
Abstract
Layered Delafossite-type Lix(M1M2M3M4M5…Mn)O2 materials, a new class of high-entropy oxides, were synthesized by nebulized spray pyrolysis and subsequent high-temperature annealing. Various metal species (M = Ni, Co, Mn, Al, Fe, Zn, Cr, Ti, Zr, Cu) could be incorporated into this structure type, and in most cases, single-phase oxides were obtained. Delafossite structures are well known and the related materials are used in different fields of application, especially in electrochemical energy storage (e.g., LiNixCoyMnzO2 [NCM]). The transfer of the high-entropy concept to this type of materials and the successful structural replication enabled the preparation of novel compounds with unprecedented properties. Here, we report on the characterization of a series of Delafossite-type high-entropy oxides by means of TEM, SEM, XPS, ICP-OES, Mössbauer spectroscopy, XRD including Rietveld refinement analysis, SAED and STEM mapping and discuss about the role of entropy stabilization. Our experimental data indicate the formation of uniform solid-solution structures with some Li/M mixing.
Similar content being viewed by others


Introduction
Recently, the utilization of the high-entropy concept for oxide materials has been enjoying rising popularity1,2,3,4,5,6,7,8. High-entropy materials can be seen as compounds comprising several different elements in a single-phase structure and according to the Gibbs–Helmholtz equation, the resulting (large) configurational entropy may have structure stabilizing effects9,10,11,12,13,14. Additionally, the materials can be tailored with regard to their elemental composition, which leads to property changes due to the interactions between the different elements. The configurational entropy is solely based on the number of different elements incorporated in a single-phase structure (e.g., different species on the cationic sublattice) and can be calculated using a Boltzmann-entropy-derived equation (Gibbs entropy, Eq. 1).
Equation 1: Calculation of configurational entropy.
In this formula, Sconfig is the configurational entropy, R the ideal gas constant and xi and xj represent the molar fraction of cations or anions on the respective sublattice. Regarding configurational entropy, an empirical value around 1.5R has been found, above which entropy-related effects appear and supposedly affect the material properties to various extents15. Although the term “high-entropy material” is formally used for every compound having a configurational entropy of > 1.5R, “entropy stabilization” can only be observed in certain high-entropy materials. A strong indication of entropy stabilization is the reversible mixing and demixing of phases upon increasing and decreasing temperature, respectively. This behavior was first reported by Rost et al. and can be used to classify materials into “high-entropy” and “entropy-stabilized” compounds9,16.
In this work, we report on a new class of high-entropy materials, namely LixMO2 (M = metals), comprising five different equimolar and homogenously distributed cations. LixMO2 materials crystallizing in the R − 3 m space group (Delafossite α-NaFeO2-type structure) are known especially for their capability to reversibly intercalate Li+ ions. This characteristic made them become the most prominent cathode materials for Li-ion battery applications, e.g., LiCoO2 (LCO) and LiNixCoyMnzO2 (NCM), to name a few17,18,19,20. However, other areas are also in the focus of both academia and industry. For example, Delafossite-type materials can be used as semiconductors in solar cells, as catalysts for the photoelectrochemical reduction of CO2 as well as for thermoelectric and lighting applications21,22,23,24,25,26.
The layered high-entropy oxides (referred to as L-HEOs) were prepared by nebulized spray pyrolysis (NSP) and subsequent high-temperature annealing. Li precursor was used in excess in the synthesis to compensate for the Li (or Li2O) loss during heating. Specifically, five different compounds were successfully produced, namely, Li(NiCoMnAlZn)1O2, Li(NiCoMnAlFe)1O2 and Li0.8Na0.2(NiCoMnAlFe)1O2, with the metal ions present in equimolar proportions, and Li(NiCoMnMgAlCrTiZrCu)1O2 and Li(NiCoMnMgAlCrTiVCu)1O2 as non-equimolar compounds. The materials were characterized in detail, especially regarding the formation of solid-solution phases. Note that non-uniform elemental distribution would decrease the configurational entropy and therefore negatively affect the entropy stabilization. Possible entropic effects and the application of Hume Rothery’s second rules for solid solutions are discussed near the end of the manuscript for better understanding of the high-entropy concept27.
Results and discussion
The L-HEO compounds were synthesized using a stepwise approach. In order to compare the structure and the structural evolution when incorporating more elements, two materials were prepared first using the same synthesis procedure. Then, additional elements were added in a stepwise manner and the resulting materials were characterized. All materials contain the metals in equimolar ratios. The study started from a typical NCM cathode material used for Li-ion battery applications, namely Li(NiCoMn)1O2 (see Fig. 1a). Then, Al was added, forming Li(NiCoMnAl)1O2, before entering into the high-entropy region with 5 or more different elements on the same cationic sublattice.

(a) XRD patterns for the different compounds, with secondary phases denoted by asterisks and number signs. (b) Crystal structure of α-NaFeO2 (drawing produced by Vesta 3, https://jp-minerals.org/). Green spheres represent lithium, red spheres oxygen and multi-colored spheres the positions of the statistically distributed metal cations. (c) Lattice parameters from Rietveld refinement analysis (color scheme as in panel a).
All samples showed a well-developed layered structure of α-NaFeO2 type (space group R − 3 m, Z = 3, Fig. 1b), which can be seen from the clear splitting of the 018 and 110 reflections in the X-ray diffraction (XRD) patterns in Fig. 1a28. Alternating layering of metal and lithium layers occurred in all cases, with the metals and Li occupying the octahedral sites in the cubic close-packed oxygen lattice. The metals statistically occupy the Wyckoff 3b and Li the 3a sites. A possible intermixing was considered using Rietveld refinement analysis. The refined patterns are shown in the Supporting Information (Figure S1). Upon addition of Al to Li(NiCoMn)1O2, the reflections shifted to larger 2θ values, thus indicating a smaller unit cell. This result coincides with the smaller ionic radius of Al3+ (0.54 Å) than low-spin Ni2+ (0.69 Å), Ni3+ (0.56 Å) and Co3+ (0.55 Å) (the ionic radius of Mn4+ is 0.53 Å)29. This is also evident from the decreasing a-axis parameter (Fig. 1c), from 2.865(1) Å for Li(NiCoMn)1O2 to 2.845(1) Å for Li(NiCoMnAl)O2. Note that the a-axis is strongly affected by the ionic radius of the respective metals30. Because V = a2 · c · (sin 60°), there is also a strong decrease in unit-cell volume, from 101.333(9) Å3 for Li(NiCoMn)1O2 to 99.97(9) Å3 for Li(NiCoMnAl)1O2.
By adding Zn, an increase in a-axis parameter and unit-cell volume as well as formation of a secondary phase were observed (denoted by an asterisk in Fig. 1a; reflections indicated with number signs could not be assigned to a specific phase). The latter phase can most probably be assigned to a spinel-type structure, similar to ZnAl2O4. Note that spinels are known to form high-entropy structures when suitable ions are incorporated31,32. Therefore, a large number of possible compositions is conceivable (i.e., Zn(M1M2M3M4M5)2O4, with M = Zn, Fe, Co, Ni and/or Mn). In addition, Zn could be replaced by one or more (different) metal ions. Rietveld refinement assuming the presence of spinel revealed a ZnAl2O4 content of 9–10%. This value has to be taken with care, though, since we doubt that the reflection stems from “pure” ZnAl2O4. To further support our hypothesis, HR-TEM and SAED measurements were performed, which also indicated the presence of a distorted ZnAl2O4-type spinel structure (Figure S2).
Moreover, the addition of Zn led to an increase in the transition metal to oxygen distance, thereby corroborating its incorporation into the lattice. However, the position of the 104 reflection changed only slightly compared to that of Li(NiCoMnAl)1O2. Hence, it seems that Zn was only partially incorporated. This result emphasizes the importance of choosing suitable elements to maintain a single-phase structure.
Substituting Fe for Zn in Li(NiCoMnAlZn)1O2 led to an increase in lattice parameters, which again indicates successful incorporation into the crystal structure. In addition, the material was single phase, with the reflections shifted toward smaller angles. It is worth mentioning that the a-axis lattice parameter value for Li(NiCoMnAlFe)1O2 is similar to that of Li(NiCoMn)1O2. Based on the ionic radii, the unit cell was expected to increase.
The partial replacement of Li+ with Na+ (here, LiNa(NiCoMnAlFe)1O2), which has a considerably larger ionic radius (0.76 Å vs 1.02 Å), had a direct effect on the lattice parameters (increase in a, c and V, see Fig. 1c). For all L-HEOs, some Li/M cation mixing was apparent (see Table 1 and Fig. 2). Especially the Al-containing materials showed a relatively higher degree of mixing, with the exception of LiNa(NiCoMnAlFe)1O2 (most likely due to the presence of Na+).

Lithium, metal and oxygen position in the Delafossite structure, as explained in Table 1 (drawing produced by Vesta 3, https://jp-minerals.org/).
The composition of the different materials was examined by inductively coupled plasma-optical emission spectroscopy (ICP-OES). The corresponding results are shown in Fig. 3 and Figure S3, supporting the assumption that the metal ions are present in mostly equimolar proportions. Note that the data were normalized to an oxygen content of 2.

Chemical composition of (a) Li(NiCoMnAlZn)1O2 and (b) Li(NiCoMnAlFe)1O2 from ICP-OES analysis. The values are given in at%.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) measurements were performed to gain more insights into the structure and the elemental distribution. Figure S4 shows SEM micrographs revealing particle sizes around 1 µm or below. These particles were agglomerated into larger aggregates. Figure 4a-c shows high-resolution TEM (HR-TEM) micrographs and selected-area electron diffraction (SAED) patterns for the different L-HEOs. The particles appear to be highly crystalline, irrespective of the composition and, for Li(NiCoMnAlZn)1O2, where a secondary phase was observed by XRD, a single-phase particle was probed. The Debye–Scherrer rings match the XRD data shown in Fig. 1a. The (003) lattice planes were also clearly detected by HR-TEM. Overall, the electron microscopy results confirm the layered structure of L-HEOs as well as the incorporation of elements into the LixMO2 lattice.

TEM of (a) Li(NiCoMnAlZn)1O2, (b) Li(NiCoMnAlFe)1O2 and (c) LiNa(NiCoMnAlFe)1O2. The HR-TEM micrographs emphasize the high degree of crystallinity and the SAED patterns corroborate the XRD results. (d) High-resolution EDX mapping of LiNa(NiCoMnAlFe)1O2, indicating uniform elemental distribution on the respective scale.
Figure 4d presents results from energy dispersive X-ray spectroscopy (EDX) analysis of LiNa(NiCoMnAlFe)1O2. The mapping indicates uniform distribution of all elements on the nanometer level. No agglomeration or segregation of single elements or secondary phases were observed. This means that the largest possible configurational entropy can be expected. Assuming even distribution of elements and equal occupation probability over all sites in the structure, the configurational entropy was calculated to be 1.75R (1.5R for the L-HEOs without Na, see entropy calculation in the Supporting Information). Table S1 lists the calculated configurational entropy of all compounds.
To probe the distribution of elements over a larger sample area and to exclude clustering of elements, SEM/EDX measurements were performed. Figure S5 shows elemental heat maps obtained on a µm-size agglomerate of LiNa(NiCoMnAlFe)1O2 particles, corroborating the TEM results.
LiNa(NiCoMnAlFe)1O2 was further used to study the chemical environment and oxidation state of Fe by Mössbauer spectroscopy. This particular material was chosen because it contains the largest number of different elements, including Fe needed for Mössbauer spectroscopy. The spectrum shown in Fig. 5 was fitted by a quadrupole doublet with an average quadrupole splitting of 0.492 mm/s. The isomer shift (0.34 mm/s) suggests the presence of Fe3+. No other components were found, which would be indicative of different oxidation states and/or magnetic interactions and therefore inhomogeneities. Overall, the Mössbauer data demonstrate that the Fe3+ ions have a uniform chemical environment in the entire sample (approx. 150 mg) and, combined with the EDX results, they provide evidence of homogenous ion distribution and solid-solution state in the LixMO2 lattice.

Mössbauer spectroscopy of LiNa(NiCoMnAlFe)1O2.
X-ray photoelectron spectroscopy (XPS) was used to determine the oxidation state(s) of metals on the surface of the L-HEOs. In Fig. 6, the multiplet deconvolution of the main Ni 2p3/2 peaks of Li(NiCoMnAlFe)1O2 and LiNa(NiCoFeMnAl)1O2 are shown in magenta and their pronounced Ni 2p satellite in cyan. According to the presented deconvolution (based on our previous publications on NCM materials), Ni ions were identified as Ni2+33,34. The Co ions in both Li(NiCoFeMnAl)1O2 and LiNa(NiCoMnAlFe)1O2 showed a minor contribution of Co2+, according to the low intensity peak detected around 785.5 eV that is considered a characteristic satellite of Co2+ ions (shown in blue in Fig. 6)33,34. The majority of Co ions were identified as Co3+ (shown in dark yellow). The ratio of Co2+/Cototal was determined as 30% and 24% for Li(NiCoMnAlFe)1O2 and LiNa(NiCoMnAlFe)1O2, respectively.

XPS of Li(NiCoMnAlFe)1O2 and LiNa(NiCoMnAlFe)1O2. The Ni, Co, Mn and Fe detailed spectra are shown.
The Mn 2p3/2 spectra could be deconvoluted with a Mn4+ multiplet structure (green color) and including the Ni LMM Auger contribution (in dark brown color)33,34. This deconvolution is the base of assignment of the 4+ state to Mn ions in L-HEOs. The use of Mn 3p and Mn 3s spectra for identification of the Mn oxidation state was hampered due to overlap of these peaks with Fe 3p and Li 1s and Fe 3s (see also Figure S6). The overlapping Ni LMM and Co LMM Auger peaks increased the background of the Fe 2p spectra and added to the difficulty of deconvoluting the spectra for oxidation state identification. However, according to the presented multiplet splitting (deconvolution of the Fe 2p3/2 spectra and the specified satellite structure, shown in blue), the Fe ions were identified mainly as Fe3+ and the contribution of the Fe2+ state was negligible (less than 8% of total Fe ions, explainable for surfaces under air and fitting difficulties because of overlapping peaks)35,36. The Al 2p spectra of L-HEOs overlapped with the Ni 3p spectra (the Al 2s also overlapped with Ni 3s). Our attempt to properly deconvolute the Al 2p spectra resulted in the peak fitting shown in Figure S6. The binding energy of Al 2p3/2 was 72.8 and 74 eV for Li(NiCoMnAlFe)1O2 and Li0.8Na0.2(NiCoMnAlFe)1O2, respectively. The differences in the binding energy of Al can be explained by the diverging cation mixing of the respective materials. Additionally, the very oxophilic nature of Al might result in the formation of oxide species on the surface, which are a bit different, depending on the composition of the layered structure37.
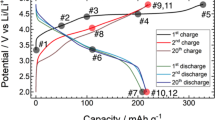
Recently, Zhao et al.38 showed for Na-ion batteries that the high-entropy concept is beneficial for achieving stable cell capacities. Because NCM-type materials are common cathodes for Li-ion batteries, in the present work, the L-HEO structures were tested regarding the possibility to reversibly intercalate Li+ ions (Fig. 7). For comparison, Li(NiCoMn)1O2 prepared under the same conditions served as a kind of reference cathode material. Most of the materials showed reasonable redox activity, but the specific capacities achieved are not yet satisfactory (Figure S7), probably because of Li/M cation mixing (note that the morphology has not yet been optimized), blocking pathways for Li transport. Figure 7a-d shows cyclic voltammetric curves at a sweep rate of 0.1 mV/s. The Li(NiCoMn)1O2 data revealed an oxidation peak around 3.8 V, in agreement with literature. The potential increased to 4.0 V for Li(NiCoMnAlZn)1O2. However, when substituting Fe3+ for Zn2+ [Li(NiCoMnAlFe)1O2], it increased even further to 4.1 V, despite the fact that the Al3+ content remained unchanged. Although this material did not show high reversibility, the increase in oxidation potential clearly demonstrates that either the incorporated elements or the configurational entropy or both exert a strong effect on the electrochemical behavior. Interestingly, when some of the Li was replaced with Na [LiNa(NiCoMnAlFe)1O2], both the specific capacity increased by a factor of more than two (Figure S7) and the reversibility improved considerably compared to Li(NiCoMnAlFe)1O2. The oxidation potential dropped by only 100 mV to 4.0 V, which seems promising for the development of high-energy-density cathode materials. Figure 7e,f shows the voltage profiles of cells using Li(NiCoMn)1O2 and LiNa(NiCoMnAlFe)O2 over the first 20 cycles, corroborating the cyclic voltammetry results.

Cyclic voltammetry and charge/discharge profiles of as prepared (a) Li(NiCoMn)1O2, (b) Li(NiCoMnAlZn)1O2, (c) Li(NiCoMnAlFe)1O2 and (d) LiNa(NiCoMnAlFe)1O2. The charge and discharge profiles for Li(NiCoMn)1O2 and LiNa(NiCoMnAlFe)1O2 are shown in (e) and (f), respectively.
In order to learn about the structural stability upon electrochemical cycling, the L-HEO materials were probed using XRD before and after 20 cycles (Fig. 8a–c). The layered structure was clearly preserved for all of the materials, thus indicating good stability with cycling. Only for Li(NiCoMnAlZn)1O2, a small reflection appeared around 23°. What is more striking is the change in intensity ratio between the 003 and 104 reflections. If the reflections do show about the same intensity, the cation mixing is low. For cycled Li(NiCoMnAlFe)1O2 and LiNa(NiCoMnAlFe)1O2, the 003 reflection showed decreased intensity, suggesting increased cation mixing, which in turn may help explain the capacity fading shown in Figs. 7 and S7.

XRD patterns of (a) Li(NiCoMnAlZn)1O2, (b) Li(NiCoMnAlFe)1O2 and (c) LiNa(NiCoMnAlFe)1O2 recorded before and after electrochemical cycling.
Taken together, the data in Figs. 7 and 8 demonstrate the versatility of the high-entropy approach—small changes in composition strongly affect the electrochemical characteristics, including cycling performance and stability. The reason for the enhanced performance of LiNa(NiCoMnAlFe)1O2 is unclear at this time, but it may well be that the larger Na+ ions somewhat widen the diffusion channels that are blocked otherwise due to cation disorder39. As somewhat expected, the L-HEO material with the lowest degree of cation mixing showed the best cyclability and delivered the largest specific capacities. This suggests that, in principle, it should be possible to design next-generation L-HEOs for battery applications38,40,41. Nevertheless, the issue of cation mixing remains to be resolved and the specific capacities need to be increased42,43,44. Overall, the impact of entropy stabilization on the cycling performance is largely unclear and is an interesting question for future studies. Common (non-HEO) cathode intercalation materials can only be delithiated to a certain extent to prevent irreversible structural changes45,46. This reduces the amount of usable Li and therefore limits the specific capacity. Overall, it would be exciting to see if L-HEO materials with a large entropy stabilization allow for higher degrees of delithiation by more effectively stabilizing the layered structure.
Making statements about possible entropy stabilization in the layered structures is difficult. Experiments to elucidate any potential entropy stabilization, like the reversible phase separation at low cooling rates, as shown by Rost et al. and others, are impeded by the Li (or Li2O) evaporation at high temperatures. This loss can lead to phase changes, as known for NCM cathode materials (the layered structure transforms into spinel and/or rock salt at high states of charge/low lithium content). Moreover, some materials are only stable because the Li+ ions compensate higher valence charges, which would break the charge neutrality of the structure, thus leading to phase separation. Nevertheless, experiments to determine the reaction(s) of the compounds when heated to a lower temperature and cooled down slowly were performed. Specifically, the L-HEO materials were heated to 750 °C for 3 h to trigger (phase) demixing and then cooled down slowly. Afterwards, the materials were heated to 900 °C for 3 h to allow the phases to mix again. Figure 9a shows the results for Li(NiCoMn)1O2, where no differences in the XRD patterns were observed. Li(NiCoMnAl)1O2 also did not show any effect of temperature and/or Li loss on the structure (Fig. 9b). These materials exhibit a configurational entropy below the 1.5R threshold and therefore do not fall under the description of high-entropy materials. The configurational entropy is too small to compensate for any mixing enthalpy penalties, so the structure itself seems to be stable (without entropy stabilization). Hence, it is not surprising that the temperature does not affect the structure. Figure 9c shows the corresponding XRD patterns for Li(NiCoMnAlZn)1O2. Upon heating to 750 °C, additional reflections appeared, which can be assigned to a distorted ZnO (Wurtzite) phase. The latter reflections did not disappear but rather grew in intensity when heating the material to 900 °C. We attribute this to demixing at 750 °C and the Li loss at 900 °C, leading to phase separation of the original material. As expected, the reflections of Li(NiCoMnAlZn)1O2 also shifted slightly toward smaller lattice parameters. Moreover, the intensity ratio between the 003 and 104 reflections changed, indicating stronger cation mixing. For Li(NiCoMnAlFe)1O2, despite being a high-entropy material, again no differences in the XRD patterns were observed (Fig. 9d). Perhaps, this is due to a significantly lower enthalpy of mixing. When Zn2+ is replaced with Fe3+, the average charge is changing toward the ideal one in the α-NaFeO2 type structure. In addition, the ionic radius of Fe3+ is closer to that of Co3+ and Ni3+, all of which probably reduces the enthalpy of mixing, so that entropy stabilization is not “required” anymore (similar to both Li(NiCoMn)1O2 and Li(NiCoMnAl)1O2). For LiNa(NiCoMnAlFe)O2, the reflections decreased in intensity and broadened significantly (Fig. 9e), indicative of major structural distortions. We attribute this to the introduction of Na and its properties. In this case, the enthalpy of mixing is clearly increased, due to the different ionic radii and electronegativities.

Heating tests of (a) Li(NiCoMn)1O2, (b) Li(NiCoMnAl)1O2, (c) Li(NiCoMnAlZn)1O2, (d) Li(NiCoMnAlFe)1O2 and (e) LiNa(NiCoMnAlFe)1O2.
The question if “only” a high-entropy or an entropy stabilization is apparent can be discussed in part using the Hume-Rothery’s second rule for solid-solution formation45. The latter can be understood as a description under which precondition elements can form a solid solution. The rule states that the ionic radius of the incorporated elements should not differ by more than about 15% and that the crystal structures, valences and electronegativities should be similar. The type of crystal structure of the L-HEO materials synthesized here is Delafossite α-NaFeO2. Many of the individual elements may show comparable stable structures in the respective lattice type (α-LiAlO2, LiCoO2, LiNiO2, LiFeO2, except for Mn and Zn). Therefore, the first precondition seems to be fulfilled for 4 out of 6 of the tested elements. Other preconditions are similar ionic radii and electronegativities. Table 2 shows the respective values. As is evident, these characteristics are similar for the trivalent transition metal ions. It is known that in NCM materials, Mn is present in the oxidation state 4+ , thereby partially reducing Ni to 2+ . The size of Mn4+ is also similar, which helps explain its successful incorporation into a single-phase structure. The radius of Zn2+ differs quite significantly and is more similar in radius to Li+. The discrepancy in Zn2+ radius and the differences in structure as explained above seem to increase the mixing enthalpy too much, such that the entropy cannot compensate it and therefore no single-phase material is formed. Nevertheless, XRD and TEM indicate that some of the Zn2+ ions are incorporated into the material, so entropy stabilization and the formation of a single-phase structure may be apparent until a certain threshold concentration in the structure is reached. Formation of a single phase at higher temperatures may be possible, but the accompanying Li loss prevents such studies. Despite its even larger radius and discrepancy in valence and electronegativity, Li+ is successfully incorporated, since it is an integral part of the layered R − 3 m structure.
In summary, application of the Hume-Rothery rules to these systems indicates good miscibility of elements for Li(NiCoMnAlFe)1O2 and LiNa(NiCoMnAlFe)1O2. In these systems, high-entropy is clearly apparent, but entropy stabilization might be questionable. The Li(NiCoMnAlZn)1O2 material showed the presence of side phases, most probably due to the different characteristics of Zn2+. Therefore, the stabilizing role of entropy remains elusive for these layered materials and the term “high-entropy” (referring to high configurational entropy) rather than “entropy stabilized” has been applied in the present work16.
Two more compositions are presented to emphasize the versatility of the high-entropy concept as well as to demonstrate how sensitive this system is toward small changes in stoichiometry. As can be seen in Fig. 10, the layered structure has clearly formed. However, upon slightly altering the Ni, Mn, Cr and Ti concentrations, a secondary phase appeared (see minor reflections between 20 and 25° 2θ, most probably lithiated V2O5 compounds, like Li0.3V2O5 or Li0.6V2O5), probably because the average valence of cations changed to some extent. This is in accordance with the Hume-Rothery’s second rule, which states that the valences should be similar. Additionally, charge neutrality has to be maintained, which forces the compound to form vacancies or to undergo structural changes. The same behavior can be assumed when Zn2+ is incorporated instead of Fe3+, as shown before. This may be a possible explanation for the multiphase nature of Li(NiCoMnAlZn)1O2. The results from heating experiments are shown in Figure S8, indicating strong cation mixing upon subsequent heating.

XRD patterns of Li(Ni0.47Co0.2Mn0.18Mg0.04Al0.04Cr0.02Ti0.02V0.015Cu0.015)O2 (in red) and Li(Ni0.46Co0.2Mn0.2Mg0.04Al0.04Cr0.015Ti0.015Zr0.015Cu0.015)O2 (in black). The inset shows the additional reflections arising from the slight change in cation stoichiometry.
In summary, a new class of high-entropy oxide materials, namely layered LixMO2, has been synthesized. The incorporated elements were found to be virtually equimolar and, more importantly, uniformly distributed on the nanometer and micrometer levels. The calculated configurational entropy amounted up to 1.75R, rendering these compounds high-entropy materials. The question if these materials are entropy-stabilized cannot be clearly answered due to Li evaporation at elevated temperatures. Moreover, reversible Li+ intercalation/extraction into/from the L-HEOs was demonstrated, but the specific capacities achieved were relatively low (when compared to state-of-the-art cathode materials). Nevertheless, given the broad application area of layered materials and the benefits arising from the high-entropy approach (e.g., versatility in composition), L-HEOs may pave the way for new technologies.
Methods
Synthesis
The compounds were synthesized by NSP (see Figure S9 and details in the Supporting Information). First, nitrate salts of the respective metals (LiNO3, Co(NO3)2·6H2O, Ni(NO3)2·6H2O, Mn(NO3)2·4H2O, Zn(NO3)2·6H2O, Fe(NO)3·9H2O, Al(NO3)3·6H2O, NaNO3; from Sigma-Aldrich) were dissolved in 200 mL of H2O to form a 0.1 M solution. 10 wt% excess LiNO3 was used to compensate for the Li (or Li2O) evaporation during heating. The resultant clear solution was sprayed as a mist and transported by means of a carrier gas into the hot zone of a tubular furnace (800 °C). The as-made powder was quenched and collected. After the NSP synthesis process, all collected powder was further calcined at 900 °C for 12 h using a heating rate of 5 °C/min and then cooled down. Li(Ni0.47Co0.2Mn0.18Mg0.04Al0.04Cr0.02Ti0.02V0.015Cu0.015)O2 and Li(Ni0.46Co0.2Mn0.2Mg0.04Al0.04Cr0.015Ti0.015Zr0.015Cu0.015)O2 were prepared by solid-state synthesis. Stoichiometric amounts of LiNO3, NiO, Co3O4, MnO, MgO, Cr(NO3)3·9H2O, Al2O3, V2O5, CuO, TiO2 and ZrC20H28O8 were thoroughly mixed by ball milling (300 rpm) for 10 h with ethanol. After drying, the mixed precursors were annealed at 900 °C for 12 h.
Electrode Processing
For the cathode preparation, 7:2:1 by weight of active material, Super C65 carbon black additive (Timcal) and PVDF binder (Solef 5130) were used. The different constituents were uniformly dispersed into N-Methyl-2-pyrrolidone (NMP) using a planetary mixer (Thinky). Next, the slurry was cast onto an Al current collector and dried in a vacuum at 80 °C for 12 h. Circular electrodes of 13 mm diameter were cut from the cathode sheet, assembled inside an Ar-filled glovebox and tested using CR2032 coin cells. 1 M LiPF6 in 3:7 by weight of ethylene carbonate (EC) and ethyl methyl carbonate (EMC; Selectilyte LP57, BASF SE), GF/C glass microfiber filter paper (Whatman) and Li metal foil (Gelon LIB Co., Ltd) were used as electrolyte, separator and counter electrode, respectively. The electrode active material loading was around 1 mg/cm2. The specific capacity was calculated on the basis of the active material mass.
Instrumentation
TEM measurements were conducted on powder samples dispersed on a lacey carbon-coated copper grid. The samples were loaded onto an FEI double tilt holder. TEM, SAED and STEM-EDX data were collected using an FEI Titan 80-300 microscope equipped with a CEOS image spherical aberration corrector, a HAADF STEM detector (Fischione model 3000), EDAX SUTW EDX detector and a Tridiem Gatan image filter. The microscope was operated at an accelerating voltage of 300 kV.
XPS measurements were performed on a K-Alpha+ instrument (Thermo Fisher Scientific) with a monochromatic Al-Kα X-ray source (1486.6 eV) and 400 μm spot size. The K-Alpha + charge compensation system was applied to prevent localized charge buildup during analysis using 8 eV electrons and low-energy Ar ions. Data acquisition and processing were carried out using the Thermo Avantage software49. The spectra were fitted with one or more Voigt profiles. The binding energies are given with respect to that of the C 1 s peak of hydrocarbons at 285.0 eV. The analyzer transmission function, Scofield sensitivity factors and effective attenuation lengths (EALs) for photoelectrons were applied for quantification50. EALs were calculated using the standard TPP-2 M formalism51. The multiplet splitting effect of Ni ions and the difficulty of identifying their oxidation state solely based on the peak barycenter have already been reported36,52,53.
SEM was performed on a ZEISS Gemini Leo 1530, equipped with an Oxford EDX detector.
XRD measurements were conducted on powder samples using either a STOE Stadi P diffractometer equipped with a Ga-jet X-ray source (Ga-Kβ radiation) or a Bruker D8 Advance diffractometer with a Cu-Kα radiation source and a LYNXEYE detector of fixed divergence slit (0.3°). Refinement of XRD patterns was performed using TOPAS Academics V5 software. Strain-free Al2O3 in a glassy matrix (particle size > 2 µm, random orientation) served as calibration sample to determine the instrumental resolution. The latter calibration sample is commercially available and was received from NIST. A linear interpolation function with 36 parameters was used for background refinement.
For ICP-OES spectroscopy (iCAP 7600 ICP-OES DUO; Thermo Fisher Scientific), an amount of about 10 mg of the samples (weighing accuracy ± 0.05 mg) was dissolved in 6 ml of hydrochloric acid, 2 ml of nitric acid and 4 ml of sulfuric acid at 513 K for 35 min in a microwave oven (Speedwave Xpert from Berghof). The analysis of elements was accomplished with 4 different calibration solutions and an internal standard (Sc). Two or three major wavelengths of elements were used for the calculation. The oxygen content was determined by the missing weight and compared to the oxygen content analyzed using the method of carrier gas hot extraction (CGHE). To this end, a commercial oxygen/nitrogen analyzer TC600 (LECO) was used. The differences were in the accuracy range of the CGHE method. The accuracy of ICP is usually very high and on average around + /− 0.2–0.5% for the metals, depending on the mass fraction of the element. Therefore, we included all metals with a mass fraction below 0.5% in a combined segment called “other elements”.
57Fe Mössbauer spectroscopy was performed in transmission geometry using a spectrometer with a moving source of 57Co in an Rh matrix and a triangular velocity variation. The isomer shift is given relative to bcc-Fe at room temperature.
Galvanostatic charge/discharge measurements were performed using an Arbin battery test system (BT-2000) at room temperature.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding authors on reasonable request.
References
Wang, Q. et al. Multi-anionic and -cationic compounds: new high entropy materials for advanced Li-ion batteries. Energy Environ. Sci. 12, 2433–2442 (2019).
Sarkar, A. et al. High-entropy oxides: fundamental aspects and electrochemical properties. Adv. Mater. 31, 1806236 (2019).
Sarkar, A. et al. Nanocrystalline multicomponent entropy stabilised transition metal oxides. J. Eur. Ceram. Soc. 37, 747–754 (2017).
Djenadic, R. et al. Multicomponent equiatomic rare earth oxides. Mater. Res. Lett. 5, 102–109 (2017).
Bérardan, D., Franger, S., Dragoe, D., Meena, A. K. & Dragoe, N. Colossal dielectric constant in high entropy oxides. Phys. Status Solidi Rapid Res. Lett. 10, 328–333 (2016).
Bérardan, D., Franger, S., Meena, A. K. K. & Dragoe, N. Room temperature lithium superionic conductivity in high entropy oxides. J. Mater. Chem. A 4, 9536–9541 (2016).
Oses, C., Toher, C. & Curtarolo, S. High-entropy ceramics. Nat. Rev. Mater. 5, 295–309 (2020).
Zhang, Y. High-Entropy Materials (Springer, Singapore, 2019). https://doi.org/10.1007/978-981-13-8526-1
Rost, C. M. et al. Entropy-stabilized oxides. Nat. Commun. 6, 8485 (2015).
Sarker, P. et al. High-entropy high-hardness metal carbides discovered by entropy descriptors. Nat. Commun. 9, 4980 (2018).
Zhang, R. Z., Gucci, F., Zhu, H., Chen, K. & Reece, M. J. Data-driven design of ecofriendly thermoelectric high-entropy sulfides. Inorg. Chem. 57, 13027–13033 (2018).
Jin, T. et al. Mechanochemical-assisted synthesis of high-entropy metal nitride via a soft urea strategy. Adv. Mater. 30, 1707512 (2018).
Gild, J. et al. High-entropy metal diborides: a new class of high-entropy materials and a new type of ultrahigh temperature ceramics. Sci. Rep. 6, 37946 (2016).
Castle, E., Csanádi, T., Grasso, S., Dusza, J. & Reece, M. Processing and properties of high-entropy ultra-high temperature carbides. Sci. Rep. 8, 8609 (2018).
Murty, B. S., Yeh, J.-W. & Ranganathan, S. High-Entropy Alloys (Butterworth-Heinemann, Oxford, 2014).
Sarkar, A., Breitung, B. & Hahn, H. High entropy oxides: the role of entropy, enthalpy and synergy. Scr. Mater. 187, 43–48 (2020).
de Biasi, L. et al. Chemical, structural, and electronic aspects of formation and degradation behavior on different length scales of Ni-rich NCM and Li-rich HE-NCM cathode materials in Li-ion batteries. Adv. Mater. 31, 1900985 (2019).
Schipper, F. et al. Review—recent advances and remaining challenges for lithium ion battery cathodes. J. Electrochem. Soc. 164, A6220–A6228 (2017).
Myung, S. T. et al. Nickel-rich layered cathode materials for automotive lithium-ion batteries: achievements and perspectives. ACS Energy Lett. 2, 196–223 (2017).
Yabuuchi, N. & Ohzuku, T. Novel lithium insertion material of LiCo1/3Ni1/3Mn1/3O2 for advanced lithium-ion batteries. J. Power Sources 119–121, 171–174 (2003).
Gu, J. et al. Mg-doped CuFeO2 photocathodes for photoelectrochemical reduction of carbon dioxide. J. Phys. Chem. C 117, 12415–12422 (2013).
Sheets, W. C. et al. Silver delafossite oxides. Inorg. Chem. 47, 2696–2705 (2008).
Jin, Y. & Chumanov, G. Solution synthesis of pure 2H CuFeO 2 at low temperatures. RSC Adv. 6, 26392–26397 (2016).
Kang, U. et al. Photosynthesis of formate from CO2 and water at 1% energy efficiency via copper iron oxide catalysis. Energy Environ. Sci. 8, 2638–2643 (2015).
Prévot, M. S., Li, Y., Guijarro, N. & Sivula, K. Improving charge collection with delafossite photocathodes: a host–guest CuAlO2 /CuFeO2 approach. J. Mater. Chem. A 4, 3018–3026 (2016).
Prévot, M. S., Guijarro, N. & Sivula, K. Enhancing the performance of a robust sol-gel-processed p-type delafossite CuFeO2 photocathode for solar water reduction. Chemsuschem 8, 1359–1367 (2015).
Otto, F., Yang, Y., Bei, H. & George, E. P. Relative effects of enthalpy and entropy on the phase stability of equiatomic high-entropy alloys. Acta Mater. 61, 2628–2638 (2013).
Zhou, P. et al. Stable layered Ni-rich LiNi0.9Co0.07Al0.03O2 microspheres assembled with nanoparticles as high-performance cathode materials for lithium-ion batteries. J. Mater. Chem. A 5, 2724–2731 (2017).
Bianchini, M., Roca-Ayats, M., Hartmann, P., Brezesinski, T. & Janek, J. There and back again—the journey of LiNiO2 as a cathode active material. Angew. Chem. Int. Ed. 58, 10434–10458 (2019).
Kondrakov, A. O. et al. Anisotropic lattice strain and mechanical degradation of high- and low-nickel NCM cathode materials for Li-Ion batteries. J. Phys. Chem. C 121, 3286–3294 (2017).
Mao, A. et al. A new class of spinel high-entropy oxides with controllable magnetic properties. J. Magn. Magn. Mater. 497, 165884 (2020).
Dąbrowa, J. et al. Synthesis and microstructure of the (Co,Cr,Fe,Mn,Ni)3O4 high entropy oxide characterized by spinel structure. Mater. Lett. 216, 32–36 (2018).
Azmi, R., Masoumi, M., Ehrenberg, H., Trouillet, V. & Bruns, M. Surface analytical characterization of LiNi0.8-yMnyCo0.2O2 (0 ≤ y ≤ 0.4) compounds for lithium-ion battery electrodes. Surf. Interface Anal. 50, 1132–1137 (2018).
Azmi, R. et al. Surface analytical approaches to reliably characterize lithium ion battery electrodes. Surf. Interface Anal. 50, 43–51 (2018).
Grosvenor, A. P., Kobe, B. A., Biesinger, M. C. & McIntyre, N. S. Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds. Surf. Interface Anal. 36, 1564–1574 (2004).
Biesinger, M. C. et al. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe Co and Ni. Appl. Surf. Sci. 257, 2717–2730 (2011).
Dahéron, L. et al. Possible explanation for the efficiency of Al-based coatings on LiCoO2: surface properties of LiCo1–xAlxO2 solid solution. Chem. Mater. 21, 5607–5616 (2009).
Zhao, C., Ding, F., Lu, Y., Chen, L. & Hu, Y.-S. High-entropy layered oxide cathodes for sodium-ion batteries. Angew. Chem. Int. Ed. 59, 264–269 (2020).
Xue, Z. et al. Sodium doping to enhance electrochemical performance of overlithiated oxide cathode materials for Li-ion batteries via Li/Na ion-exchange method. ACS Appl. Mater. Interfaces 10, 27141–27149 (2018).
Sarkar, A. et al. High entropy oxides for reversible energy storage. Nat. Commun. 9, 3400 (2018).
Qiu, N. et al. A high entropy oxide (Mg0.2Co0.2Ni0.2Cu0.2Zn0.2O) with superior lithium storage performance. J. Alloys Compd. 777, 767–774 (2019).
Zhang, X. et al. Minimization of the cation mixing in Li1+x(NMC)1-xO2 as cathode material. J. Power Sources 195, 1292–1301 (2010).
Abdellahi, A., Urban, A., Dacek, S. & Ceder, G. The effect of cation disorder on the average Li intercalation voltage of transition-metal oxides. Chem. Mater. 28, 3659–3665 (2016).
Li, D., Sasaki, Y., Kobayakawa, K. & Sato, Y. Morphological, structural, and electrochemical characteristics of LiNi0.5Mn0.4M0.1O2 (M = Li, Mg, Co, Al). J. Power Sources 157, 488–493 (2006).
de Biasi, L. et al. Between scylla and charybdis: balancing among structural stability and energy density of layered NCM cathode materials for advanced lithium-ion batteries. J. Phys. Chem. C 121, 26163–26171 (2017).
Yin, S. C., Rho, Y. H., Swainson, I. & Nazar, L. F. X-ray/neutron diffraction and electrochemical studies of lithium De/Re-intercalation in Li1-xCo1/3Ni1/3Mn1/3O2 (x = 0 → 1). Chem. Mater. 18, 1901–1910 (2006).
Li, K. & Xue, D. Estimation of electronegativity values of elements in different valence states. J. Phys. Chem. A 110, 11332–11337 (2006).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 32, 751–767 (1976).
Parry, K. L. et al. ARXPS characterisation of plasma polymerised surface chemical gradients. Surf. Interface Anal. 38, 1497–1504 (2006).
Scofield, J. H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron. Spectros. Relat. Phenomena 8, 129–137 (1976).
Tanuma, S., Powell, C. J. & Penn, D. R. Calculations of electron inelastic mean free paths. IX. Data for 41 elemental solids over the 50 eV to 30 keV range. Surf. Interface Anal. 43, 689–713 (2011).
Biesinger, M. C., Payne, B. P., Lau, L. W. M., Gerson, A. & Smart, R. S. C. X-ray photoelectron spectroscopic chemical state quantification of mixed nickel metal, oxide and hydroxide systems. Surf. Interface Anal. 41, 324–332 (2009).
Grosvenor, A. P., Biesinger, M. C., Smart, R. S. C. & McIntyre, N. S. New interpretations of XPS spectra of nickel metal and oxides. Surf. Sci. 600, 1771–1779 (2006).
Acknowledgements
A.S. and H.H. acknowledge financial support from the Helmholtz Association and the Deutsche Forschungsgemeinschaft (HA 1344/43‐1). A.S., Q.W., D.S., B.B. and H.H. appreciate the support of EnABLES, a project funded by the European Union’s Horizon 2020 research and innovation program under grant agreement no. 730957. J.W, K.W., X.H. and Y.C. acknowledge financial support from the China Scholarship Council (CSC). The work was partly performed with the support of the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Q.W. and S.S.B. developed the material synthesis and supported the synthesis efforts. J.W. and Y.C. synthesized the materials, fabricated the electrodes and performed the electrochemical experiments and analysis. K.W. and X.H. conducted the HRTEM and EDX measurements. T.Be. conducted and D.S. supported the ICP-OES measurements. R.A. performed the XPS measurements. A.S. and R.K. performed the Mössbauer spectroscopy measurements. S.S. conducted the XRD experiments and performed the Rietveld refinement analysis. H.H., T.B. and B.B. supervised the synthesis and experiments, directed the project and co-wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, J., Cui, Y., Wang, Q. et al. Lithium containing layered high entropy oxide structures. Sci Rep 10, 18430 (2020). https://doi.org/10.1038/s41598-020-75134-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-75134-1
This article is cited by
-
Compositionally complex doping for zero-strain zero-cobalt layered cathodes
Nature (2022)
-
Novel P2-type layered medium-entropy ceramics oxide as cathode material for sodium-ion batteries
Journal of Advanced Ceramics (2022)
-
Deep eutectic solvent assisted facile synthesis of low-dimensional hierarchical porous high-entropy oxides
Nano Research (2022)
-
Operando acoustic emission monitoring of degradation processes in lithium-ion batteries with a high-entropy oxide anode
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
