viral loads, ART use or duration of infection in the HIV positive women included in this study and these may have affected our findings for HIV positive women.
In summary, we found that HIV negative women with a high relative abundance of Lactobacillus species in the vaginal microbiota had lower odds of persistent hrHPV compared to HIV negative women with lower relative abundance of Lactobacillus species. There was also an inverse relationship between the relative abundance of Lactobacillus species and HIV infection, such that women who were HIV-positive had lower odds of a Lactobacillus specie dominant vaginal microbiota. Further work is required to understand the functional profile of the vaginal microbial communities and how this interacts with host factors to modulate risk of hrHPV persistence in the pathogenesis of cervical cancer.
Methods
Study population
We enrolled 1,020 women from our cervical cancer screening studies into a prospective study of host and viral factors associated with persistent hrHPV infection in Abuja, Nigeria between 2012 and 201353,54. We included non-pregnant women who were at least 18 years old, had engaged in vaginal sexual intercourse and had no previous history of hysterectomy or cervical cancer. Trained nurses collected information on sociodemographic characteristics, self-reported HIV status, lifestyle factors, sexual behaviour and reproductive history from all participants. Participants were scheduled for a follow-up visit six months after the baseline visit. At both baseline and follow-up visits, nurses performed gynaecological examinations and collected biological specimens.
For this analysis, we used a nested case control design restricted to 211 women selected as cases and controls. Details of patient selection are provided in Fig. 1. Of the 1020 women in the parent cohort, 23 HIV negative women and 71 HIV positive women were persistently positive for any HPV. All of these 94 women were selected as cases in this nested study. For each case we identified either one (for HIV positive cases) or two (for HIV negative cases) controls from the cohort, matched by age (± 5 years) and study enrolment date (± 5 days) resulting in 117 controls in total. These 211 women provided data at two clinic visits resulting in 422 visits.
Sample collection and processing
We collected mid-vaginal swabs from all participants using the Elution Swab system (Copan, Italy) which was inserted in one ml Amies’ Transport media (Copan, Italy) and stored at − 80 °C till further processing. Whole genomic bacterial DNA was extracted from the swabs using the MoBio Powersoil kit, as previously described18.
Bacterial 16S rRNA gene sequencing and analysis
A dual barcode system with fusion primers 338F and 806R was used to amplify the V3–V4 hypervariable regions of the 16S rRNA gene as previously described55,56. Purified amplicons were sequenced on an Illumina Miseq instrument (Illumina, San Diego, CA) using a 300 bp paired end protocol at the Institute for Genome Sciences, University of Maryland School of Medicine. Raw reads were pre-processed to remove the first three and last three bases if their Phred score was lower than 3. Read ends were trimmed if the average Phred quality score of four consecutive bases was below 15 and only retained if their length was at least 75% of their original length after trimming. QIIME (v1.8.0)57 was used to perform quality control of the sequence reads. Fast Length Adjustment of Short reads (FLASH)58 was used to assemble paired reads, with an overlap of ~ 90 bp on average. De-multiplexing by binning sequences with the same barcode was performed in QIIME (v1.8.0). Chimera detection was done using de novo and referenced-based methods in UCHIME (v5.1) with the Greengenes database of 16S rRNA gene sequences (August 2013) as a reference59,60. The processed 16S rRNA gene amplicon sequences were assigned to genera and species, using PECAN that uses fifth-order Markov Chain model for precise species-level assignments and a pre-compiled database that contains all known microbes in the vaginal microbiota. Raw, demultiplexed, sequence files are available at the NBCI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with accession code PRJNA668516. Hierarchical clustering analysis based on Jensen Shannon distances using ward linkage was used to cluster samples into Community State Types (CSTs) using the vegan package in R23.
HPV detection
We extracted DNA from ectocervical cells and used the SPF10LiPA25 system for HPV DNA detection61. Briefly, we performed polymerase chain reaction on DNA isolates using biotin labeled SPF10 primers which generated small amplicons of 65 bp. The amplified PCR products were tested using DNA Enzyme Immunoassay (DEIA), a probe hybridization assay containing a cocktail of conservative probes that allows the recognition of at least 65 HPV genotypes. The amplimers of the DEIA HPV DNA positive samples were subsequently analyzed by the reverse hybridization line probe assay, LiPA25, Version 1 (Laboratory Biomedical Products, Rijswijk, The Netherlands) which can identify 25 HPV types, 12 of which are high-risk: 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59. In this analysis, we use group persistence as the definition of persistent hrHPV, that is, the presence of at least one hrHPV type at baseline and one hrHPV type at the follow up visit as opposed to type specific persistence, which is the presence of identical hrHPV types at the two visits.
Covariates
We collected information on potential confounders such as menopausal state, time since last menstrual period, douching practice, total number of sexual partners in past year, body mass index (BMI), vaginal pH, sexual activity within 24 h of sampling, use of condoms and hormonal contraceptive use. Vaginal pH was assessed as an ordinal variable with three categories: < 4.5, 4.5–5.5 and > 5.5. These covariates were assessed at the baseline visit and updated at the follow up visit. They were included in the models as time varying exposures. The vaginal microbiota was also modelled as a time varying exposure. In addition to CST based models, we conducted analysis based on the relative abundance of Lactobacillus (at the genus level) and L. crispatus. We specifically investigated L.crispatus at the species level because, of all the species of the Lactobacillus genus, commonly identified in the vaginal microbiota, communities with high relative abundance of L.crispatus have the lowest vaginal pH values, a marker for important vaginal defence mechanisms20,33,62.
Statistical analysis
We compared the characteristics of participants at baseline by HIV status. We used generalized estimating equation (GEE) logistic regression models with robust variance estimation and the cluster option, which accounts for repeated measures, to investigate the relationship between potential risk factors and the vaginal microbiota; and the association between the vaginal microbiota and persistent hrHPV infection. Variables considered for inclusion in multivariable regression models were selected using a two-step approach. First, sociodemographic characteristics and potential risk factors associated with persistent HPV infection at a nominal p < 0.10 in the parent cohort of this study were identified from previously published results25. The variables identified from this step were BMI, marital status, education, socioeconomic status, age at sexual initiation, total lifetime sex partners, vaginal douching, vaginal pH and HIV status. Next, we examined potential risk factors and its association with vaginal microbiota in this cohort of participants. Variables that were significantly associated with the vaginal microbiota at p < 0.10 were identified. Finally, variables that were associated with both persistent HPV infection and the vaginal microbiota were selected for inclusion in multivariable models.
In modelling the vaginal microbiota, we considered three approaches: dichotomising based on L. crispatus relative abundance (using a cut off of 70%), dichotomising based on all Lactobacillus species relative abundance (using a cut off of 70%) and using CST assignments. To select the cut-off point, we used the mean of the relative abundance of L. crispatus in CST I because L. crispatus is often considered a marker of a healthy microbiome19,63. We investigated the sensitivity of results to various cut points by varying the cut points in increments of 5% from 50 to 80%. The upper and lower bounds for these sensitivity analyses were selected to ensure that we reached a point where further dichotomization would become uninformative.
We used Markov chain models generated by inferring transition probabilities from our data to evaluate vaginal community dynamics over the two time periods, the baseline visit and the follow up visit.
We used Linear Discriminant Analysis (LDA) effect size (LEfSe) algorithm to identify bacterial taxa that were differentially present in women with persistent hrHPV infections64.
Of the 422 clinical visits included in this study, results for 69 vaginal microbiota samples were missing. We performed univariate analysis for missingness at baseline and at the follow up visit to determine whether missingness was associated with important covariates such as HIV status, persistent hrHPV, age, socioeconomic status, education, menopausal state, time since last menstrual period, douching practice, BMI, sex within the last 24 h prior to sample collection and total number of sexual partners in the past one year. Furthermore, to minimize the potential for selection bias, we imputed missing microbiota data using a data augmentation algorithm in a Markov Chain Monte Carlo (MCMC) procedure creating 10 datasets in 1000 iterations. We included three variables as auxiliary predictor variables (socioeconomic status, study visit and HIV status) identified from the univariate analysis of missingness. Next, we implemented GEE logistic regression models in the imputed dataset and compared results obtained from the imputed data with results obtained from complete case analysis.
Based on previous studies that have provided evidence of increased microbial diversity and a higher risk of persistent hrHPV infections among HIV positive women compared to HIV negative women25,27,65, we considered the possibility of HIV modifying the relationship between the vaginal microbiota and persistent hrHPV infection, a priori. We tested for this statistically by fitting an interaction term in the GEE logistic regression model and comparing the fitted model to a model without the interaction term using likelihood ratio tests. The interaction term was statistically significant (p = 0.02). Therefore, we present results by HIV strata.
All analyses were conducted using Stata/SE 15 (StataCorp, College Station, Texas) and R version 3.4.0 (R Core Team, Vienna, Australia).
Ethics
All methods were carried out in accordance with relevant guidelines and regulations. We obtained ethical approval to conduct this study from the National Health Research Ethics Committee of Nigeria and the University of Maryland School of Medicine Institutional Review Board. All participants provided written informed consent prior to enrolment.
References
Ferlay, J. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer 144, 1941–1953. https://doi.org/10.1002/ijc.31937 (2019).
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. A Review of Human Carcinogens. Part B: Biological agents. (Lyon, France, 2009).
Baseman, J. G. & Koutsky, L. A. The epidemiology of human papillomavirus infections. J. Clin. Vitol. 32(Suppl 1), S16-24. https://doi.org/10.1016/j.jcv.2004.12.008 (2005).
Doorbar, J. Host control of human papillomavirus infection and disease. Best Pract. Res. Clin. Obstet. Gynaecol. https://doi.org/10.1016/j.bpobgyn.2017.08.001 (2017).
Doorbar, J., Egawa, N., Griffin, H., Kranjec, C. & Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 25(Suppl 1), 2–23. https://doi.org/10.1002/rmv.1822 (2015).
Mitra, A. et al. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: What do we know and where are we going next?. Microbiome. 4, 58. https://doi.org/10.1186/s40168-016-0203-0 (2016).
van de Wijgert, J. H. et al. The vaginal microbiota: What have we learned after a decade of molecular characterization?. PLoS ONE 9, e105998. https://doi.org/10.1371/journal.pone.0105998 (2014).
Hillier, S. L., Krohn, M. A., Klebanoff, S. J. & Eschenbach, D. A. The relationship of hydrogen peroxide-producing lactobacilli to bacterial vaginosis and genital microflora in pregnant women. Obstet. Gynecol. 79, 369–373 (1992).
Gong, Z., Luna, Y., Yu, P. & Fan, H. Lactobacilli inactivate Chlamydia trachomatis through lactic acid but not H2O2. PLoS ONE 9, e107758. https://doi.org/10.1371/journal.pone.0107758 (2014).
O’Hanlon, D. E., Moench, T. R. & Cone, R. A. In vaginal fluid, bacteria associated with bacterial vaginosis can be suppressed with lactic acid but not hydrogen peroxide. BMC Infect. Dis. 11, 200. https://doi.org/10.1186/1471-2334-11-200 (2011).
Doerflinger, S. Y., Throop, A. L. & Herbst-Kralovetz, M. M. Bacteria in the vaginal microbiome alter the innate immune response and barrier properties of the human vaginal epithelia in a species-specific manner. J. Infect. Dis. 209, 1989–1999. https://doi.org/10.1093/infdis/jiu004 (2014).
Gaiha, G. D. et al. Surfactant protein A binds to HIV and inhibits direct infection of CD4+ cells, but enhances dendritic cell-mediated viral transfer. J. Immunol. 181, 601–609. https://doi.org/10.4049/jimmunol.181.1.601 (2008).
Lee, J. E. et al. Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS ONE 8, e63514. https://doi.org/10.1371/journal.pone.0063514 (2013).
Borgdorff, H. et al. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. 9, 621–633. https://doi.org/10.1038/mi.2015.86 (2016).
Anahtar, M. N. et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity 42, 965–976. https://doi.org/10.1016/j.immuni.2015.04.019 (2015).
Shannon, B. et al. Association of HPV infection and clearance with cervicovaginal immunology and the vaginal microbiota. Mucosal Immunol. 10, 1310–1319. https://doi.org/10.1038/mi.2016.129 (2017).
Bermudez-Morales, V. H., Gutierrez, L. X., Alcocer-Gonzalez, J. M., Burguete, A. & Madrid-Marina, V. Correlation between IL-10 gene expression and HPV infection in cervical cancer: A mechanism for immune response escape. Cancer Invest. 26, 1037–1043. https://doi.org/10.1080/07357900802112693 (2008).
Dareng, E. O. et al. Prevalent high-risk HPV infection and vaginal microbiota in Nigerian women. Epidemiol. Infect. https://doi.org/10.1017/s0950268815000965 (2015).
Reimers, L. L. et al. The Cervicovaginal microbiota and its associations with human papillomavirus detection in HIV-infected and HIV-uninfected women. J. Infect. Dis. 214, 1361–1369. https://doi.org/10.1093/infdis/jiw374 (2016).
Borgdorff, H. et al. Lactobacillus-dominated cervicovaginal microbiota associated with reduced HIV/STI prevalence and genital HIV viral load in African women. ISME J. 8, 1781–1793. https://doi.org/10.1038/ismej.2014.26 (2014).
Di Paola, M. et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk human Papillomavirus infection. Sci. Rep. 7, 10200. https://doi.org/10.1038/s41598-017-09842-6 (2017).
Norenhag, J. et al. The vaginal microbiota, human papillomavirus and cervical dysplasia: A systematic review and network meta-analysis. BJOG https://doi.org/10.1111/1471-0528.15854 (2019).
Gajer, P. et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 4, 132ra152. https://doi.org/10.1126/scitranslmed.3003605 (2012).
Brotman, R. M. et al. Interplay between the temporal yynamics of the vaginal microbiota and human Papillomavirus detection. J. Infect. Dis. 210, 1723–1733. https://doi.org/10.1093/infdis/jiu330 (2014).
Adebamowo, S. N. et al. Persistent low-risk and high-risk human papillomavirus infections of the uterine cervix in HIV-negative and HIV-positive women. Front. Public Health 5, 178. https://doi.org/10.3389/fpubh.2017.00178 (2017).
Adebamowo, S. N. et al. Clearance of type-specific, low-risk, and high-risk cervical human papillomavirus infections in HIV-negative and HIV-positive women. J. Glob. Oncol. 4, 1–12. https://doi.org/10.1200/jgo.17.00129 (2018).
Klein, C. et al. Relationship between the cervical microbiome, HIV status, and precancerous lesions. mBio https://doi.org/10.1128/mBio.02785-18 (2019).
Mitra, A. et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 5, 16865. https://doi.org/10.1038/srep16865 (2015).
Oh, H. Y. et al. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin. Microbiol. Infect 21(674), e671-679. https://doi.org/10.1016/j.cmi.2015.02.026 (2015).
Piyathilake, C. J. et al. Cervical microbiota associated with higher grade cervical intraepithelial neoplasia in women infected with high-risk human papillomaviruses. Cancer Prev. Res. (Phila) 9, 357–366. https://doi.org/10.1158/1940-6207.capr-15-0350 (2016).
Rose, W. A. 2nd. et al. Commensal bacteria modulate innate immune responses of vaginal epithelial cell multilayer cultures. PLoS ONE 7, e32728. https://doi.org/10.1371/journal.pone.0032728 (2012).
Giannini, S. L. et al. Cytokine expression in squamous intraepithelial lesions of the uterine cervix: Implications for the generation of local immunosuppression. Clin. Exp. Immunol. 113, 183–189. https://doi.org/10.1046/j.1365-2249.1998.00639.x (1998).
Audirac-Chalifour, A. et al. Cervical microbiome and cytokine profile at various stages of cervical cancer: A pilot study. PLoS ONE 11, e0153274. https://doi.org/10.1371/journal.pone.0153274 (2016).
Jespers, V. et al. A longitudinal analysis of the vaginal microbiota and vaginal immune mediators in women from sub-Saharan Africa. Sci. Rep. 7, 11974. https://doi.org/10.1038/s41598-017-12198-6 (2017).
Macklaim, J. M., Gloor, G. B., Anukam, K. C., Cribby, S. & Reid, G. At the crossroads of vaginal health and disease, the genome sequence of Lactobacillus iners AB-1. Proc. Natl. Acad. Sci. U.S.A. 108, 4688–4695. https://doi.org/10.1073/pnas.1000086107 (2011).
Atashili, J., Poole, C., Ndumbe, P. M., Adimora, A. A. & Smith, J. S. Bacterial vaginosis and HIV acquisition: A meta-analysis of published studies. AIDS 22, 1493–1501. https://doi.org/10.1097/QAD.0b013e3283021a37 (2008).
Mitchell, C. et al. Interaction between lactobacilli, bacterial vaginosis-associated bacteria, and HIV type 1 RNA and DNA genital shedding in US and Kenyan women. AIDS Res. Hum. Retroviruses 29, 13–19. https://doi.org/10.1089/aid.2012.0187 (2013).
Borgdorff, H. et al. The association between ethnicity and vaginal microbiota composition in Amsterdam, the Netherlands. PLoS ONE 12, e0181135. https://doi.org/10.1371/journal.pone.0181135 (2017).
Jespers, V. et al. The significance of Lactobacillus crispatus and L. vaginalis for vaginal health and the negative effect of recent sex: a cross-sectional descriptive study across groups of African women. BMC Infect. Dis. 15, 115. https://doi.org/10.1186/s12879-015-0825-z (2015).
McCarthy, F.N.-A., Nii-Trebi, N. I., Musah, B. O. & Asmah, R. H. Intravaginal practices and lactobacilli colonization among women in Accra, Ghana. BMC Women’s Health 15, 46. https://doi.org/10.1186/s12905-015-0205-2 (2015).
Low, N. et al. Intravaginal practices, bacterial vaginosis, and HIV infection in women: Individual participant data meta-analysis. PLoS Med. 8, e1000416. https://doi.org/10.1371/journal.pmed.1000416 (2011).
Delwiche, E. A., Pestka, J. J. & Tortorello, M. L. The veillonellae: Gram-negative cocci with a unique physiology. Annu. Rev. Microbiol. 39, 175–193. https://doi.org/10.1146/annurev.mi.39.100185.001135 (1985).
Han, M., Liu, G., Chen, Y., Wang, D. & Zhang, Y. Comparative genomics uncovers the genetic diversity and characters of veillonella atypica and provides insights into its potential applications. Front. Microbiol. 11, 1219. https://doi.org/10.3389/fmicb.2020.01219 (2020).
Vesth, T. et al. Veillonella, firmicutes: microbes disguised as gram negatives. Stand. Genomic. Sci. 9, 431–448. https://doi.org/10.4056/sigs.2981345 (2013).
Mashima, I. & Nakazawa, F. The interaction between Streptococcus spp. and Veillonella tobetsuensis in the early stages of oral biofilm formation. J. Bacteriol. 197, 2104–2111. https://doi.org/10.1128/jb.02512-14 (2015).
Tuominen, H., Rautava, S., Syrjänen, S., Collado, M. C. & Rautava, J. HPV infection and bacterial microbiota in the placenta, uterine cervix and oral mucosa. Sci. Rep. 8, 9787. https://doi.org/10.1038/s41598-018-27980-3 (2018).
van den Bogert, B., Meijerink, M., Zoetendal, E. G., Wells, J. M. & Kleerebezem, M. Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PLoS ONE 9, e114277. https://doi.org/10.1371/journal.pone.0114277 (2014).
Koshiol, J. et al. Persistent human papillomavirus infection and cervical neoplasia: A systematic review and meta-analysis. Am. J. Epidemiol. 168, 123–137. https://doi.org/10.1093/aje/kwn036 (2008).
Kjaer, S. K. et al. Type specific persistence of high risk human papillomavirus (HPV) as indicator of high grade cervical squamous intraepithelial lesions in young women: Population based prospective follow up study. BMJ 325, 572. https://doi.org/10.1136/bmj.325.7364.572 (2002).
Gage, J. C., Schiffman, M., Solomon, D., Wheeler, C. M. & Castle, P. E. Comparison of measurements of human papillomavirus persistence for postcolposcopic surveillance for cervical precancerous lesions. Cancer Epidemiol. Biomark. Prev. 19, 1668–1674. https://doi.org/10.1158/1055-9965.Epi-09-1286 (2010).
Marks, M. A., Castle, P. E., Schiffman, M. & Gravitt, P. E. Evaluation of any or type-specific persistence of high-risk human papillomavirus for detecting cervical precancer. J. Clin. Micriobiol. 50, 300–306. https://doi.org/10.1128/jcm.05979-11 (2012).
Hernán, M. A., Hernández-Díaz, S. & Robins, J. M. A structural approach to selection bias. Epidemiology 15, 615–625. https://doi.org/10.1097/01.ede.0000135174.63482.43 (2004).
Adebamowo, S. N. et al. Persisent low risk and high risk HPV Infections of the uterine cervix in HIV negative and HIV positive women. Front. Public Health 2, 2 (2017).
Dareng, E. O. et al. Age, HIV status, and research context determined attrition in a longitudinal cohort in Nigeria. J. Clin. Epidemiol. 100, 32–43. https://doi.org/10.1016/j.jclinepi.2018.04.012 (2018).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. https://doi.org/10.1038/ismej.2012.8 (2012).
Adebamowo, S. N. et al. Mycoplasma hominis and Mycoplasma genitalium in the Vaginal microbiota and persistent high-risk human papillomavirus infection. Front. Public Health 5, 140. https://doi.org/10.3389/fpubh.2017.00140 (2017).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. https://doi.org/10.1038/nmeth.f.303 (2010).
Magoc, T. & Salzberg, S. L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. https://doi.org/10.1093/bioinformatics/btr507 (2011).
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C. & Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. https://doi.org/10.1093/bioinformatics/btr381 (2011).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618. https://doi.org/10.1038/ismej.2011.139 (2012).
Geraets, D. T. et al. The original SPF LiPA algorithm is more sensitive and suitable for epidemiologic HPV research than the SPF INNO-LiPA Extra. J. Virol. Methods https://doi.org/10.1016/j.jviromet.2015.01.001 (2015).
Brusselaers, N., Shrestha, S., van de Wijgert, J. & Verstraelen, H. Vaginal dysbiosis and the risk of human papillomavirus and cervical cancer: Systematic review and meta-analysis. Am. J Obstet. Gynecol. 221, 9-18.e18. https://doi.org/10.1016/j.ajog.2018.12.011 (2019).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U.S.A. 108, 4680–4687. https://doi.org/10.1073/pnas.1002611107 (2010).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. https://doi.org/10.1186/gb-2011-12-6-r60 (2011).
Price, J. T. et al. Highly diverse anaerobe-predominant vaginal microbiota among HIV-infected pregnant women in Zambia. PLoS ONE 14, e0223128. https://doi.org/10.1371/journal.pone.0223128 (2019).
Oksanen, J. et al. in R CRAN package (2011).
Spedicato, G. A., Kang, T. S., Yalamanchi, S. B., Yadav, D. & Cordón, I. The markovchain package: a package for easily handling Discrete Markov Chains in R. Accessed Dec (2016).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544. https://doi.org/10.1093/nar/gky379 (2018).
Acknowledgements
The authors would like to thank all the participants of the study and research assistants that helped with data collection. This work was supported by the UM-Capacity Development for Research in AIDS Associated Malignancy Grant (NIH/NCI 1D43CA153792-01); African Collaborative Center for Microbiome and Genomics Research Grant (NIH/NHGRI 1U54HG006947) from the the Office Of The Director, National Institutes Of Health (OD) and the National Human Genome Research Institute (NHGRI); and funds through the Maryland Department of Health's Cigarette Restitution Fund Program. We thank other members of the ACCME group and the H3Africa Consortium for their contribution to this study.
Author information
Authors and Affiliations
Contributions
E.O.D., S.N.A., A.F., C.A.A. contributed to study design, implementation and data collection; E.O.D., B.M. analysed the data and generated figures and tables; E.O.D., B.M., S.N.A., J.R., P.P.P. and C.A.A. contributed to data interpretation; E.O.D. drafted the manuscript. C.A.A. conceived the project and obtained funding for the project. All authors critically reviewed the manuscript and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dareng, E.O., Ma, B., Adebamowo, S.N. et al. Vaginal microbiota diversity and paucity of Lactobacillus species are associated with persistent hrHPV infection in HIV negative but not in HIV positive women. Sci Rep 10, 19095 (2020). https://doi.org/10.1038/s41598-020-76003-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76003-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
